Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics

 , , , , and
, , , , and
Abstract
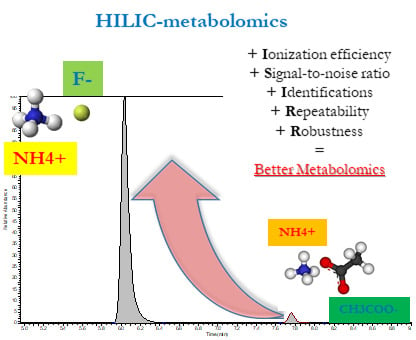
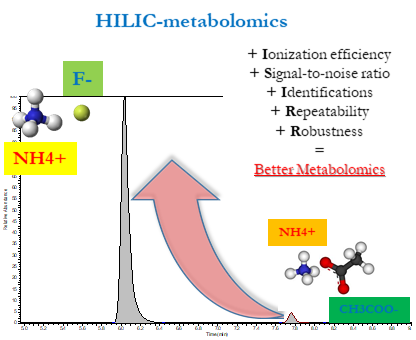
:
1. Introduction
2. Materials and Methods
2.1. Solvents and Reagents
2.2. Standards Preparation
2.3. Sample Preparation
2.4. Liquid Chromatography–High-Resolution Mass Spectrometry
2.5. Instrumental Calibration and Performance Control
2.6. Chromatographic Columns (Figure 1)
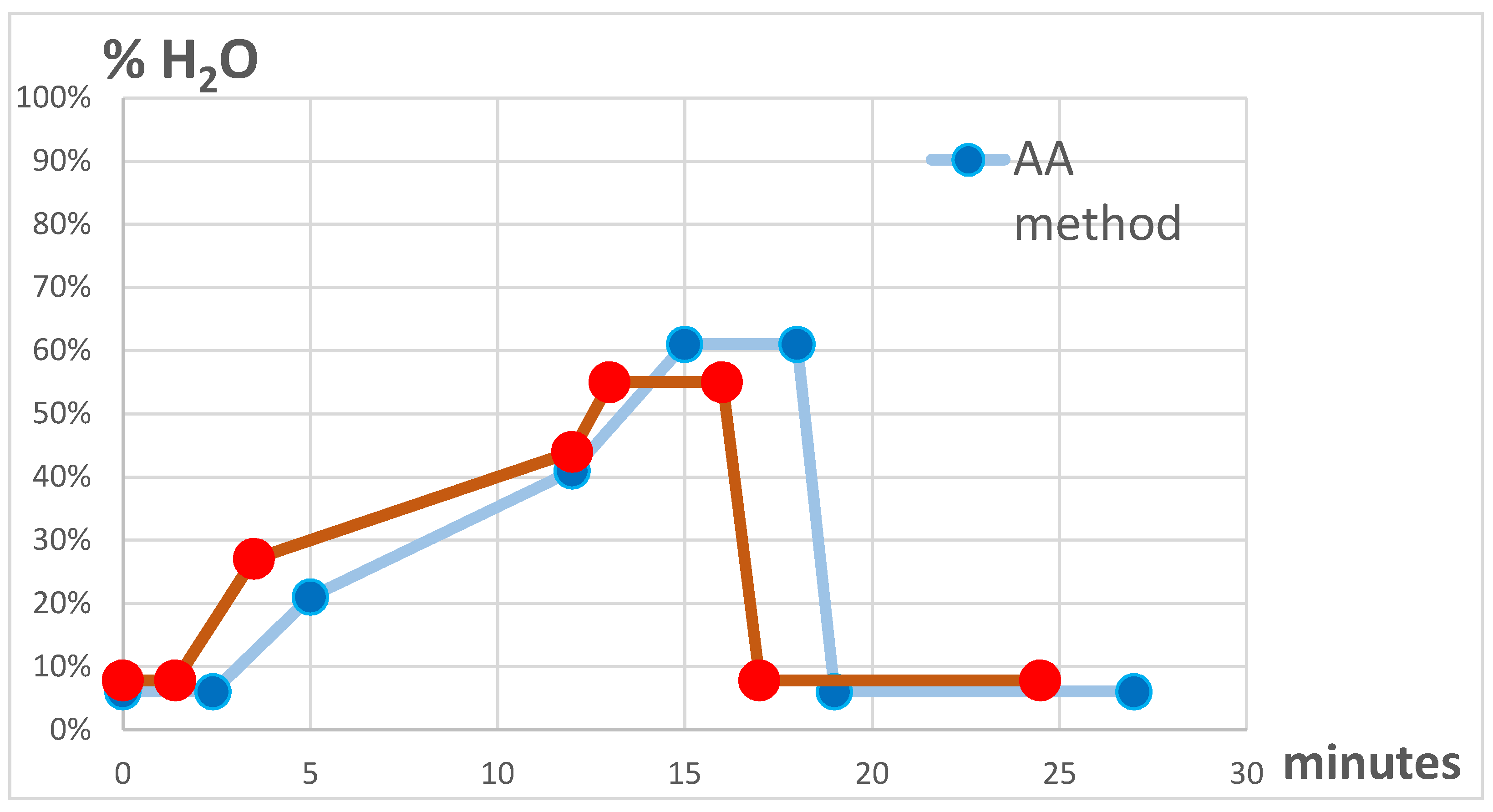
2.7. Chromatographic Conditions
2.7.1. Ammonium Acetate Method (AA)
2.7.2. Ammonium Fluoride Method (AF)
2.8. Evaluation Process
3. Results
3.1. Pre-Experimental Settings: Column Choice and Gradient Development
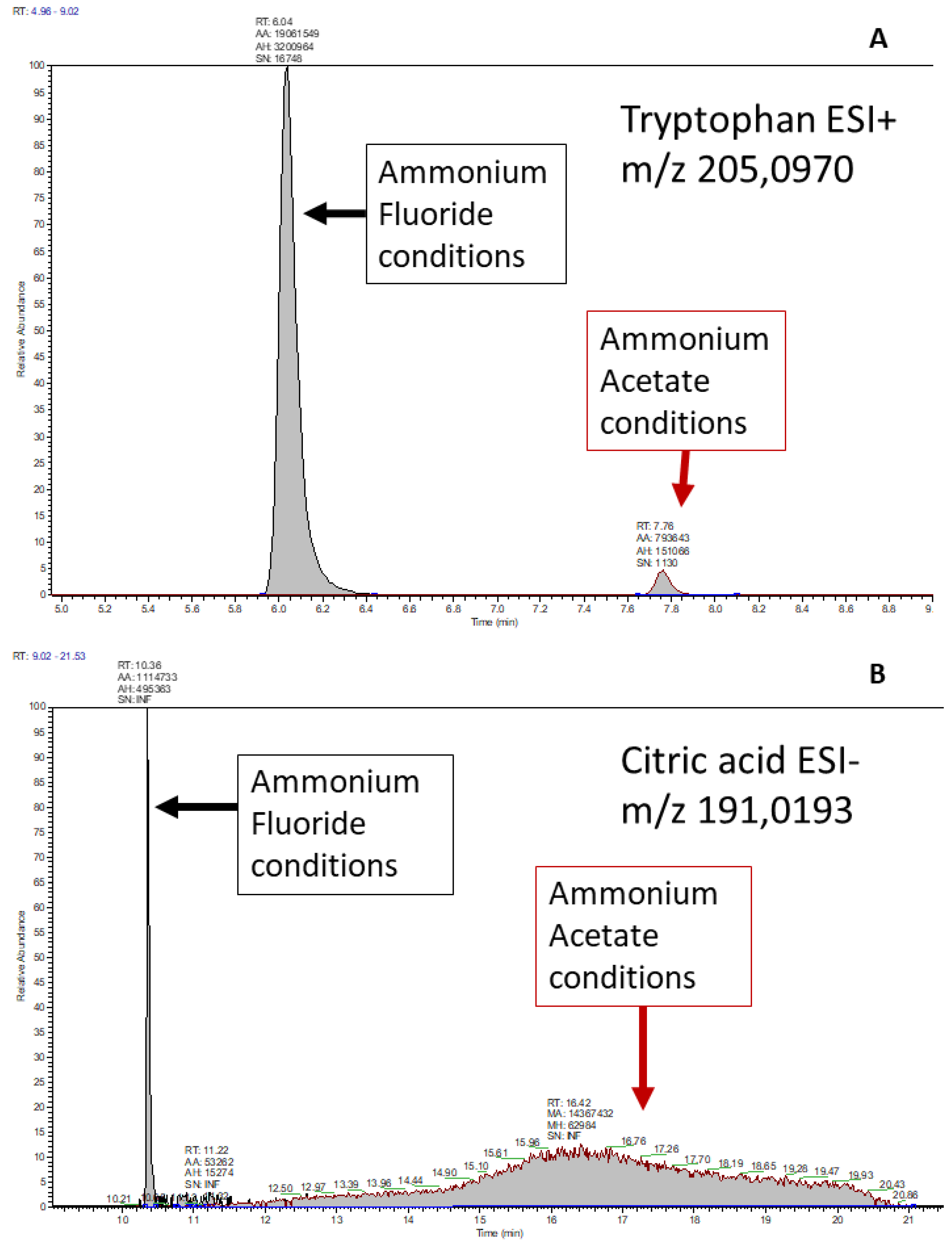
3.2. Method Comparison: Standard Injection Evaluation
3.3. Repeatability and Robustness Test: Pooled Sample Injections Evaluation.
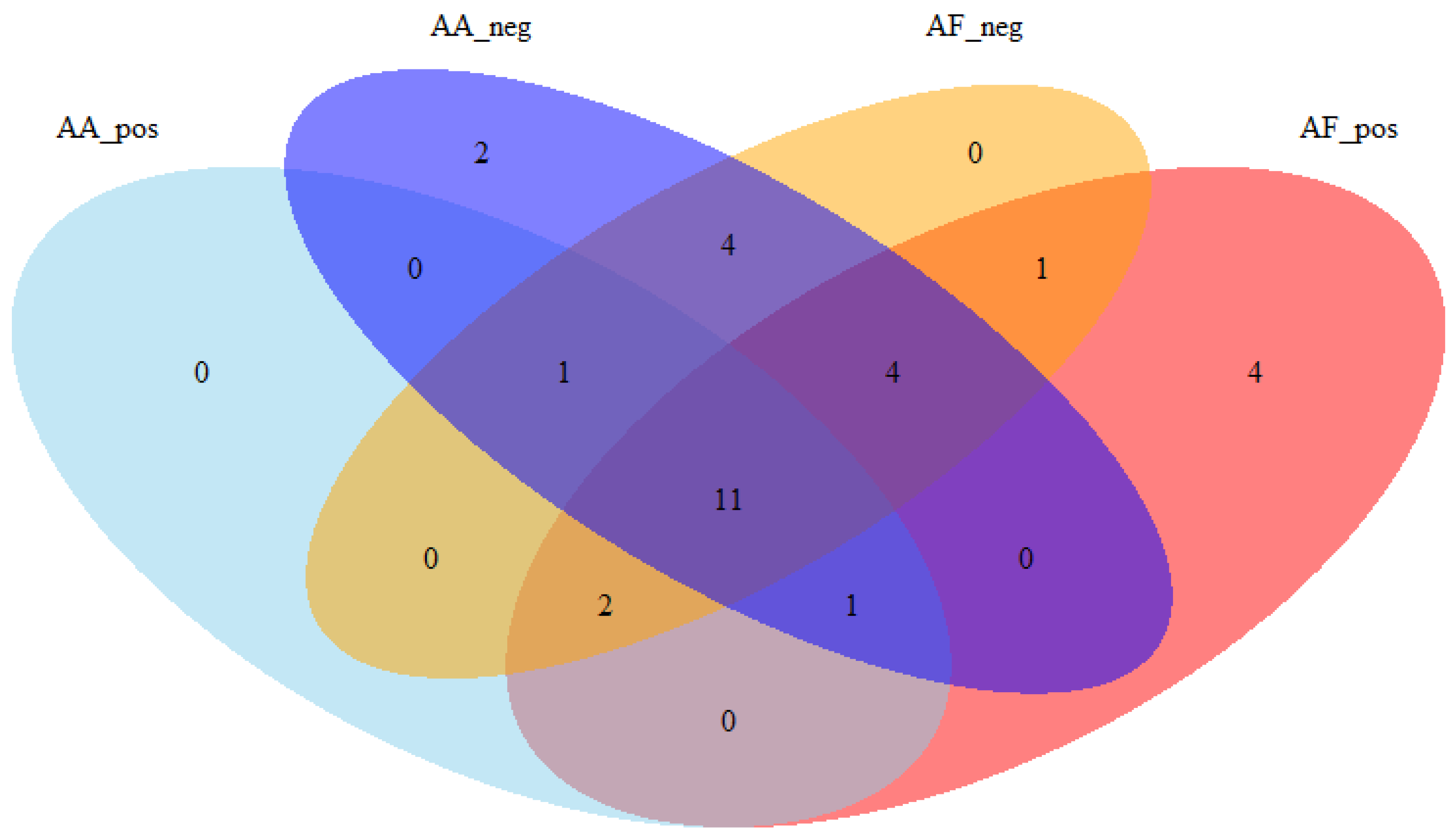
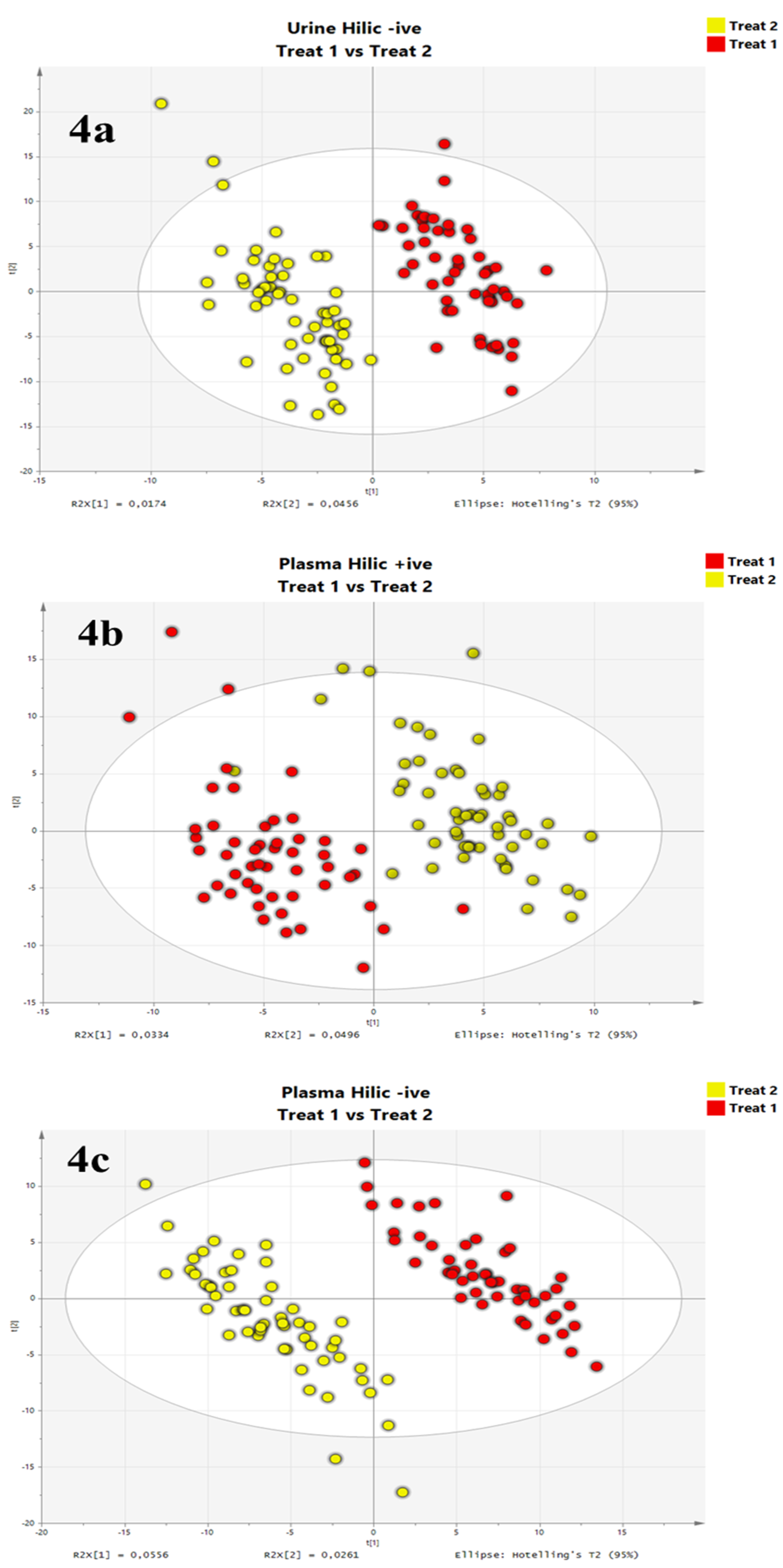
3.4. Evaluation on Real Datasets: Urine and Plasma Metabolomics
