Metabolomic Profiling of Cerebral Palsy Brain Tissue Reveals Novel Central Biomarkers and Biochemical Pathways Associated with the Disease: A Pilot Study

 ,
, 

Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Sample Preparation
4.3. Data Collection and Metabolic Profiling
4.4. Statistical Analysis
4.5. Metabolite Pathway Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blair, E.; Watson, L. Epidemiology of cerebral palsy. Semin. Fetal Neonatal Med. 2006, 11, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wimalasundera, N.; Stevenson, V.L. Cerebral palsy. Pract. Neurol. 2016, 16, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Hoon, A.H., Jr. Cerebral palsy. Neuromol. Med. 2006, 8, 435–450. [Google Scholar] [CrossRef]
- Drougia, A.; Giapros, V.; Krallis, N.; Theocharis, P.; Nikaki, A.; Tzoufi, M.; Andronikou, S. Incidence and risk factors for cerebral palsy in infants with perinatal problems: A 15-year review. Early Hum. Dev. 2007, 83, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.B.; Blair, E. Prenatal factors in singletons with cerebral palsy born at or near term. N. Engl. J. Med. 2015, 373, 946–953. [Google Scholar] [CrossRef]
- Boyle, A.K.; Rinaldi, S.F.; Norman, J.E.; Stock, S.J. Preterm birth: Inflammation, fetal injury and treatment strategies. J. Reprod. Immunol. 2017, 119, 62–66. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, A.H.; Thompson, S.C.; Gecz, J. Cerebral palsy: Causes, pathways, and the role of genetic variants. Am. J. Obstet. Gynecol. 2015, 213, 779–788. [Google Scholar] [CrossRef]
- Keogh, J.M.; Badawi, N. The origins of cerebral palsy. Curr. Opin. Neurol. 2006, 19, 129–134. [Google Scholar] [CrossRef]
- Tonni, G.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C. Pathology of perinatal brain damage: Background and oxidative stress markers. Arch. Gynecol. Obstet. 2014, 290, 13–20. [Google Scholar] [CrossRef]
- Korzeniewski, S.J.; Slaughter, J.; Lenski, M.; Haak, P.; Paneth, N. The complex aetiology of cerebral palsy. Nat. Rev. Neurol. 2018, 14, 528–543. [Google Scholar] [CrossRef]
- Spedding, M.; Gressens, P. Neurotrophins and cytokines in neuronal plasticity. Novartis Found. Symp. 2008, 289, 222–233; discussion 233–240. [Google Scholar] [PubMed]
- Marret, S.; Vanhulle, C.; Laquerriere, A. Pathophysiology of cerebral palsy. Handb. Clin. Neurol. 2013, 111, 169–176. [Google Scholar] [PubMed]
- Denihan, N.M.; Boylan, G.B.; Murray, D.M. Metabolomic profiling in perinatal asphyxia: A promising new field. Biomed. Res. Int. 2015, 2015, 254076. [Google Scholar] [CrossRef] [PubMed]
- Yli, B.M.; Kjellmer, I. Pathophysiology of foetal oxygenation and cell damage during labour. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 30, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Hoon, A.H., Jr.; Vasconcellos Faria, A. Pathogenesis, neuroimaging and management in children with cerebral palsy born preterm. Dev. Disabil. Res. Rev. 2010, 16, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, C.M.; Ozen, M.; Burd, I. Perinatal brain injury: Mechanisms, prevention, and outcomes. Clin. Perinatol. 2018, 45, 357–375. [Google Scholar] [CrossRef]
