Amino Acids as Building Blocks for Carbonic Anhydrase Inhibitors


Abstract
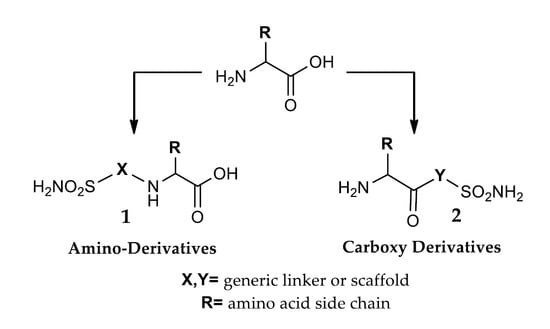
:
1. Introduction
2. Carbonic Anhydrase Inhibitors (CAIs)
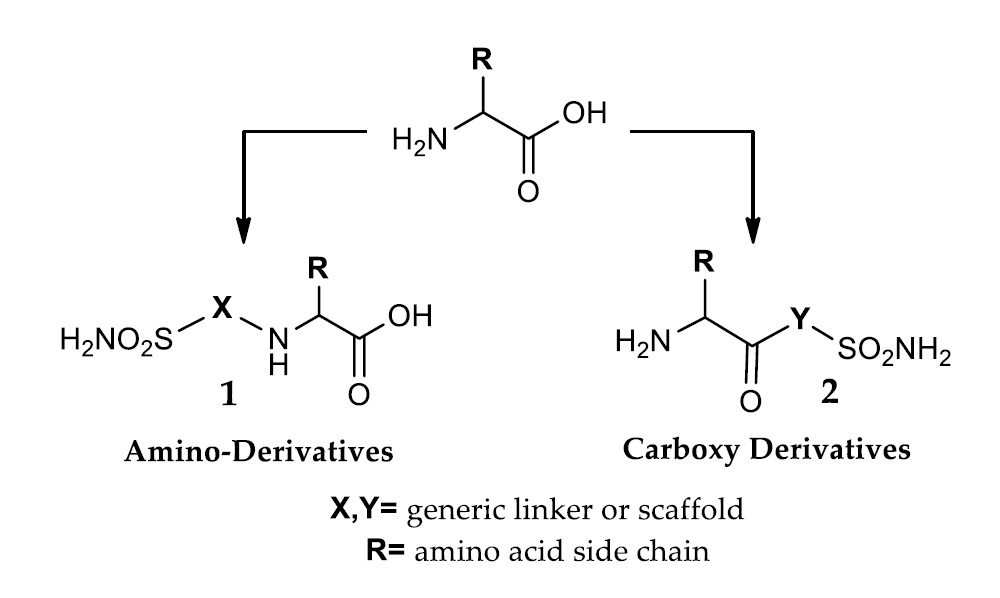
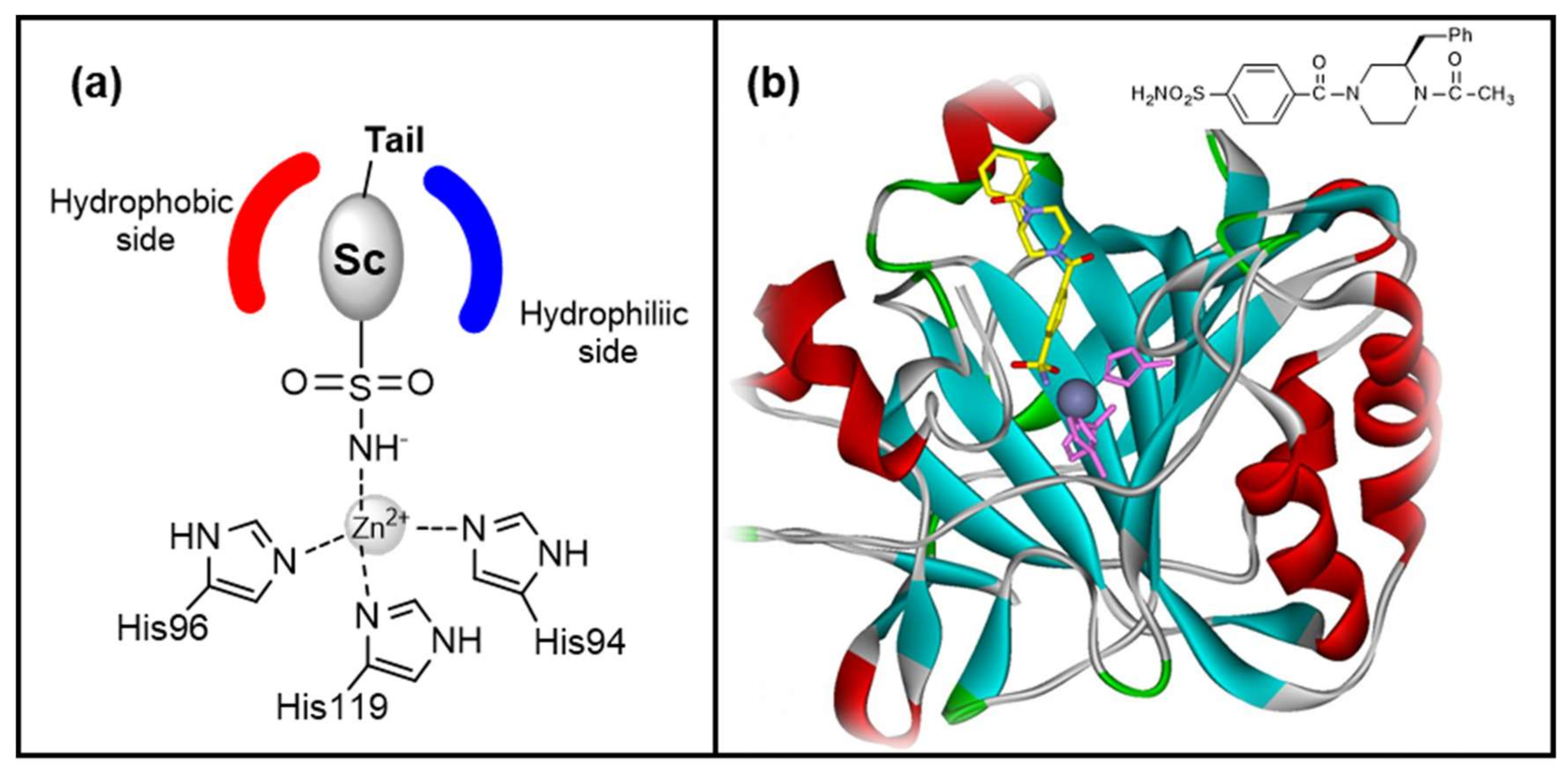
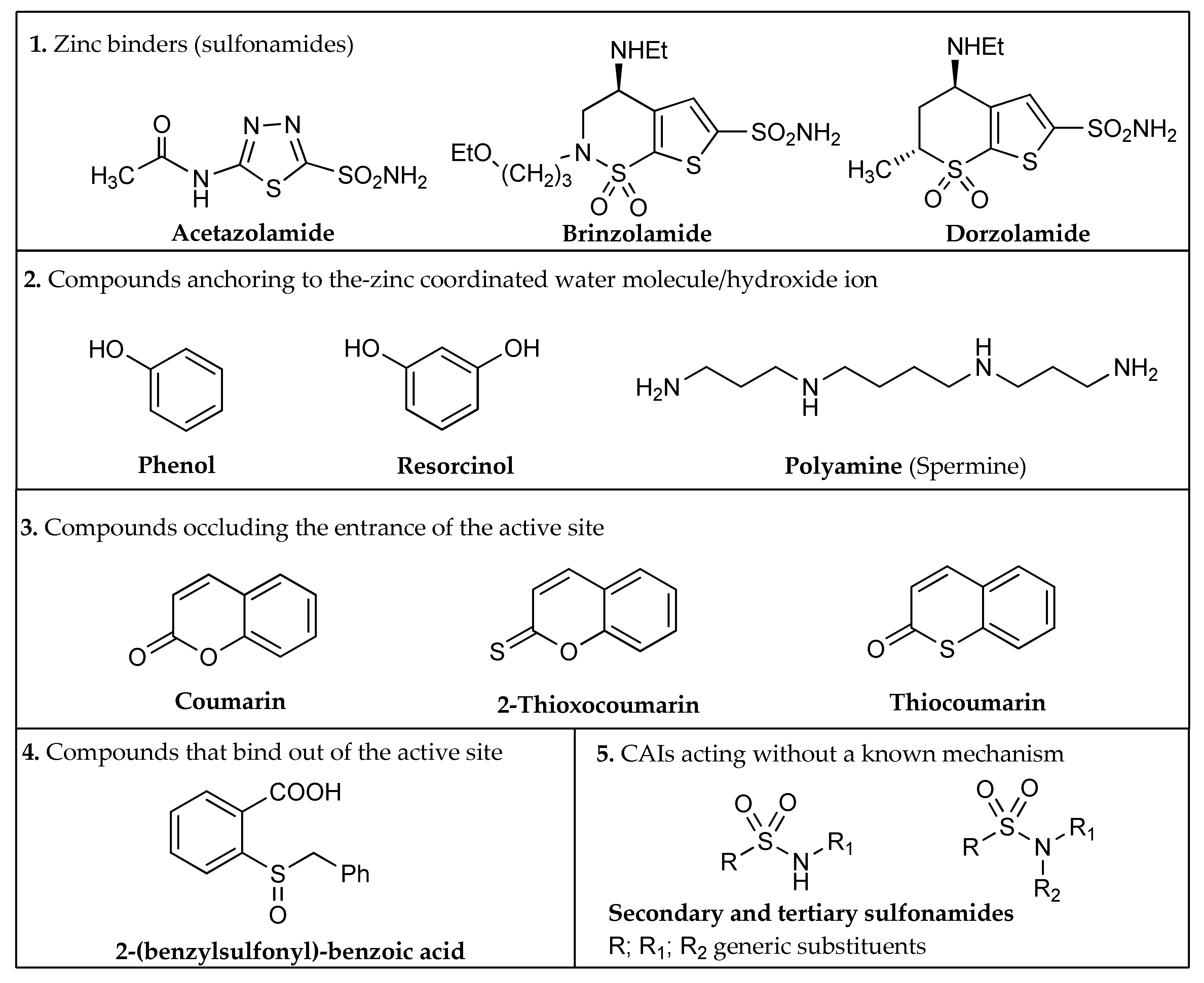
- Zinc binders, i.e., compounds that chelates the bivalent metal ion of the active site. This interaction interrupts the coordination between the Zn2+ atom and the water molecule/hydroxide ion and consequently blocks the enzymatic activity [21,24,25]. The mechanism is schematized in Figure 1a: the scaffold of these molecules (reported as “Sc”) may interact with one or both the halves of the active site, stabilizing the interaction with the ion in a tetrahedral geometry. This is the most important class of inhibitors, to which belong sulfonamides and their isosteres (sulfamates or sulfamides), dithiocarbamates, hydroxamate, etc. [3,24]. Sulfonamides are the most widely studied CAIs with at least 20 compounds in clinical use for decades [24]. Some examples (acetazolamide, brinzolamide and dorzolamide) are shown in Figure 2.