3.5. Tip and Tricks: Method Adjustments and Important Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Tang, D.-Q.; Zou, L.; Yin, X.-X.; Ong, C.N. Hilic-ms for metabolomics: an attractive and complementary approach to rplc-ms. Mass Spectrom. Rev. 2016, 35, 574–600. [Google Scholar] [CrossRef] [PubMed]
- Kohler, I.; Derks, R.J.E.; Giera, M. The rise of hydrophilic interaction chromatography in untargeted clinical metabolomics. LC-GC Eur. 2016, 29, 60–75. [Google Scholar]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)-a powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef]
- Konermann, L. Addressing a Common Misconception: Ammonium Acetate as Neutral pH “Buffer” for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1827–1835. [Google Scholar] [CrossRef]
- Greco, G.; Grosse, S.; Letzel, T. Study of the retention behavior in zwitterionic hydrophilic interaction chromatography of isomeric hydroxy- and aminobenzoic acids. J. Chromatogr. A 2012, 1235, 60–67. [Google Scholar] [CrossRef]
- Schellinger, A.P.; Carr, P.W. Solubility of Buffers in Aqueous – Organic Eluents for Reversed-Phase Liquid Chromatography. Liq. Chromatogr. Gas Chromatogr. North Am. 2004, 22, 544–548. [Google Scholar]