- Fahey, M.C.; Maclennan, A.H.; Kretzschmar, D.; Gecz, J.; Kruer, M.C. The genetic basis of cerebral palsy. Dev. Med. Child Neurol. 2017, 59, 462–469. [Google Scholar] [CrossRef]
- Pascal, A.; Govaert, P.; Oostra, A.; Naulaers, G.; Ortibus, E.; Van den Broeck, C. Neurodevelopmental outcome in very preterm and very-low-birthweight infants born over the past decade: A meta-analytic review. Dev. Med. Child Neurol. 2018, 60, 342–355. [Google Scholar] [CrossRef]
- Shankaran, S. Prevention, diagnosis, and treatment of cerebral palsy in near-term and term infants. Clin. Obstet. Gynecol. 2008, 51, 829–839. [Google Scholar] [CrossRef]
- Rizzotti, A.; Bas, J.; Cuestas, E. Efficacy and securyty of therapeutic hypothermia for hypoxic ischemic encephalopathy: A meta-analysis. Rev. Fac. Cienc. Med. 2010, 67, 15–23. [Google Scholar]
- Shepherd, E.; Salam, R.A.; Middleton, P.; Han, S.; Makrides, M.; McIntyre, S.; Badawi, N.; Crowther, C.A. Neonatal interventions for preventing cerebral palsy: An overview of cochrane systematic reviews. Cochrane Database Syst. Rev. 2018, 6, Cd012409. [Google Scholar] [CrossRef] [PubMed]
- Chollat, C.; Sentilhes, L.; Marret, S. Protection of brain development by antenatal magnesium sulphate for infants born preterm. Dev. Med. Child Neurol. 2019, 61, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Rey, N.L.; Yilmaz, A.; Kumar, P.; Madaj, Z.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Biochemical profiling of the brain and blood metabolome in a mouse model of prodromal parkinson’s disease reveal distinct metabolic profiles. J. Proteome Res. 2018, 17, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Chevallier, O.P.; Kumar, P.; Turkoglu, O.; Bahado-Singh, R.O. Metabolomic profiling of brain from infants who died from sudden infant death syndrome reveals novel predictive biomarkers. J. Perinatol. 2017, 37, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Geddes, T.; Han, B.; Bahado-Singh, R.O.; Wilson, G.D.; Imam, K.; Maddens, M.; Graham, S.F. Diagnostic biomarkers of alzheimer’s disease as identified in saliva using 1h nmr-based metabolomics. J. Alzheimers Dis. 2017, 58, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Holscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic profiling of bile acids in clinical and experimental samples of alzheimer’s disease. Metabolites 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Graham, S.F.; Turkoglu, O.; Beauchamp, K.; Bjorndahl, T.C.; Han, B.; Mandal, R.; Pantane, J.; Kowalenko, T.; Wishart, D.S.; et al. Identification of candidate biomarkers of brain damage in a mouse model of closed head injury: A metabolomic pilot study. Metabolomics 2016, 12, 42. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.; Bahado-Singh, R.O.; Robinson, A.; Mann, D.; Green, B.D. Novel metabolite biomarkers of huntington’s disease (hd) as detected by high resolution mass spectrometry. J. Proteome Res. 2016, 15, 1592–1601. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite panel predicts preclinical transition to clinical stages of alzheimer’s disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.A. Advanced neuroimaging and its role in predicting neurodevelopmental outcomes in very preterm infants. Semin. Perinatol. 