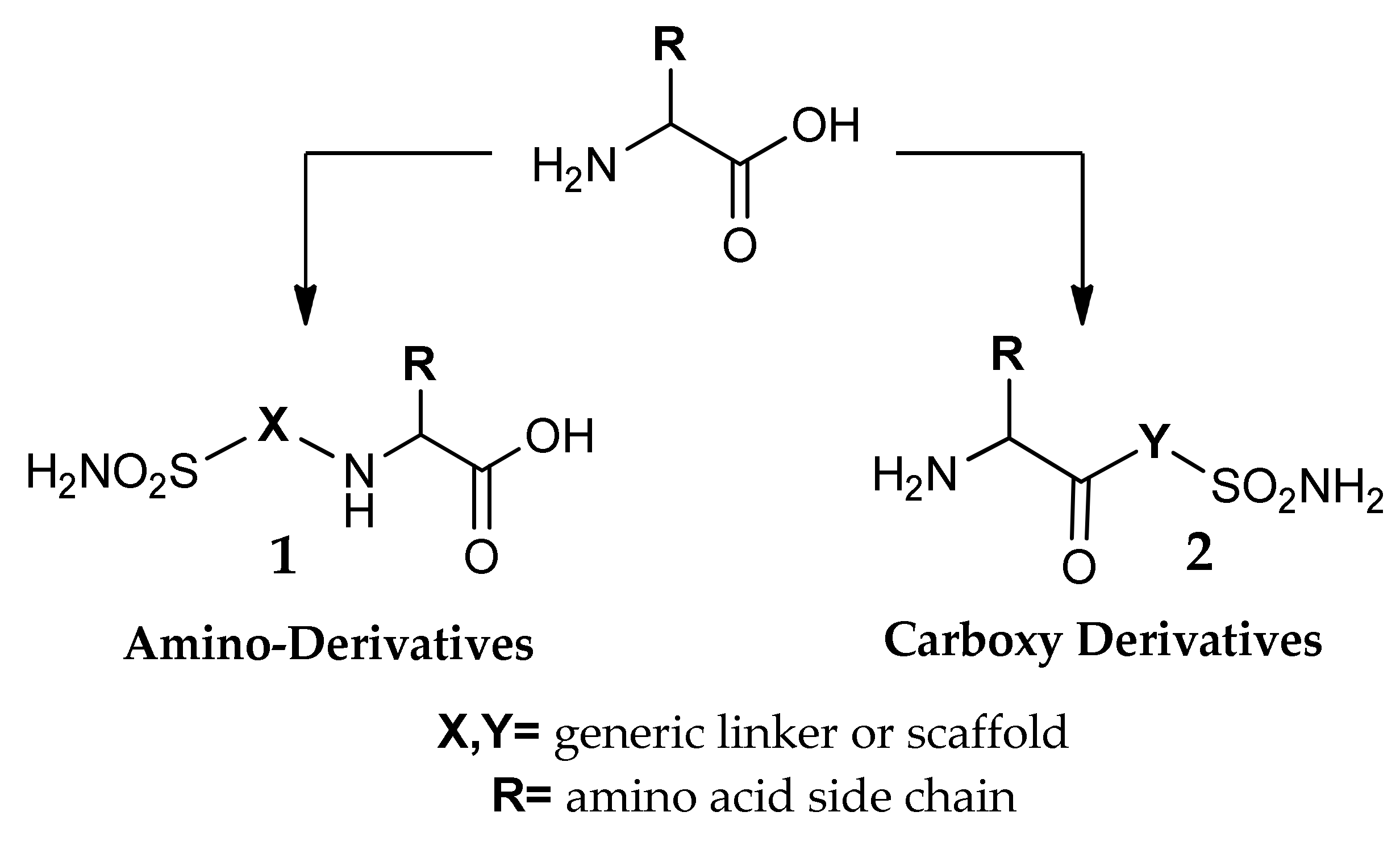
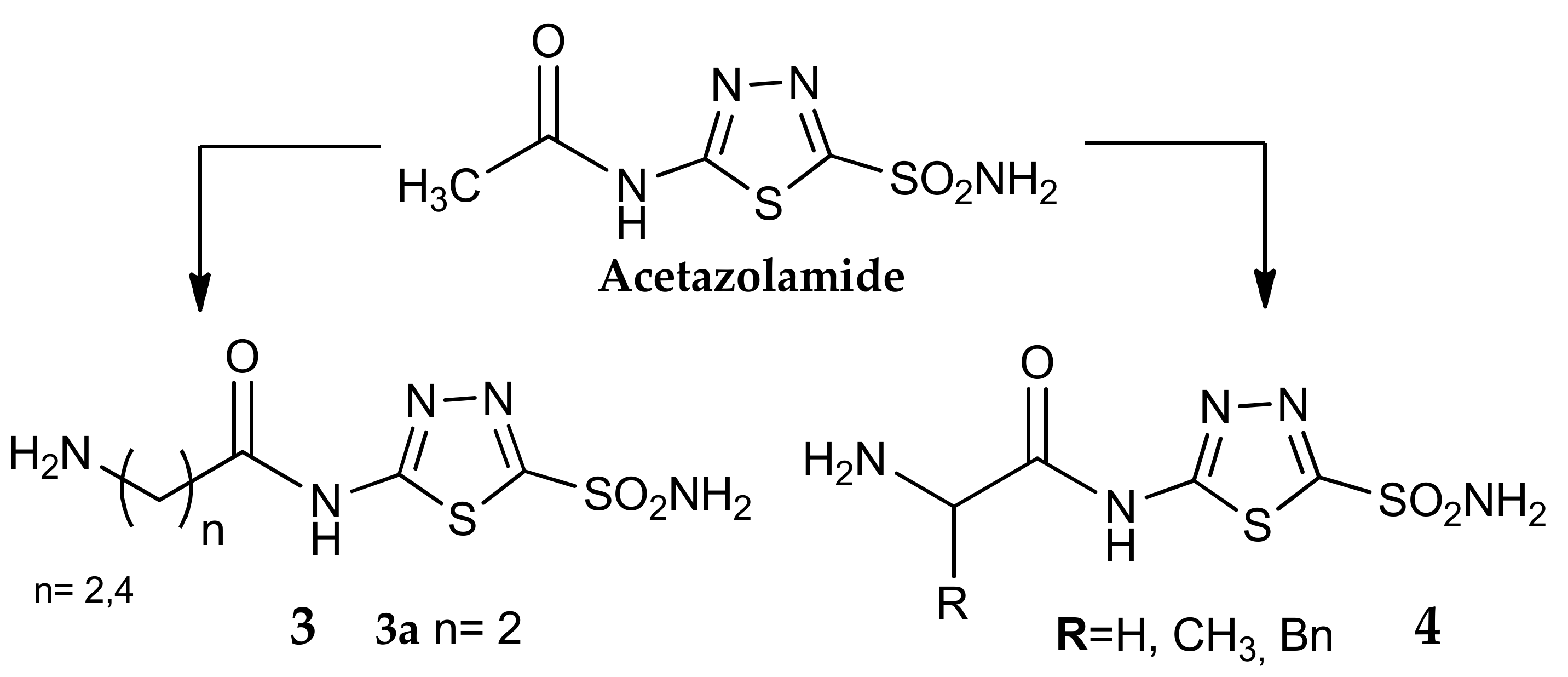
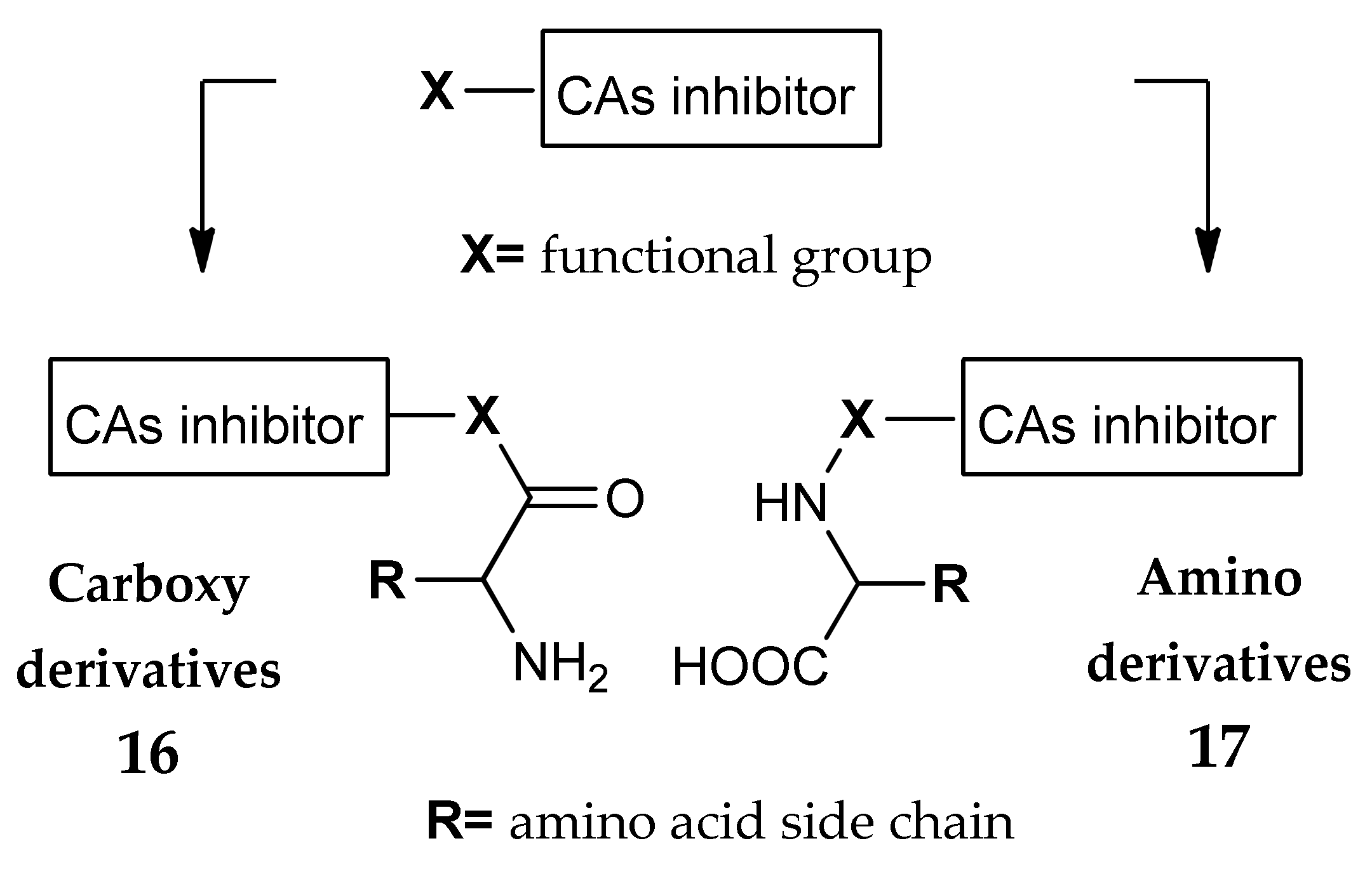
3. Amino Acyl as a Water-Solubilizing Tail
4. The Tail Approach to Carbonic Anhydrases Inhibitors Using Amino Acids
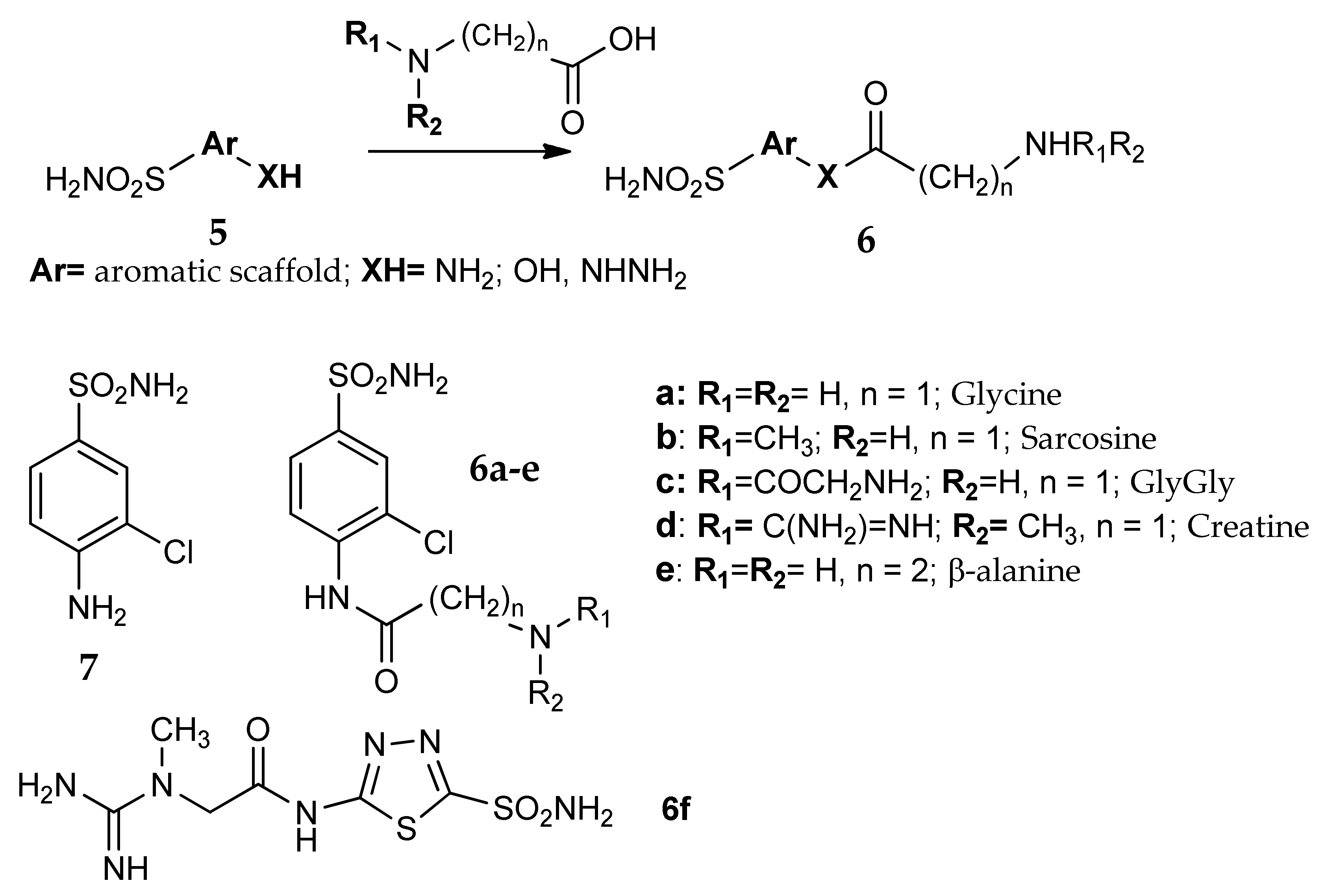
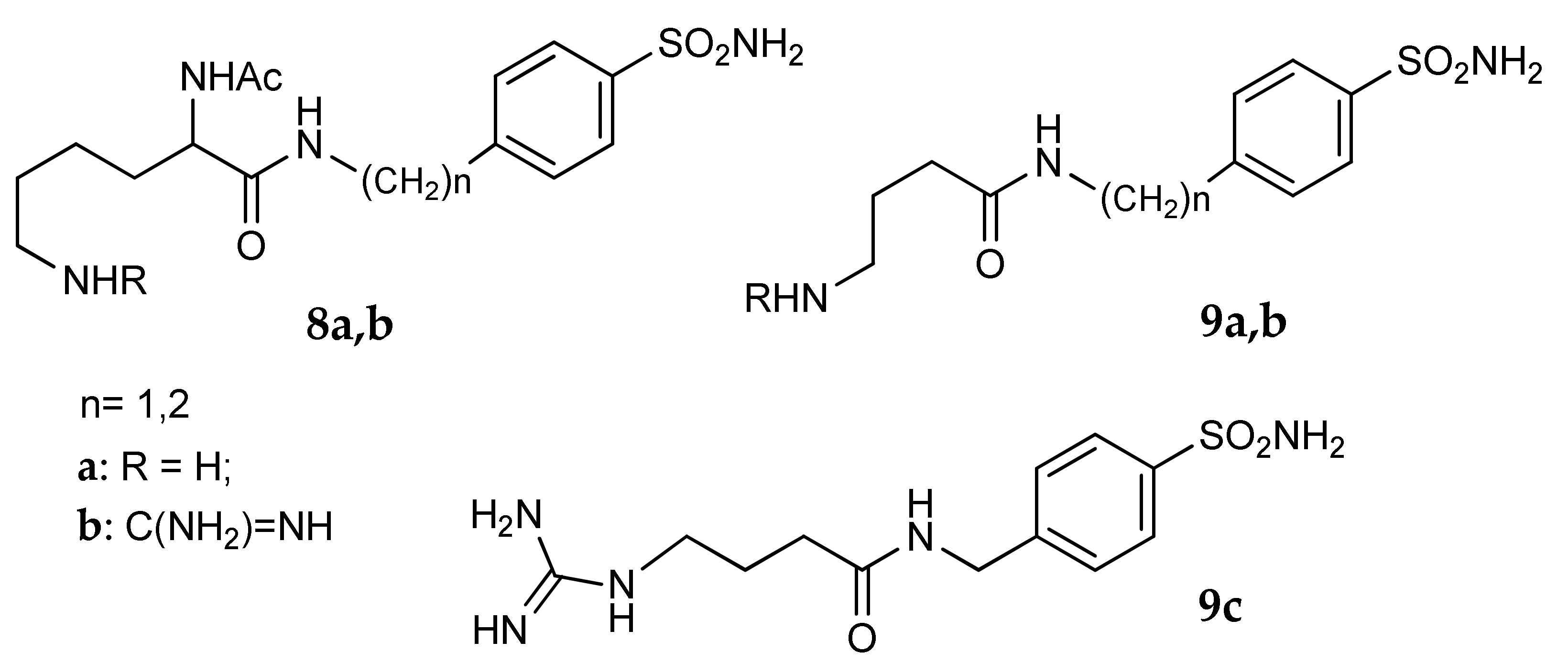
5. Amino Acids as Linkers
6. Dual Carbonic Anhydrase (CA) and Matrix Metalloproteinase (MMP) Inhibitors
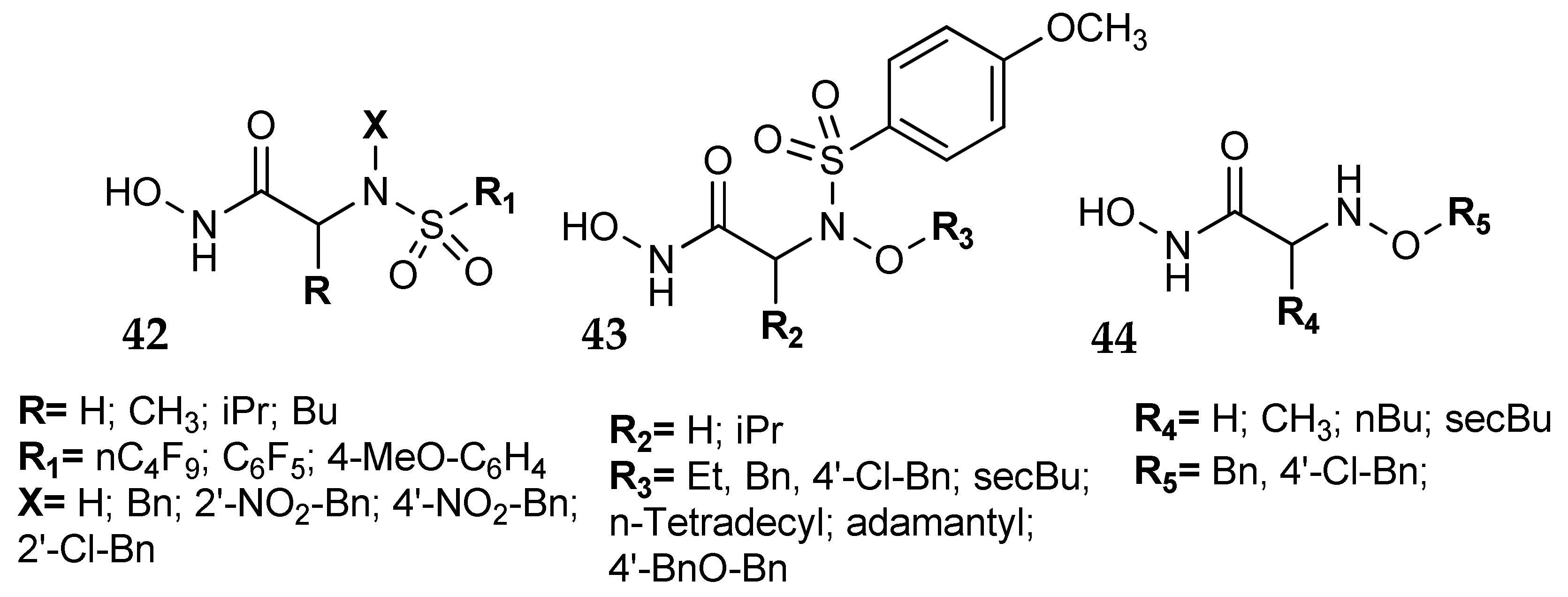
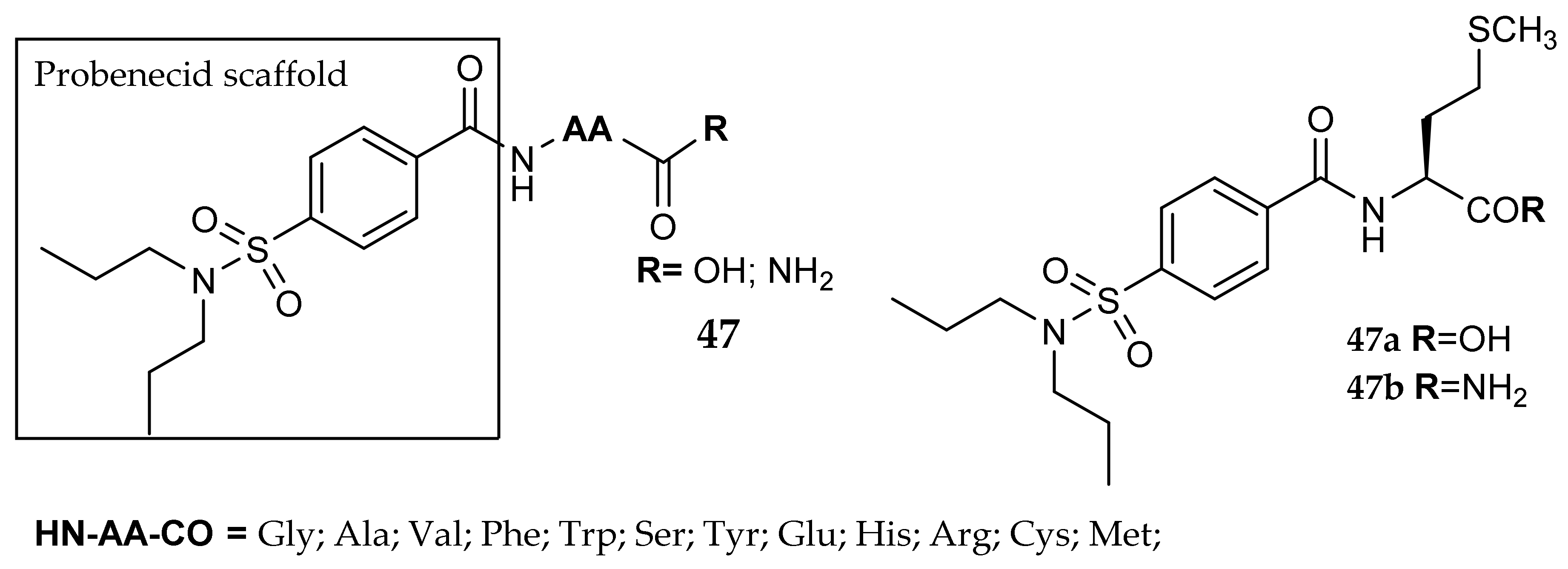
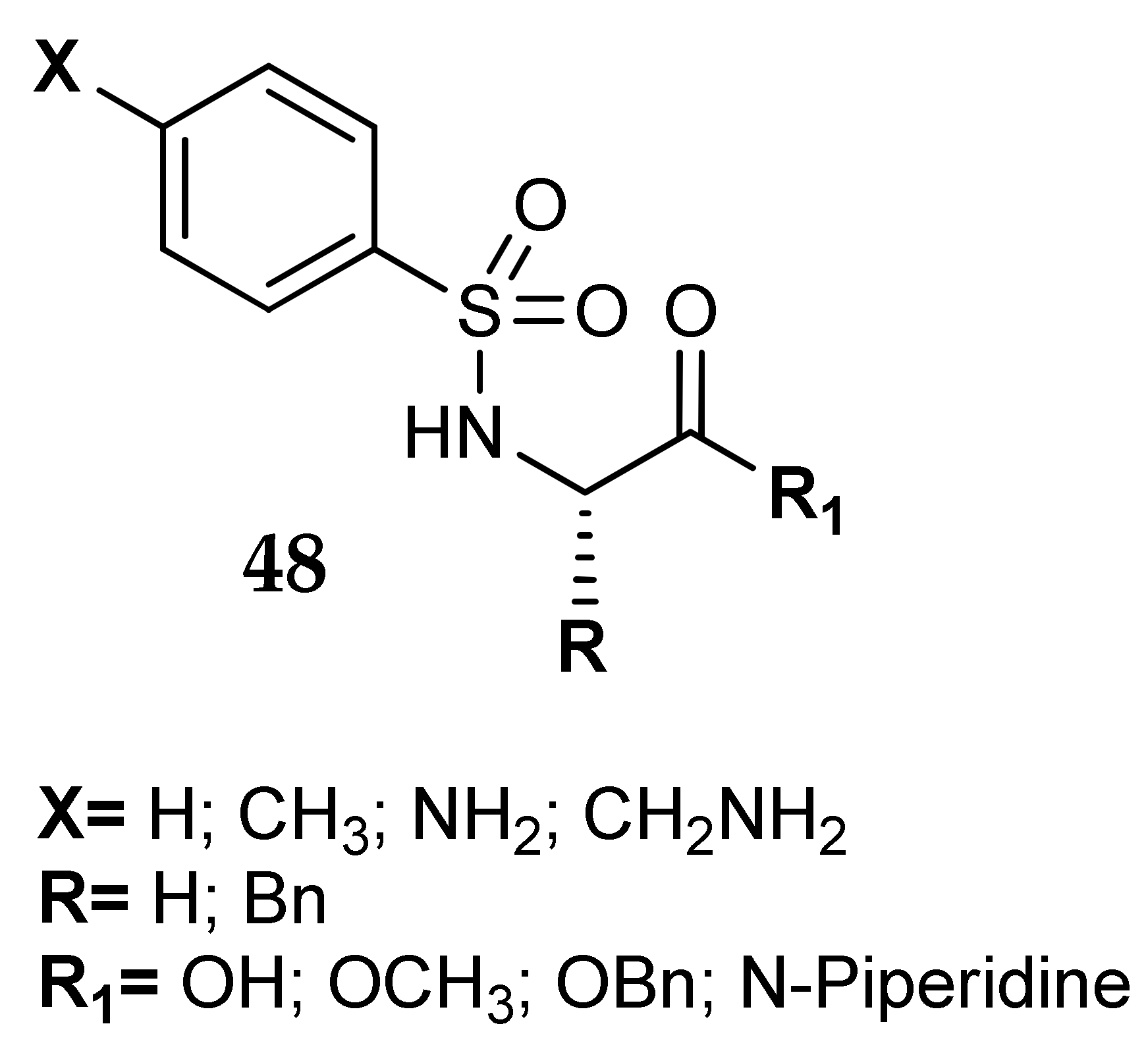
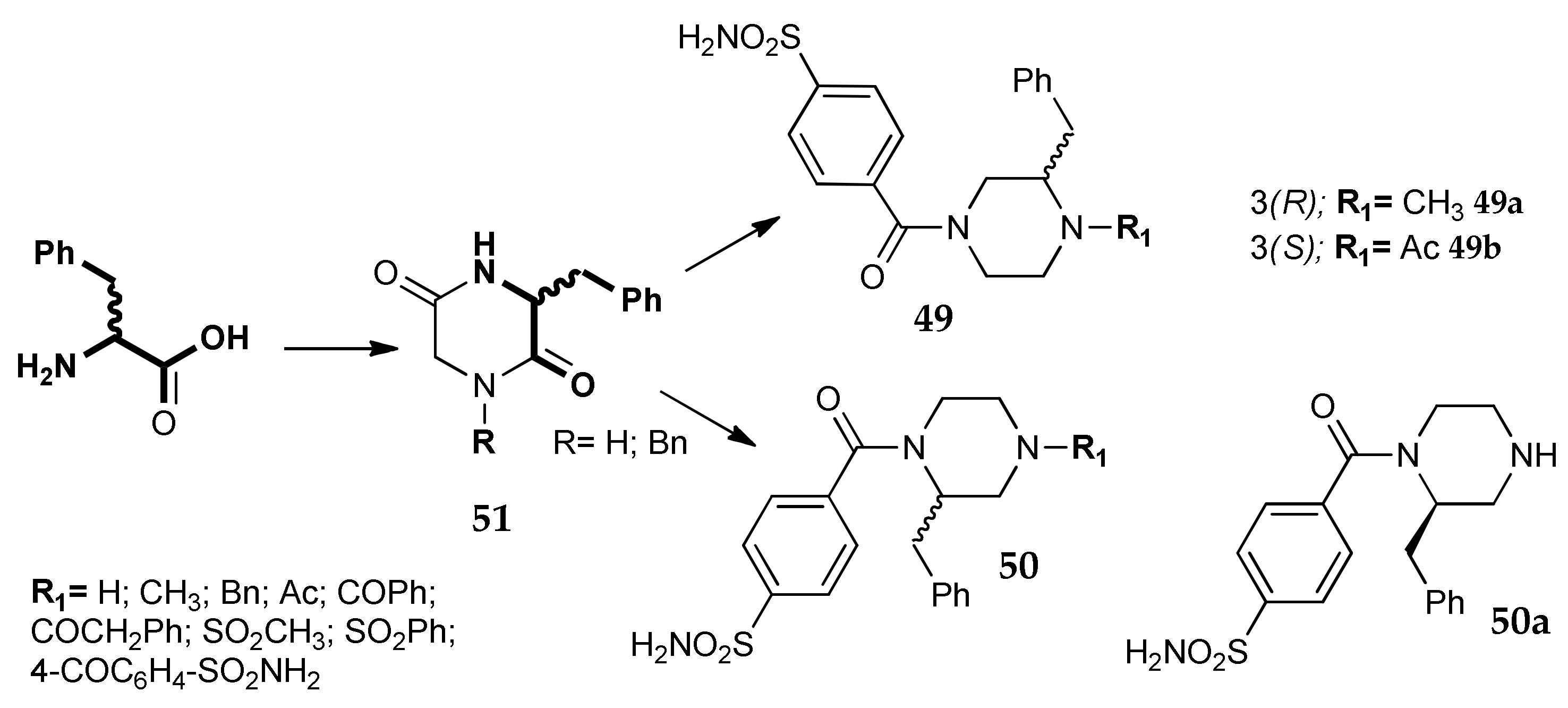
7. Other Approaches
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CA | carbonic anhydrase |
| hCA | human carbonic anhydrase |
| bCA | bovine carbonic anhydrase |
| CAIs | carbonic anhydrases inhibitors |
| MMPs | matrix metalloproteinases |
| IOP | intraocular pressure |
| Ala | alanine |
| Asn | asparagine |
| Asp | aspartic acid |
| Arg | arginine |
| β-Ala | beta-alanine |
| β-PhSer | beta-phenylserine |
| Boc | tert-butyloxycarbonyl |
| Cys | cysteine |
| DOPA | (3′,4′-dihydroxy)phenylalanine |
| Fmoc | fluorenylmethyloxycarbonyl |
| GABA | γ-aminobutyric acid |
| Gly | glycine |
| Gln | glutamine |
| Glu | glutamic acid |
| Ile | isoleucine |
| Kcat | turnover number |
| Kd | dissociation constant |
| Ki | inhibition constant |
| Leu | leucine |
| Lys | lysine |
| Met | Methionine |
| PDB | protein data bank |
| PET | positron emission tomography |
| Phe | phenylalanine |
| PhGly | phenylglycine |
| Pro | proline |
| Ser | serine |
| Thr | threonine |
| Trp | tryptophan |
| Tyr | tyrosine |
| Val | valine |
| ZBG | zinc binding group |
References