- Mallet, C.R.; Lu, Z.; Mazzeo, J.R. A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts. Rapid Commun. Mass Spectrom. 2004, 18, 49–58. [Google Scholar] [CrossRef]
- Koch, W.; Forcisi, S.; Lehmann, R.; Schmitt-Kopplin, P. Sensitivity improvement in hydrophilic interaction chromatography negative mode electrospray ionization mass spectrometry using 2-(2-methoxyethoxy)ethanol as a post-column modifier for non-targeted metabolomics. J. Chromatogr. A 2014, 1361, 209–216. [Google Scholar] [CrossRef]
- Yu, T.; Jones, D.P. Improving peak detection in high-resolution LC/MS metabolomics data using preexisting knowledge and machine learning approach. Bioinformatics 2014, 30, 2941–2948. [Google Scholar] [CrossRef]
- Spalding, J.L.; Naser, F.J.; Mahieu, N.G.; Johnson, S.L.; Patti, G.J. Trace phosphate improves ZIC-pHILIC peak shape, sensitivity, and coverage for untargeted metabolomics. J. Proteome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, F.E.; Apffel, A.; Fischer, S.M.; Goldberg, G.; Goodley, P.C. Signal enhancement for gradient reverse-phase high-performance liquid chromatography-electrospray ionization mass spectrometry analysis with trifluoroacetic and other strong acid modifiers by postcolumn addition of propionic acid and isopropanol. J. Am. Soc. Mass Spectrom. 1995, 6, 1221–1225. [Google Scholar] [CrossRef]