2016, 40, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, E.; Byblow, W.D.; Feys, H.; Wenderoth, N. The corticospinal tract: A biomarker to categorize upper limb functional potential in unilateral cerebral palsy. Front. Pediatr. 2015, 3, 112. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Molloy, E.J. Biomarkers of multiorgan injury in neonatal encephalopathy. Biomark. Med. 2015, 9, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Shalak, L.F.; Perlman, J.M. Infection markers and early signs of neonatal encephalopathy in the term infant. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Londono, I.; Mallard, C.; Lodygensky, G.A. New means to assess neonatal inflammatory brain injury. J. Neuroinflamm. 2015, 12, 180. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Chevallier, O.P.; Kumar, P.; Türkoğlu, O.; Bahado-Singh, R.O. High resolution metabolomic analysis of asd human brain uncovers novel biomarkers of disease. Metabolomics 2016, 12, 62. [Google Scholar] [CrossRef]
- Graham, S.F.; Chevallier, O.P.; Roberts, D.; Hölscher, C.; Elliott, C.T.; Green, B.D. Investigation of the human brain metabolome to identify potential markers for early diagnosis and therapeutic targets of alzheimer’s disease. Anal. Chem. 2013, 85, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Holscher, C.; Green, B.D. Metabolic signatures of human alzheimer’s disease (ad): 1h nmr analysis of the polar metabolome of post-mortem brain tissue. Metabolomics 2014, 10, 744–753. [Google Scholar] [CrossRef]
- Graham, S.F.; Holscher, C.; McClean, P.; Elliott, C.T.; Green, B.D. 1 h nmr metabolomics investigation of an alzheimer’s disease (ad) mouse model pinpoints important biochemical disturbances in brain and plasma. Metabolomics 2013, 9, 974–983. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.K.; Bjorndahl, T.; Han, B.; Yilmaz, A.; Sherman, E.; Bahado-Singh, R.O.; Wishart, D.; Mann, D.; Green, B.D. Metabolic signatures of huntington’s disease (hd): (1)h nmr analysis of the polar metabolome in post-mortem human brain. Biochim. Biophys. Acta 2016, 1862, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Pan, X.; Yilmaz, A.; Macias, S.; Robinson, A.; Mann, D.; Green, B.D. Targeted biochemical profiling of brain from huntington’s disease patients reveals novel metabolic pathways of interest. Biochim. Biophys. Acta 2018, 1864, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Turkoglu, O.; Kumar, P.; Yilmaz, A.; Bjorndahl, T.C.; Han, B.; Mandal, R.; Wishart, D.S.; Bahado-Singh, R.O. Targeted metabolic profiling of post-mortem brain from infants who died from sudden infant death syndrome. J. Proteome Res. 2017, 16, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Foster, A.; Lanthorn, T. An overview of glutamate as a neurotransmitter. Adv. Biochem. Psychopharmacol. 1981, 27, 1–27. [Google Scholar] [PubMed]
- Beal, M.F. Role of excitotoxicity in human neurological disease. Curr. Opin. Neurobiol. 1992, 2, 657–662. [Google Scholar] [CrossRef]