- Kupriyanova, E.; Pronina, N.; Los, D. Carbonic anhydrase—A universal enzyme of the carbon-based life. Photosynthetica 2017, 55, 3–19. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; De Simone, G. Carbonic Anhydrases: An Overview. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Elsevier: Amsterdam, Netherlands, 2015; pp. 3–13. [Google Scholar]
- D’Ambrosio, K.; De Simone, G.; Supuran, C.T. Human Carbonic Anhydrases: Catalytic Properties, Structural Features, and Tissue Distribution. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Elsevier: Amsterdam, Netherlands, 2015; pp. 17–30. [Google Scholar]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C. Carbonic Anhydrases and Metabolism. Metabolites 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.S.; Jakubzick, C.; Whittam, T.S.; Ferry, J.G. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. USA 1999, 96, 15184–15189. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [PubMed]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Silverman, D.N.; Lindskog, S. The Catalytic Mechanism of Carbonic Anhydrase: Implications of a Rate-Limiting Protolysis of Water. Acc. Chem. Res. 1988, 21, 30–36. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Carta, F.; Scozzafava, A.; Supuran, C.T. Antiglaucoma carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.; Mahon, B.; McKenna, R.; Frost, S. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Anticancer carbonic anhydrase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Supuran, C.T.; Carta, F. Antiobesity carbonic anhydrase inhibitors: A literature and patent review. Expert Opin. Ther. Pat. 2013, 23, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Alterio, V.; Supuran, C.T. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2013, 8, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, N.; Bua, S.; Ferraroni, M.; Nocentini, A.; Bonardi, A.; Bartolucci, G.; Durante, M.; Lucarini, L.; Chiapponi, D.; Dei, S.; et al. 2-Benzylpiperazine: A new scaffold for potent human carbonic anhydrase inhibitors. Synthesis, enzyme inhibition, enantioselectivity, computational and crystallographic studies and in vivo activity for a new class of intraocular pressure lowering agents. Eur. J. Med. Chem. 2018, 151, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzym. Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Karioti, A.; Carta, F.; Supuran, C.T. Phenols and polyphenols as Carbonic anhydrase inhibitors. Molecules 2016, 21, 1649. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, K.; Carradori, S.; Monti, S.M.; Buonanno, M.; Secci, D.; Vullo, D.; Supuran, C.T.; De Simone, G. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem. Commun. 2015, 51, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, R.; Schlüter, H. Amino acids: Chemistry, functionality and selected non-enzymatic post-translational modifications. J. Proteomics 2012, 75, 2275–2296. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, D.M.M. Thirteen decades of peptide synthesis: Key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, D.; Schmetterer, L.; Garhöfer, G.; Popa-Cherecheanu, A. Pharmacotherapy of Glaucoma. J. Ocul. Pharmacol. Ther. 2015, 31, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, M.F. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog. Retin. Eye Res. 2000, 19, 87–112. [Google Scholar] [CrossRef]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Development of Sulfonamide Carbonic Anhydrase Inhibitors. In Carbonic Anhydrase; CRC Press: Boca Raton, FL, USA, 2004; pp. 67–147. [Google Scholar]