- Yamaguchi, J.I.; Ohmichi, M.; Jingu, S.; Ogawa, N.; Higuchi, S. Utility of postcolumn addition of 2-(2-methoxyethoxy)ethanol, a signal- enhancing modifier, for metabolite screening with liquid chromatography and negative ion electrospray ionization mass spectrometry. Anal. Chem. 1999, 71, 5386–5390. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Tautenhahn, R.; Patti, G.J.; Siuzdak, G. Expanding Coverage of the Metabolome for Global Metabolite Profiling. Anal Chem 2011, 83, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Takkis, K.; Aro, R.; Kõrgvee, L.T.; Varendi, H.; Lass, J.; Herodes, K.; Kipper, K. Signal Enhancement in the HPLC-ESI-MS/MS analysis of spironolactone and its metabolites using HFIP and NH4F as eluent additives. Anal. Bioanal. Chem. 2017, 409, 3145–3151. [Google Scholar] [CrossRef] [PubMed]
- Petrie, B.; Youdan, J.; Barden, R.; Kasprzyk-Hordern, B. Multi-residue analysis of 90 emerging contaminants in liquid and solid environmental matrices by ultra-high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1431, 64–78. [Google Scholar] [CrossRef]
- Creydt, M.; Fischer, M. Plant Metabolomics: Maximizing Metabolome Coverage by Optimizing Mobile Phase Additives for Nontargeted Mass Spectrometry in Positive and Negative Electrospray Ionization Mode. Anal. Chem. 2017, 89, 10474–10486. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T. Ammonium fluoride as a mobile phase additive in aqueous normal phase chromatography. J. Chromatogr. A 2015, 1401, 69–74. [Google Scholar] [CrossRef]
- Kulsing, C.; Nolvachai, Y.; Marriott, P.J.; Boysen, R.I.; Matyska, M.T.; Pesek, J.J.; Hearn, M.T.W. Insights into the origin of the separation selectivity with silica hydride adsorbents. J. Phys. Chem. B 2015, 119, 3063–3069. [Google Scholar] [CrossRef]
- Royer, A.; Gallart-ayala, H.; Antignac, J.; Dervilly-pinel, G.; Monteau, F.; Bizec, B. Le A versatile multi-analytical platform for serum metabolomics and lipidomics profiling. In Proceedings of the RFMF, Villeurbanne, France, 19–21 May 2014; p. 2014. [Google Scholar]
- Jacob, C.C.; Dervilly-Pinel, G.; Biancotto, G.; Monteau, F.; Le Bizec, B. Global urine fingerprinting by LC-ESI(+)-HRMS for better characterization of metabolic pathway disruption upon anabolic practices in bovine. Metabolomics 2014, 11, 184–197. [Google Scholar] [CrossRef]
- Peng, T.; Royer, A.L.; Guitton, Y.; Le Bizec, B.; Dervilly-Pinel, G. Serum-based metabolomics characterization of pigs treated with ractopamine. Metabolomics 2017, 13, 1–15. [Google Scholar] [CrossRef]
- Pezzatti, J.; González-Ruiza, V.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Guillarme, D.; Schappler, J.; Picard, D.; Boccard, J.; Rudaz, S. A scoring approach for multi-platform acquisition in metabolomics. J. Chromatogr. A 2018, 1592, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, J.; Yang, Q.; Li, S.; Cui, X.; Li, Y.; Chen, Y.; Xue, W.; Li, X.; Zhu, F. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017, 45, 162–170. [Google Scholar] [CrossRef]
- Heckendorf, A.; Alpert, A. Electrostatic Repulsion–Hydrophilic Interaction Chromatography: Using One Mode to Tune Retention from a Second Mode. LC-GC Eur. 2011, 29, 606–610. [Google Scholar]
- Narduzzi, L.; Dervilly, G.; Marchand, A.; Audran, M.; Le, B.; Buisson, C. A metabolomics approach to detect growth hormone administration in athletes. Submitted.