- Battaglia, G.; Bruno, V. Metabotropic glutamate receptor involvement in the pathophysiology of amyotrophic lateral sclerosis: New potential drug targets for therapeutic applications. Curr. Opin. Pharmacol. 2018, 38, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of glutamate and nmda receptors in alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Walker, J.E. Glutamate, gaba, and cns disease: A review. Neurochem. Res. 1983, 8, 521–550. [Google Scholar] [CrossRef]
- Choi, D.W.; Rothman, S.M. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990, 13, 171–182. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Back, S.A.; Craig, A.; Kayton, R.J.; Luo, N.L.; Meshul, C.K.; Allcock, N.; Fern, R. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J. Cereb. Blood Flow Metab. 2007, 27, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Daher, I.; Le Dieu-Lugon, B.; Dourmap, N.; Lecuyer, M.; Ramet, L.; Gomila, C.; Ausseil, J.; Marret, S.; Leroux, P.; Roy, V.; et al. Magnesium sulfate prevents neurochemical and long-term behavioral consequences of neonatal excitotoxic lesions: Comparison between male and female mice. J. Neuropathol. Exp. Neurol. 2017, 76, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Hermansson, M.; Hokynar, K.; Somerharju, P. Mechanisms of glycerophospholipid homeostasis in mammalian cells. Prog. Lipid Res. 2011, 50, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem. Phys. Lipids 2000, 106, 1–29. [Google Scholar] [CrossRef]
- Fuchs, B.; Schiller, J.; Cross, M.A. Apoptosis-associated changes in the glycerophospholipid composition of hematopoietic progenitor cells monitored by 31p nmr spectroscopy and maldi-tof mass spectrometry. Chem. Phys. Lipids 2007, 150, 229–238. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A.; Farooqui, T. Interactions between neural membrane glycerophospholipid and sphingolipid mediators: A recipe for neural cell survival or suicide. J. Neurosci. Res. 2007, 85, 1834–1850. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, M.; Fairn, G.D. Phospholipid subcellular localization and dynamics. J. Biol. Chem. 2018, 293, 6230–6240. [Google Scholar] [CrossRef]
- Frisardi, V.; Panza, F.; Seripa, D.; Farooqui, T.; Farooqui, A.A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in alzheimer’s disease pathology. Prog. Lipid Res. 2011, 50, 313–330. [Google Scholar] [CrossRef]
- Muller, C.P.; Reichel, M.; Muhle, C.; Rhein, C.; Gulbins, E.; Kornhuber, J. Brain membrane lipids in major depression and anxiety disorders. Biochim. Biophys. Acta 2015, 1851, 1052–1065. [Google Scholar] [CrossRef] [Green Version]
- Villamil-Ortiz, J.G.; Barrera-Ocampo, A.; Arias-Londono, J.D.; Villegas, A.; Lopera, F.; Cardona-Gomez, G.P. Differential pattern of phospholipid profile in the temporal cortex from e280a-familiar and sporadic alzheimer’s disease brains. J. Alzheimers Dis. 2018, 61, 209–219. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High performance liquid chromatography-mass spectrometry (lc-ms) based quantitative lipidomics study of ganglioside-nana-3 plasma to establish its association with parkinson’s disease patients. Med Sci. Monit. Int. Med J. Exp. Clin. Res. 2017, 23, 5345–5353. [Google Scholar] [CrossRef]
- Walsh, B.H.; Broadhurst, D.I.; Mandal, R.; Wishart, D.S.; Boylan, G.B.; Kenny, L.C.; Murray, D.M. The metabolomic profile of umbilical cord blood in neonatal hypoxic ischaemic encephalopathy. PLoS ONE 2012, 7, e50520. [Google Scholar] [CrossRef] [PubMed]