- Maren, T.H. Carbonic anhydrase: Chemistry, physiology, and inhibition. Physiol. Rev. 1967, 47, 595–781. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Scozzafava, A.; Mincione, F.; Menabuoni, L.; Ilies, M.A.; Supuran, C.T. Carbonic anhydrase inhibitors: Water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J. Med. Chem. 2000, 43, 4884–4892. [Google Scholar] [CrossRef] [PubMed]
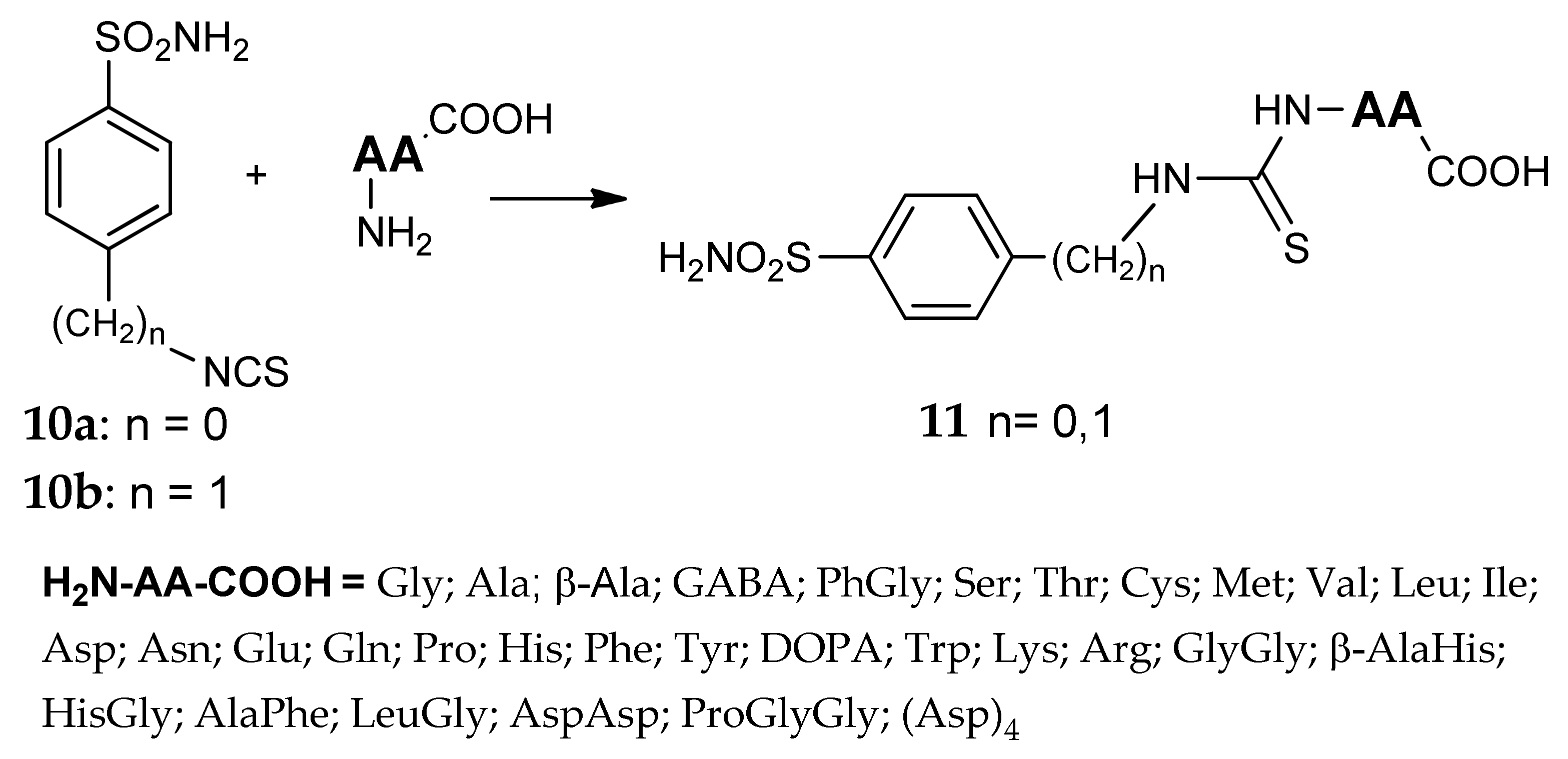
- Scozzafava, A.; Briganti, F.; Mincione, G.; Menabuoni, L.; Mincione, F.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis of water-soluble, aminoacyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J. Med. Chem. 1999, 42, 3690–3700. [Google Scholar] [CrossRef] [PubMed]
- Antonaroli, S.; Bianco, A.; Brufani, M.; Cellai, L.; Baido, G.L.; Potier, E.; Bonomi, L.; Perfetti, S.; Fiaschi, A.I.; Segre, G. Acetazolamide-like Carbonic Anhydrase Inhibitors with Topical Ocular Hypotensive Activity. J. Med. Chem. 1992, 35, 2697–2703. [Google Scholar] [CrossRef] [PubMed]
- Jayaweera, G.D.S.A.; MacNeil, S.A.; Trager, S.F.; Blackburn, G.M. Synthesis of 2-substituted-1,3,4-thiadiazole-5-sulphonamides as novel water-soluble inhibitors of carbonic anhydrase. Bioorg. Med. Chem. Lett. 1991, 1, 407–410. [Google Scholar] [CrossRef]
- Barboiu, M.; Supuran, C.T.; Menabuoni, L.; Scozzafava, A.; Mincione, F.; Briganti, F.; Mincione, G. Carbonic anhydrase inhibitors. Synthesis of topically effective intraocular pressure lowering agents derived from 5-(omega-aminoalkylcarboxamido)-1,3,4-thiadiazole-2-sulfonamide. J. Enzym. Inhib. 1999, 15, 23–46. [Google Scholar] [PubMed]