- Wernisch, S.; Pennathur, S. Evaluation of coverage, retention patterns and selectivity of seven liquid chromatographic methods for metabolomics. Anal. Bioanal. Chem. 2016, 408, 6079–6091. [Google Scholar] [CrossRef]
- Wold, S.; Sjostrom, M.; Eriksson, L. PLS-regression: a basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Dolan, J.W. LC troubleshooting-Autosampler Carryover. LC-GC Eur. 2001, 3. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | logP | Mass Da | Class |
|---|---|---|---|
| sorbitol | −4.7 | 182.0790 | sugar |
| mannitol | −4.7 | 182.0790 | sugar |
| betaine | −3.3 | 117.0790 | amino acids |
| nicotinamide n-oxide | −2.7 | 138.0430 | pirimidine |
| taurine | −2.5 | 125.0147 | amino acids |
| myo-inositol | −2.1 | 180.0634 | sugar |
| inosine | −1.9 | 268.0808 | purine |
| cis-4-hydroxy-d-proline | −1.8 | 131.0583 | amino acids |
| d-fructose-6-phosphate | −1.8 | 260.0297 | sugar |
| citric acid | −1.7 | 192.0270 | acid |
| uridine | −1.7 | 244.0695 | pirimidine |
| 2’-deoxyguanosine-5-phosphate | −1.4 | 345.0485 | purine |
| d,l-pantotheic acid | −0.9 | 219.1107 | vitamin |
| glu-val-phe | −0.9 | 393.1900 | amino acids |
| uracil | −0.7 | 112.0273 | pirimidine |
| hydroxy-hippuric acid | 0.0 | 195.0532 | phenolic acid |
| fumaric acid | 0.0 | 116.0110 | acid |
| adipic acid | 0.1 | 146.0579 | lipid |
| biotin | 0.1 | 244.0882 | vitamin |
| valine | 0.2 | 117.0790 | amino acids |
| 5-hydroxy-indole acetic acid | 0.3 | 190.0504 | indole |
| 2-hydroxy-indole acetic acid | 0.3 | 190.0504 | indole |
| hippuric acid | 0.3 | 179.0582 | phenolic acid |
| leucine | 0.7 | 131.0947 | amino acids |
| isoleucine | 0.7 | 131.0947 | amino acids |
| l-tryptophan | 1.0 | 204.0880 | amino acids |
| l-3-phenyllactic acid | 1.1 | 166.0630 | phenolic acid |
| l-kynurenine | 1.1 | 208.0848 | phenolic acid |
| l-phenylalanine | 1.1 | 165.0791 | amino acids |
| 3-indole acetic acid | 1.4 | 175.0633 | indole |
| d-homophenylalanine | 1.5 | 179.0946 | amino acids |
| oxooctanoyl homoserine lactone | 2.0 | 297.1940 | amino acids |
| glycourso-deoxycholic acid | 3.5 | 449.3135 | bile acid |
| tetradecanoic acid | 6.1 | 228.2089 | lipid |
| Compounds | Pos. Ionization Si Score | Neg. Ionization Si Score | ||||
|---|---|---|---|---|---|---|
| AA | AF | Δ Score | AA | AF | Δ Score | |
| betaine | 0.06 | 0.58 | 0.52 | 0.00 | 0.00 | 0.00 |
| valine | 0.04 | 0.35 | 0.32 | 0.13 | 0.57 | 0.44 |
| leucine | 0.05 | 0.39 | 0.34 | 0.10 | 0.36 | 0.26 |
| isoleucine | 0.07 | 0.36 | 0.29 | 0.10 | 0.43 | 0.33 |
| l-phenyllactic acid | 0.00 | 0.00 | 0.00 | 0.14 | 0.10 | –0.04 |
| myo-inositol | 0.00 | 0.00 | 0.00 | 0.65 | 0.64 | –0.01 |
| fumaric acid | 0.00 | 0.00 | 0.00 | 0.42 | 0.36 | –0.06 |
| fructose-6-phosphate | 0.23 | 0.43 | 0.20 | 0.39 | 0.23 | –0.16 |
| citric acid | 0.00 | 0.00 | 0.00 | 0.00 | 0.28 | 0.28 |
| uracil | 0.00 | 0.07 | 0.07 | 0.38 | 0.04 | –0.34 |