- Handley, R.R.; Reid, S.J.; Brauning, R.; Maclean, P.; Mears, E.R.; Fourie, I.; Patassini, S.; Cooper, G.J.S.; Rudiger, S.R.; McLaughlan, C.J.; et al. Brain urea increase is an early huntington’s disease pathogenic event observed in a prodromal transgenic sheep model and hd cases. Proc. Natl. Acad. Sci. USA 2017, 114, E11293–E11302. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; Hollywood, K.A.; Jullig, M.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.; Unwin, R.D.; et al. Graded perturbations of metabolism in multiple regions of human brain in alzheimer’s disease: Snapshot of a pervasive metabolic disorder. Biochim. Biophys. Acta 2016, 1862, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Summar, M.L.; Mew, N.A. Inborn errors of metabolism with hyperammonemia: Urea cycle defects and related disorders. Pediatr. Clin. N. Am. 2018, 65, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.L.; Jaishankar, G.B. Urea Cycle Disorders; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Baluarte, J.H. Neurological complications of renal disease. Semin. Pediatr. Neurol. 2017, 24, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Li, X.; Ding, Y.; Liu, Y.; Song, J.; Wang, Q.; Li, M.; Qin, Y.; Yang, Y. Seven patients of argininemia with spastic tetraplegia as the first and major symptom and prenatal diagnosis of two fetuses with high risk. Zhonghua Er Ke Za Zhi = Chin. J. Pediatr. 2015, 53, 425–430. [Google Scholar]
- Gao, J.; Zhao, B.; He, L.; Sun, M.; Yu, X.; Wang, L. Risk of cerebral palsy in chinese children: A n:M matched case control study. J. Paediatr. Child Health 2017, 53, 464–469. [Google Scholar] [CrossRef]
- Schoendorfer, N.C.; Obeid, R.; Moxon-Lester, L.; Sharp, N.; Vitetta, L.; Boyd, R.N.; Davies, P.S. Methylation capacity in children with severe cerebral palsy. Eur. J. Clin. Investig. 2012, 42, 768–776. [Google Scholar] [CrossRef]
- Nabiuni, M.; Rasouli, J.; Parivar, K.; Kochesfehani, H.M.; Irian, S.; Miyan, J.A. In vitro effects of fetal rat cerebrospinal fluid on viability and neuronal differentiation of pc12 cells. Fluids Barriers CNS 2012, 9, 8. [Google Scholar] [CrossRef]
- Chan, A.; Tchantchou, F.; Graves, V.; Rozen, R.; Shea, T.B. Dietary and genetic compromise in folate availability reduces acetylcholine, cognitive performance and increases aggression: Critical role of s-adenosyl methionine. J. Nutr. Health Aging 2008, 12, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Lovati, C.; Galimberti, D.; Pomati, S.; Capiluppi, E.; Dolci, A.; Scapellato, L.; Rosa, S.; Mailland, E.; Suardelli, M.; Vanotti, A.; et al. Serum folate concentrations in patients with cortical and subcortical dementias. Neurosci. Lett. 2007, 420, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Botez, M.I.; Peyronnard, J.M.; Berube, L.; Labrecque, R. Relapsing neuropathy, cerebral atrophy and folate deficiency. A close association. Appl. Neurophysiol. 1979, 42, 171–183. [Google Scholar] [PubMed]