- Supuran, C.T.; Briganti, F.; Menabuoni, L.; Mincione, G.; Mincione, F.; Scozzafava, A. Carbonic anhydrase inhibitors—Part 78. Synthesis of water-soluble sulfonamides incorporating β-alanyl moieties, possessing long lasting- intraocular pressure lowering properties via the topical route. Eur. J. Med. Chem. 2000, 35, 309–321. [Google Scholar] [CrossRef]
- Maren, T.H.; Brechue, W.F.; Bar-Ilan, A. Relations among IOP reduction, ocular disposition and pharmacology of the carbonic anhydrase inhibitor ethoxzolamide. Exp. Eye Res. 1992, 55, 73–79. [Google Scholar] [CrossRef]
- Brechue, W.F.; Maren, T.H. pH and drug ionization affects ocular pressure lowering of topical carbonic anhydrase inhibitors. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2581–2587. [Google Scholar]
- Ceruso, M.; Del Prete, S.; Alothman, Z.; Osman, S.M.; Scozzafava, A.; Capasso, C.; Supuran, C.T. Synthesis of sulfonamides with effective inhibitory action against Porphyromonas gingivalis γ-carbonic anhydrase. Bioorg. Med. Chem. Lett. 2014, 24, 4006–4010. [Google Scholar] [CrossRef] [PubMed]
- Ceruso, M.; Bragagni, M.; Alothman, Z.; Osman, S.M.; Supuran, C.T. New series of sulfonamides containing amino acid moiety act as effective and selective inhibitors of tumor-associated carbonic anhydrase XII. J. Enzym. Inhib. Med. Chem. 2015, 30, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Casin, A.; Scozzafava, A.; Mincione, F.; Menabuoni, L.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis of water soluble sulfonamides incorporating a 4-sulfamoylphenylmethylthiourea scaffold, with potent intraocular pressure lowering properties. J. Enzym. Inhib. Med. Chem. 2002, 17, 333–343. [Google Scholar] [CrossRef] [PubMed]