| nicotinamide-n-oxide | 0.12 | 0.20 | 0.09 | 0.55 | 0.39 | –0.16 |
| uridine | 0.06 | 0.20 | 0.14 | 0.33 | 0.43 | 0.11 |
| 5-hydroxy-indole acetic acid | 0.00 | 0.29 | 0.29 | 0.43 | 0.90 | 0.47 |
| taurine | 0.17 | 0.35 | 0.18 | 0.72 | 0.36 | –0.35 |
| mannitol | 0.00 | 0.00 | 0.00 | 0.00 | 0.88 | 0.88 |
| sorbitol | 0.00 | 0.07 | 0.07 | 0.09 | 0.88 | 0.80 |
| hydroxy-hippuric acid | 0.00 | 0.00 | 0.00 | 0.05 | 0.20 | 0.15 |
| tetradecanoic acid | 0.00 | 0.00 | 0.00 | 0.00 | 0.10 | 0.10 |
| 3-indole acetic acid | 0.00 | 0.00 | 0.00 | 0.53 | 0.06 | –0.46 |
| hippuric acid | 0.01 | 0.14 | 0.14 | 0.67 | 0.30 | –0.37 |
| glycourso-dexoycholic acid | 0.07 | 0.35 | 0.28 | 0.28 | 0.83 | 0.55 |
| inosine | 0.09 | 0.65 | 0.56 | 0.14 | 0.73 | 0.58 |
| d-homophenylalanine | 0.18 | 0.73 | 0.56 | 0.21 | 0.74 | 0.54 |
| l-phenylalanine | 0.15 | 0.70 | 0.55 | 0.20 | 0.74 | 0.54 |
| l-kynurenine | 0.16 | 0.71 | 0.55 | 0.16 | 0.63 | 0.47 |
| biotin | 0.14 | 0.66 | 0.52 | 0.14 | 0.65 | 0.51 |
| d-,l-pantotheic acid | 0.00 | 0.00 | 0.00 | 0.29 | 0.72 | 0.43 |
| glu-val-phe | 0.16 | 0.38 | 0.22 | 0.23 | 0.70 | 0.47 |
| l-tryptophan | 0.17 | 0.69 | 0.52 | 0.00 | 0.74 | 0.74 |
| d,l-pantotheic acid | 0.11 | 0.62 | 0.51 | 0.00 | 0.00 | 0.00 |
| adipic acid | 0.00 | 0.00 | 0.00 | 0.38 | 0.10 | –0.28 |
| cis-4-hydroxy-d-proline | 0.16 | 0.75 | 0.59 | 0.57 | 0.37 | –0.19 |
| 2-hydroxy-indole acetic acid | 0.00 | 0.67 | 0.67 | 0.00 | 0.00 | 0.00 |
| deoxyguanosine-5-phosphate | 0.00 | 0.00 | 0.00 | 0.44 | 0.58 | 0.14 |
| Chromatographic Conditions | AA | AF | ||
|---|---|---|---|---|
| Ionization Mode | ESI+ | ESI– | ESI+ | ESI– |
| median area | 5,234,608 | 10,394,744 | 42,179,020 | 35,910,970 |
| ratio neg/pos | 2.0 | 0.9 | ||
| ratio AFpos/AApos | - | - | 8.1 | - |
| ratio AFneg/AAneg | - | - | - | 3.5 |
| Ionization Mode | ESI+ | ESI– | ||
|---|---|---|---|---|
| Additive | AA | AF | AA | AF |
| ND** peaks | 14 | 7 | 11 | 11 |
| Median peak area | 692,901 | 1,036,833 | 663,922 | 2,994,235 |
| Median area RSD* | 29 max–min 49–8 | 10 max–min 53–2.8 | 17 max–min 90–6 | 8.7 max–min 45–5 |
| Mean RT shift | 11 s | 10 s | 16 s | 9 s |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narduzzi, L.; Royer, A.-L.; Bichon, E.; Guitton, Y.; Buisson, C.; Le Bizec, B.; Dervilly-Pinel, G. Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics. Metabolites 2019, 9, 292. https://doi.org/10.3390/metabo9120292
Narduzzi L, Royer A-L, Bichon E, Guitton Y, Buisson C, Le Bizec B, Dervilly-Pinel G. Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics. Metabolites. 2019; 9(12):292. https://doi.org/10.3390/metabo9120292
Chicago/Turabian StyleNarduzzi, Luca, Anne-Lise Royer, Emmanuelle Bichon, Yann Guitton, Corinne Buisson, Bruno Le Bizec, and Gaud Dervilly-Pinel. 2019. "Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics" Metabolites 9, no. 12: 292. https://doi.org/10.3390/metabo9120292