- Mortensen, P.B.; Clausen, M.R. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand. J. Gastroenterol. Suppl. 1996, 216, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Conn, A.R.; Fell, D.I.; Steele, R.D. Characterization of alpha-keto acid transport across blood-brain barrier in rats. Am. J. Physiol. 1983, 245, E253–E260. [Google Scholar] [CrossRef]
- Thomas, R.H.; Meeking, M.M.; Mepham, J.R.; Tichenoff, L.; Possmayer, F.; Liu, S.; MacFabe, D.F. The enteric bacterial metabolite propionic acid alters brain and plasma phospholipid molecular species: Further development of a rodent model of autism spectrum disorders. J. Neuroinflamm. 2012, 9, 153. [Google Scholar] [CrossRef]
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially produced imidazole propionate impairs insulin signaling through mtorc1. Cell 2018, 175, 947–961. [Google Scholar] [CrossRef]
- Morland, C.; Froland, A.S.; Pettersen, M.N.; Storm-Mathisen, J.; Gundersen, V.; Rise, F.; Hassel, B. Propionate enters gabaergic neurons, inhibits gaba transaminase, causes gaba accumulation and lethargy in a model of propionic acidemia. Biochem. J. 2018, 475, 749–758. [Google Scholar] [CrossRef]
- Hoyles, L.; Snelling, T.; Umlai, U.K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-host systems interactions: Protective effects of propionate upon the blood-brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- van den Berge, M.; Jonker, M.R.; Miller-Larsson, A.; Postma, D.S.; Heijink, I.H. Effects of fluticasone propionate and budesonide on the expression of immune defense genes in bronchial epithelial cells. Pulm. Pharmacol. Ther. 2018, 50, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Pluciennik, F.; Verrecchia, F.; Bastide, B.; Herve, J.C.; Joffre, M.; Deleze, J. Reversible interruption of gap junctional communication by testosterone propionate in cultured sertoli cells and cardiac myocytes. J. Membr. Biol. 1996, 149, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41r–47r. [Google Scholar] [CrossRef] [PubMed]
- MacFabe, D.F.; Cain, D.P.; Rodriguez-Capote, K.; Franklin, A.E.; Hoffman, J.E.; Boon, F.; Taylor, A.R.; Kavaliers, M.; Ossenkopp, K.P. Neurobiological effects of intraventricular propionic acid in rats: Possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav. Brain Res. 2007, 176, 149–169. [Google Scholar] [CrossRef] [PubMed]
- MacFabe, D.F.; Cain, N.E.; Boon, F.; Ossenkopp, K.P.; Cain, D.P. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: Relevance to autism spectrum disorder. Behav. Brain Res. 2011, 217, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Shultz, S.R.; Macfabe, D.F.; Martin, S.; Jackson, J.; Taylor, R.; Boon, F.; Ossenkopp, K.P.; Cain, D.P. Intracerebroventricular injections of the enteric bacterial metabolic product propionic acid impair cognition and sensorimotor ability in the long-evans rat: Further development of a rodent model of autism. Behav. Brain Res. 2009, 200, 33–41. [Google Scholar] [CrossRef]
- Shultz, S.R.; MacFabe, D.F.; Ossenkopp, K.P.; Scratch, S.; Whelan, J.; Taylor, R.; Cain, D.P. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: Implications for an animal model of autism. Neuropharmacology 2008, 54, 901–911. [Google Scholar] [CrossRef]