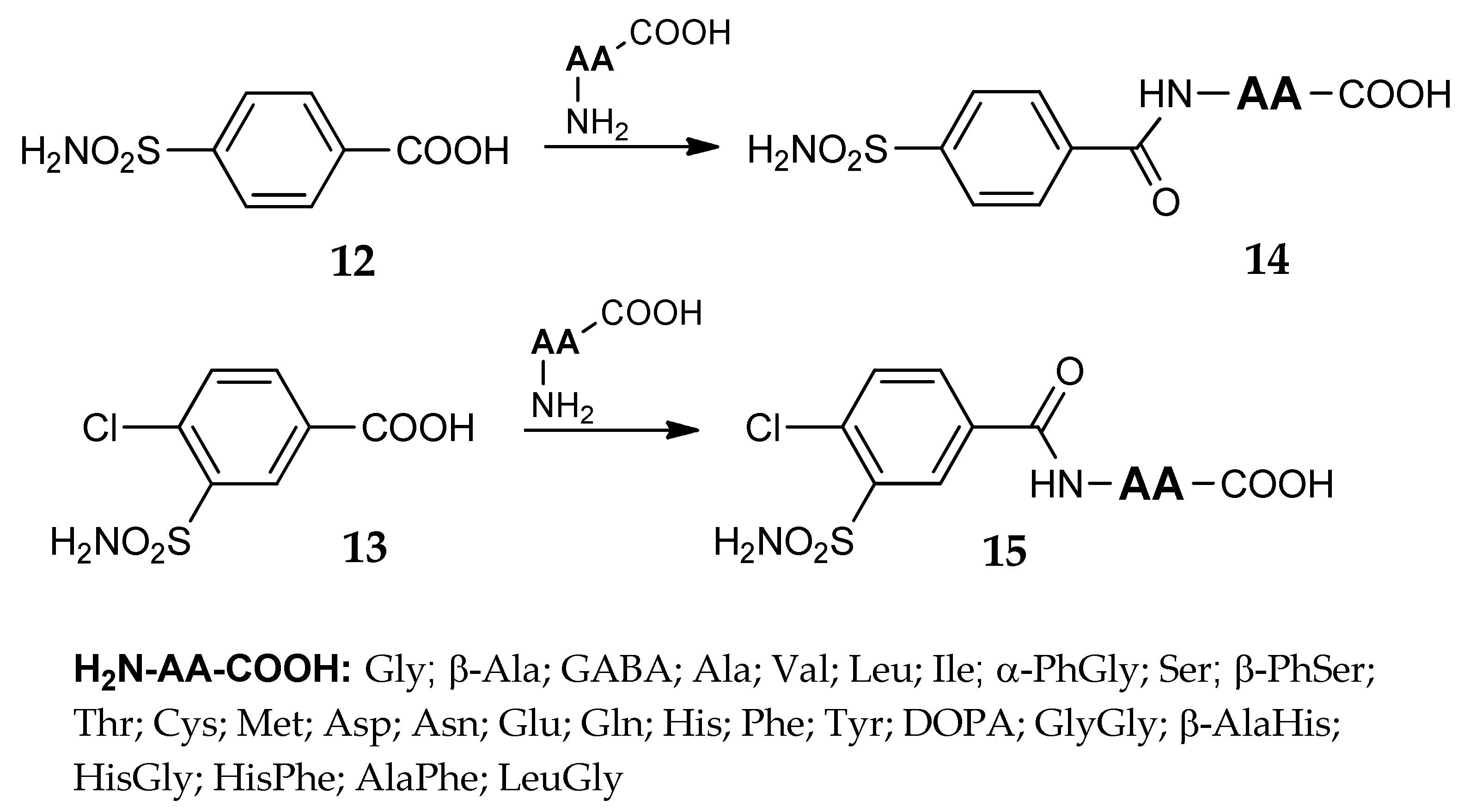
- Mincione, F.; Starnotti, M.; Menabuoni, L.; Scozzafava, A.; Casini, A.; Supuran, C.T. Carbonic anhydrase inhibitors: 4-Sulfamoyl-benzenecarboxamides and 4-chloro-3-sulfamoyl-benzenecarboxamides with strong topical antiglaucoma properties. Bioorg. Med. Chem. Lett. 2001, 11, 1787–1791. [Google Scholar] [CrossRef]
- Boriack, P.A.; Christianson, D.W.; Kingery-Wood, J.; Whitesides, G.M. Secondary Interactions Significantly Removed from the Sulfonamide Binding Pocket of Carbonic Anhydrase II Influence Inhibitor Binding Constants. J. Med. Chem. 1995, 38, 2286–2291. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Huang, S.G.; Whitesides, G.M. Lack of Effect of the Length of Oligoglycine- and Oligo(ethylene glycol)-Derived para-Substituents on the Affinity of Benzenesulfonamides for Carbonic Anhydrase II in Solution. J. Am. Chem. Soc. 1994, 116, 5057–5062. [Google Scholar] [CrossRef]
- Jain, A.; Whitesides, G.M.; Alexander, R.S.; Christianson, D.W. Identification of Two Hydrophobic Patches in the Active-Site Cavity of Human Carbonic Anhydrase II by Solution-Phase and Solid-State Studies and Their Use in the Development of Tight-Binding Inhibitors. J. Med. Chem. 1994, 37, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
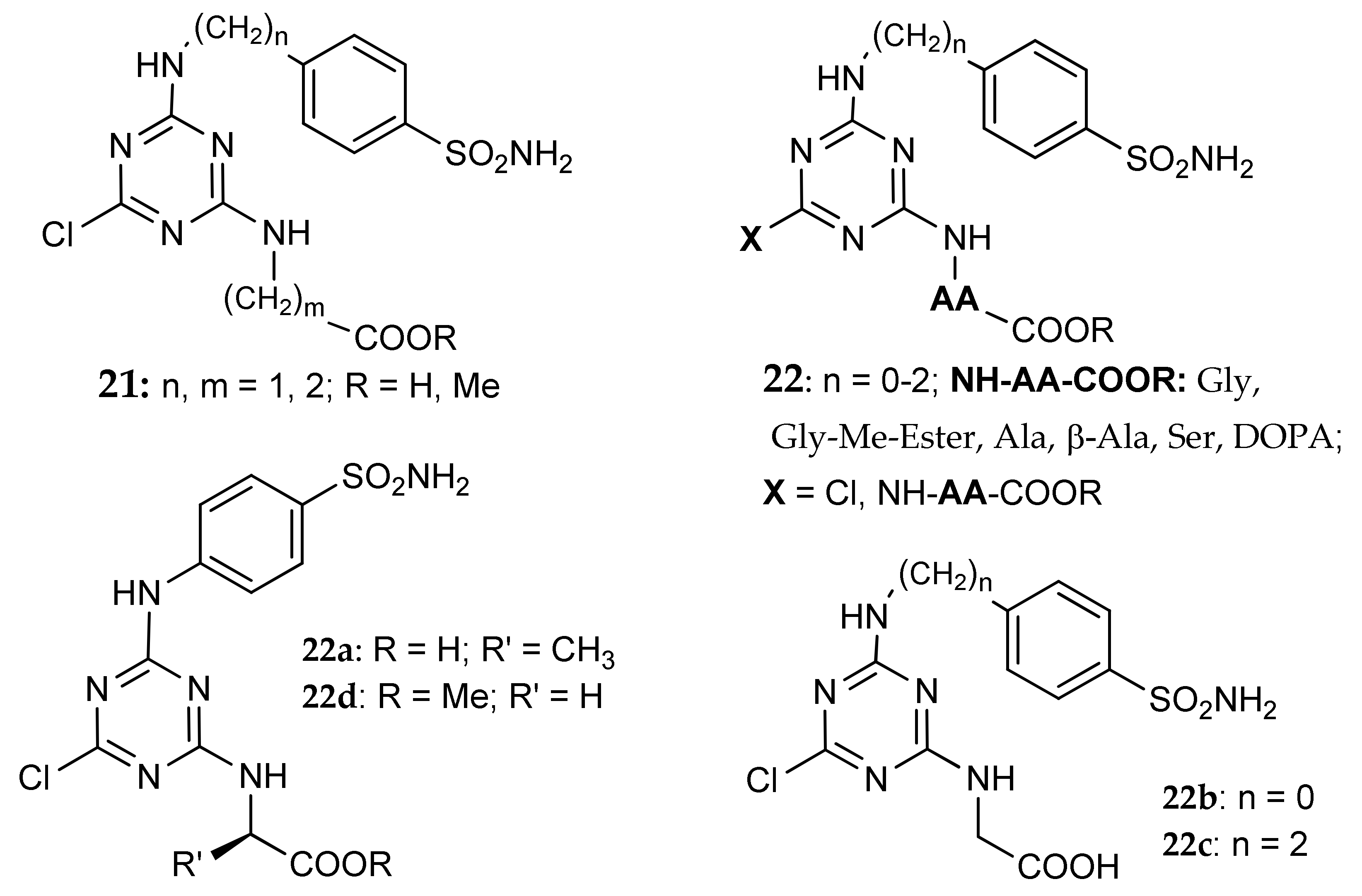
- Garaj, V.; Puccetti, L.; Fasolis, G.; Winum, J.Y.; Montero, J.L.; Scozzafava, A.; Vullo, D.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg. Med. Chem. Lett. 2005, 15, 3102–3108. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Garaj, V.; Maresca, A.; Wagner, J.; Avvaru, B.S.; Robbins, A.H.; Scozzafava, A.; McKenna, R.; Supuran, C.T. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: Solution and X-ray crystallographic studies. Bioorg. Med. Chem. 2011, 19, 3105–3119. [Google Scholar] [CrossRef] [PubMed]