- Broeder, C.E.; Brenner, M.; Hofman, Z.; Paijmans, I.J.; Thomas, E.L.; Wilmore, J.H. The metabolic consequences of low and moderate intensity exercise with or without feeding in lean and borderline obese males. Int. J. Obes. 1991, 15, 95–104. [Google Scholar]
- Gerstner, B.; Lee, J.; DeSilva, T.M.; Jensen, F.E.; Volpe, J.J.; Rosenberg, P.A. 17beta-estradiol protects against hypoxic/ischemic white matter damage in the neonatal rat brain. J. Neurosci. Res. 2009, 87, 2078–2086. [Google Scholar] [CrossRef]
- Zisk, J.L.; Genen, L.H.; Kirkby, S.; Webb, D.; Greenspan, J.; Dysart, K. Do premature female infants really do better than their male counterparts? Am. J. Perinatol. 2011, 28, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Zhao, J.; Li, S. Estrogen receptors’ neuroprotective effect against glutamate-induced neurotoxicity. Neurol. Sci. 2014, 35, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; McBride, P.A.; Hertzig, M.E.; Snow, M.E.; Hall, L.M.; Ferrari, P.; Cohen, D.J. Plasma androgens in autism. J. Autism Dev. Disord. 1995, 25, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjordahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, fully-automated nmr spectral profiling for metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [PubMed]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton nmr spectra. J. Biomol. NMR 2011, 49, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. Metaboanalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building predictive models in r using the caret package. J. Stat. Softw. 2008, 28, 26. [Google Scholar] [CrossRef]
- Angermueller, C.; Parnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical power analyses using g*power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]



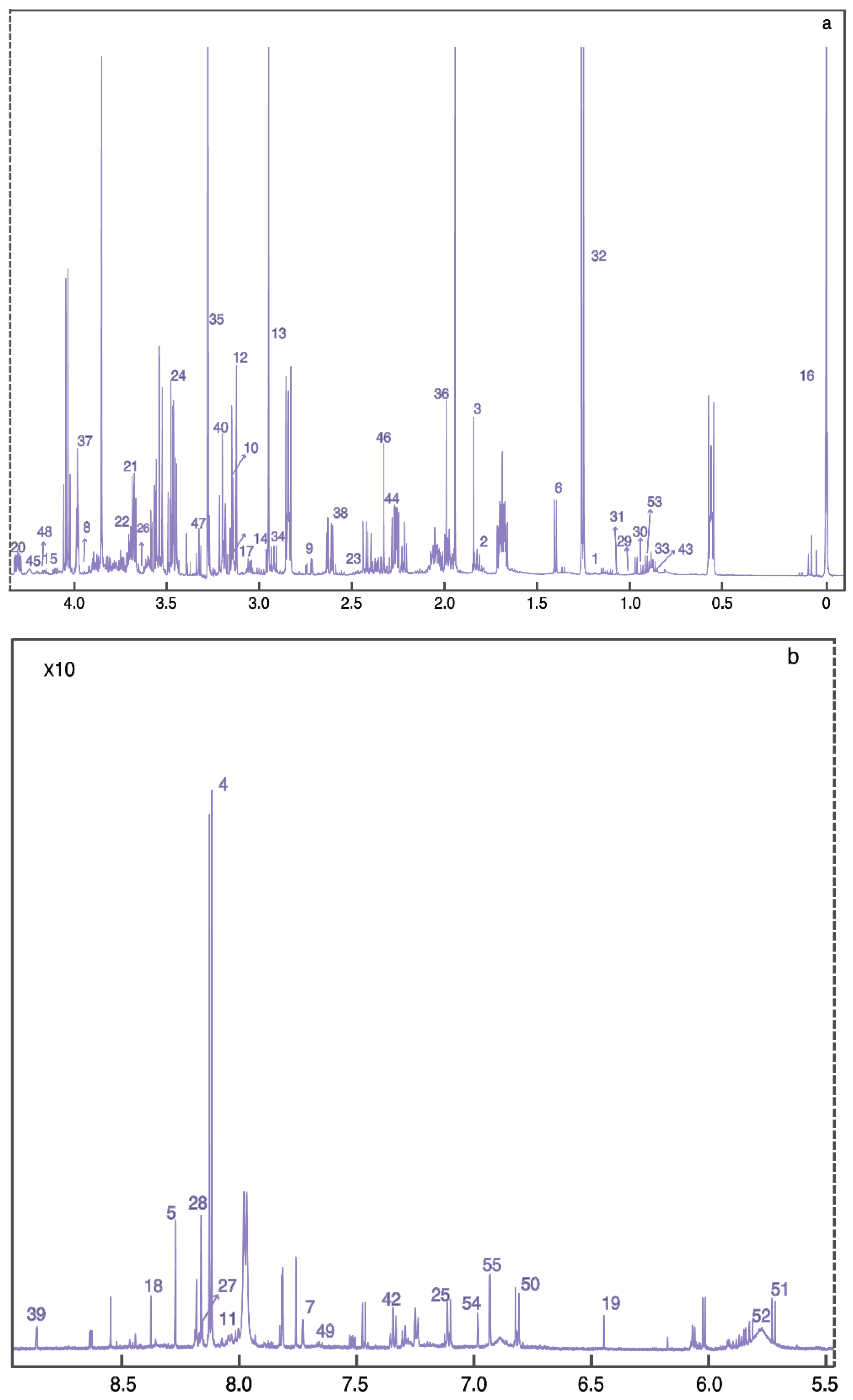{kind=link}
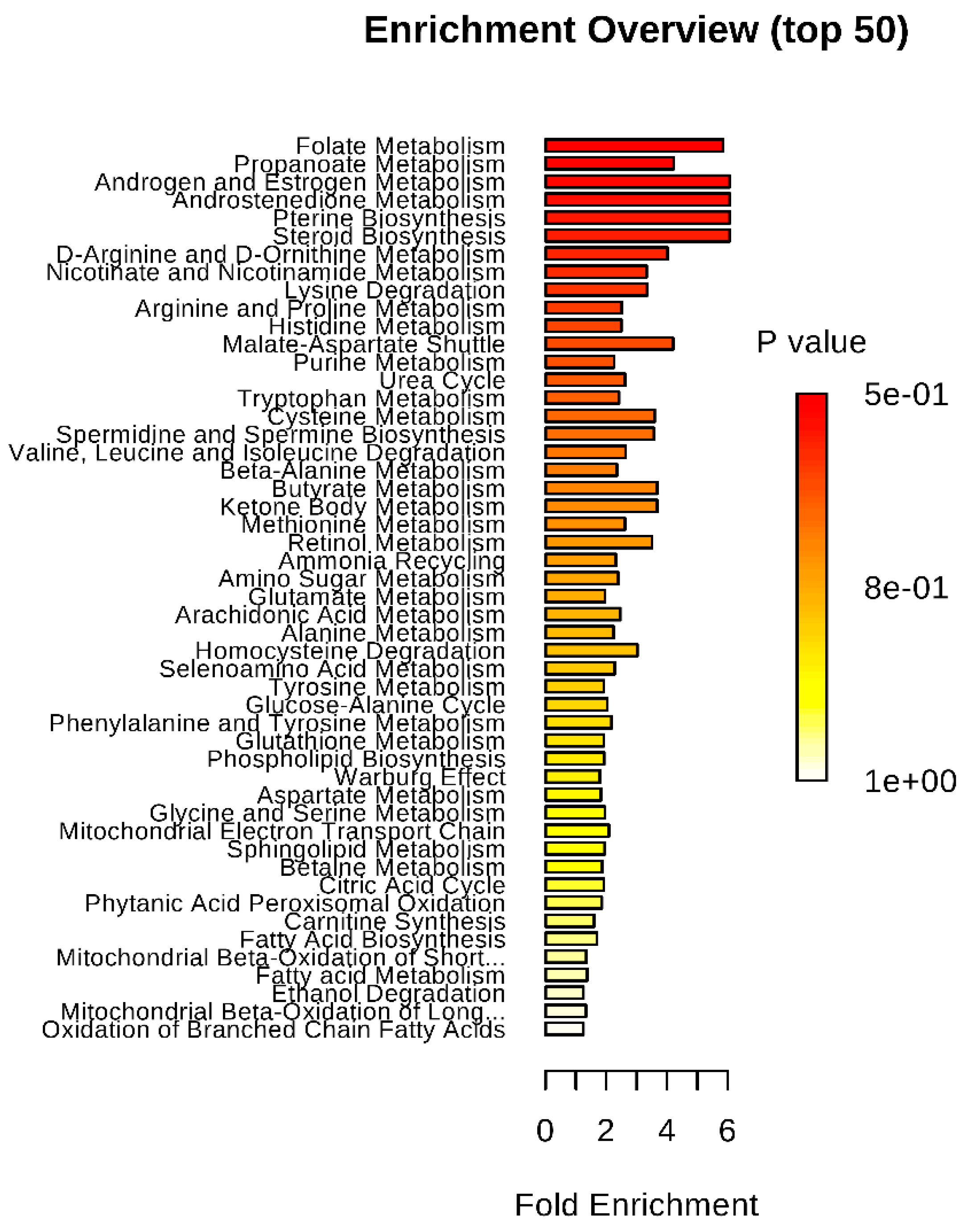{kind=link}
| HMDB | Compound ID | Mean (SD) of Control (μM) | Mean (SD) of CP (μM) | p-Value | q-Value (FDR) | Fold Change |
|---|---|---|---|---|---|---|
| HMDB00294 | Urea | 59.236 (37.499) | 184.144 (14.774) | 0.0074 (W) | 0.299 | −3.11 |
| HMDB00148 | L-Glutamic acid | 499.627 (15.680) | 6.767 (13.764) | 0.0106 (W) | 0.299 | 73.83 |
| HMDB13456 | PC(o-22:2(13Z,16Z)/22:3(10Z,13Z,16Z)) | 1.187 (0.902) | 0.335 (0.379) | 0.0125 (W) | 0.299 | 3.54 |
| HMDB08276 | PC(20:0/20:2(11Z,14Z)) | 0.265 (0.190) | 0.051 (0.110) | 0.0166 (W) | 0.299 | 5.16 |
| HMDB13450 | PC(o-22:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 0.847 (0.710) | 0.231 (0.404) | 0.0166 (W) | 0.299 | 3.66 |
| HMDB00195 | Inosine | 8.082 (4.627) | 14.333 (6.338) | 0.0201 | 0.299 | −1.77 |
| HMDB13333 | 3-Hydroxy-9-hexadecenoylcarnitine | 0.061 (0.062) | 0.129 (0.076) | 0.0204 (W) | 0.299 | -2.13 |
| HMDB10379 | LysoPC(14:0) | 5.237 (1.153) | 4.151 (0.665) | 0.0224 | 0.299 | 1.26 |
| HMDB13433 | PC(o-18:1(9Z)/22:0) | 1.334 (0.714) | 0.638 (0.487) | 0.023 | 0.299 | 2.09 |
| HMDB13453 | PC(o-22:1(13Z)/22:3(10Z,13Z,16Z)) | 0.281 (0.180) | 0.133 (0.069) | 0.0248 | 0.299 | 2.12 |
| HMDB07991 | PC(16:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 55.251 (4.352) | 19.532 (5.971) | 0.0249 | 0.299 | 2.83 |
| HMDB08055 | PC(18:0/22:5(4Z,7Z,10Z,13Z,16Z)) | 9.151 (6.281) | 3.871 (2.773) | 0.0249 | 0.299 | 2.36 |
| HMDB06083 | Troxerutin | 188.555 (18.953) | 432.889 (25.759) | 0.0250 (W) | 0.299 | −2.3 |
| HMDB08048 | PC(18:0/20:4(5Z,8Z,11Z,14Z)) | 114.082 (59.935) | 56.311 (43.130) | 0.0264 | 0.299 | 2.03 |
| HMDB00142 | Formic acid | 4.718 (2.078) | 7.489 (3.055) | 0.0269 | 0.299 | −1.59 |
| HMDB08057 | PC(18:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 23.314 (15.829) | 11.438 (6.380) | 0.0275 (W) | 0.299 | 2.04 |
| HMDB07892 | PC(14:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 0.405 (0.338) | 0.139 (0.090) | 0.028 | 0.299 | 2.91 |
| HMDB0029205 | LysoPC(26:0) | 0.227 (0.197) | 0.456 (0.235) | 0.0293 | 0.299 | −2.01 |
| HMDB07874 | PC(14:0/18:2(9Z,12Z)) | 3.462 (3.478) | 0.558 (0.715) | 0.0297 (W) | 0.299 | 6.21 |
| HMDB03334 | Symmetric dimethylarginine | 0.638 (0.399) | 1.405 (0.802) | 0.0310 (W) | 0.299 | −2.2 |
| HMDB10394 | LysoPC(20:3(8Z,11Z,14Z)) | 1.213 (0.902) | 0.492 (0.500) | 0.0310 (W) | 0.299 | 2.46 |
| HMDB08288 | PC(20:0/22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 0.367 (0.230) | 0.186 (0.100) | 0.0332 | 0.299 | 1.98 |
| HMDB11151 | PC(O-16:0/18:2(9Z,12Z)) | 10.915 (6.853) | 5.759 (2.592) | 0.0381 | 0.299 | 1.9 |
| HMDB13469 | SM(d18:0/24:1(15Z)(OH)) | 1.353 (0.764) | 2.168 (1.131) | 0.0402 (W) | 0.299 | −1.6 |
| HMDB13458 | PC(o-24:0/18:3(6Z,9Z,12Z)) | 0.909 (0.441) | 0.536 (0.290) | 0.0428 | 0.299 | 1.7 |
| HMDB08138 | PC(18:2(9Z,12Z)/18:2(9Z,12Z)) | 189.522 (12.500) | 60.640 (6.755) | 0.0465 (W) | 0.299 | 3.13 |
| HMDB13411 | PC(o-16:1(9Z)/16:1(9Z)) | 0.720 (0.496) | 0.362 (0.212) | 0.048 | 0.299 | 1.99 |
| Models | Selected Features |
|---|---|
| LR | PC ae C44:5, Urea |
| SVM | PC ae C44:5, Urea, C9 |
| PLS-DA | PC ae C44:5, Urea, C9, PC aa C40:6, PC ae C40:1, PC ae C44:6 |
| RF | PC ae C44:5, Urea, C9, PC aa C40:6, PC ae C40:1 |
| PAM | Urea, PC ae C44:5, PC ae C44:6, C9, PC aa C40:6, PC ae C40:1 |
| DL | C9, PC ae C40:1, Urea, PC ae C44:6, PC ae C44:5 |
| LR | SVM | PLS-DA | RF | PAM | DL | |
|---|---|---|---|---|---|---|
| AUC (95% CI) | 0.861 (0.688–1) | 0.925 (0.73–1) | 0.929 (0.8–1) | 0.899 (0.6–1) | 0.93 (0.8–1) | 0.937 (0.8–1) |
| Sensitivity | 0.842 | 0.778 | 0.870 | 0.889 | 0.899 | 0.833 |
| Specificity | 0.909 | 0.625 | 0.725 | 0.850 | 0.855 | 0.667 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpay Savasan, Z.; Yilmaz, A.; Ugur, Z.; Aydas, B.; Bahado-Singh, R.O.; Graham, S.F. Metabolomic Profiling of Cerebral Palsy Brain Tissue Reveals Novel Central Biomarkers and Biochemical Pathways Associated with the Disease: A Pilot Study. Metabolites 2019, 9, 27. https://doi.org/10.3390/metabo9020027
Alpay Savasan Z, Yilmaz A, Ugur Z, Aydas B, Bahado-Singh RO, Graham SF. Metabolomic Profiling of Cerebral Palsy Brain Tissue Reveals Novel Central Biomarkers and Biochemical Pathways Associated with the Disease: A Pilot Study. Metabolites. 2019; 9(2):27. https://doi.org/10.3390/metabo9020027
Chicago/Turabian StyleAlpay Savasan, Zeynep, Ali Yilmaz, Zafer Ugur, Buket Aydas, Ray O. Bahado-Singh, and Stewart F. Graham. 2019. "Metabolomic Profiling of Cerebral Palsy Brain Tissue Reveals Novel Central Biomarkers and Biochemical Pathways Associated with the Disease: A Pilot Study" Metabolites 9, no. 2: 27. https://doi.org/10.3390/metabo9020027