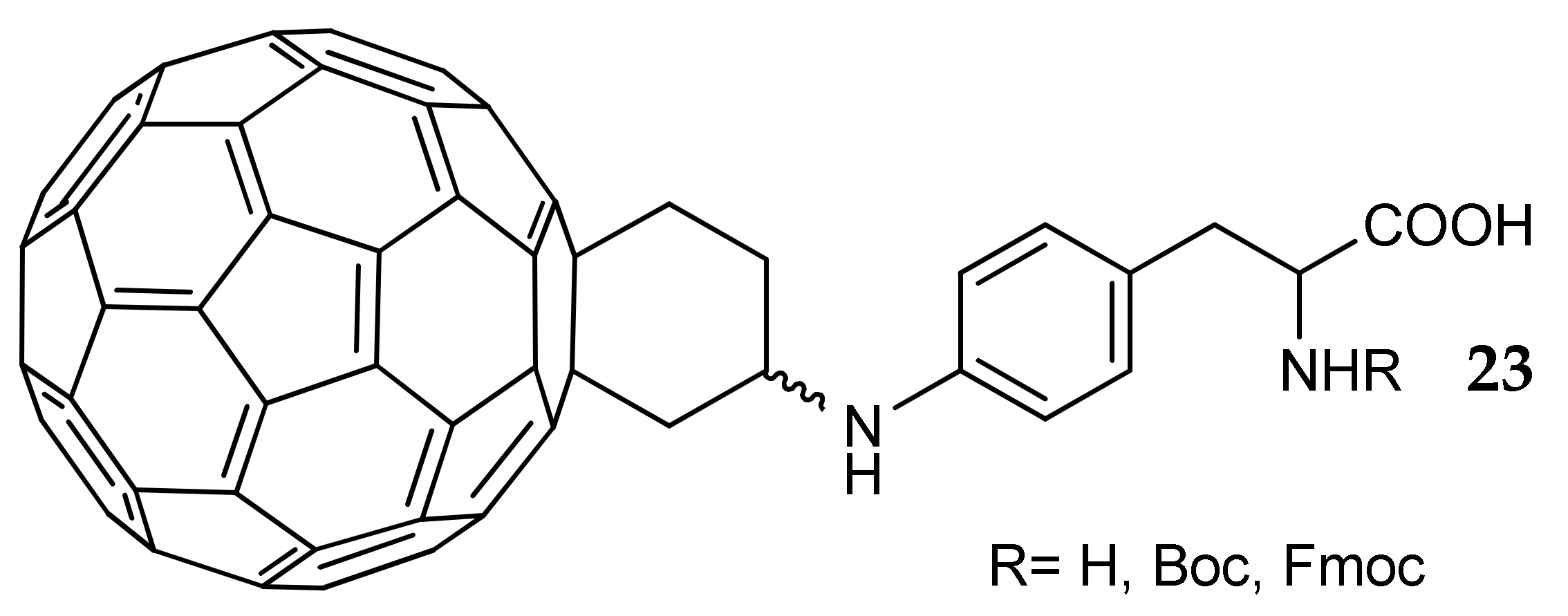
- Innocenti, A.; Durdagi, S.; Doostdar, N.; Amanda Strom, T.; Barron, A.R.; Supuran, C.T. Nanoscale enzyme inhibitors: Fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg. Med. Chem. 2010, 18, 2822–2828. [Google Scholar] [CrossRef] [PubMed]
- Nosik, D.N.; Lyalina, I.K.; Kalnlna, L.B.; Lobach, O.A.; Chataeva, M.S.; Rasnetsov, L.D. The antiretroviral agent Fullevir. Vopr. Virusol. 2009, 54, 41–43. [Google Scholar] [PubMed]
- Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J.A.; Wilkins, C.; Friedman, S.H.; DeCamp, D.L.; Kenyon, G.L. Synthesis of a Fullerene Derivative for the Inhibition of HIV Enzymes. J. Am. Chem. Soc. 1993, 115, 6510–6512. [Google Scholar] [CrossRef]
- Kang, B.; Yu, D.; Dai, Y.; Chang, S.; Chen, D.; Ding, Y. Cancer-cell targeting and photoacoustic therapy using carbon nanotubes as “bomb” agents. Small 2009, 5, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Fukuhara, A.; Saito, Y.; Yoshida, Y.; Sato, H.; Ohi, H.; Obata, M.; Mikata, Y.; Yano, S.; Niki, E. Antioxidant action of sugar-pendant C60 fullerenes. Bioorg. Med. Chem. Lett. 2009, 19, 5902–5904. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Paraskar, A.; Soni, S.; Mashelkar, R.A.; Sengupta, S. Fullerenol-cytotoxic conjugates for cancer chemotherapy. ACS Nano 2009, 3, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Flore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. X-ray Crystallography of Carbonic Anhydrase Inhibitors and Its Importance in Drug Design; Wiley: Hoboken, NJ, USA, 2009; pp. 73–138. [Google Scholar]
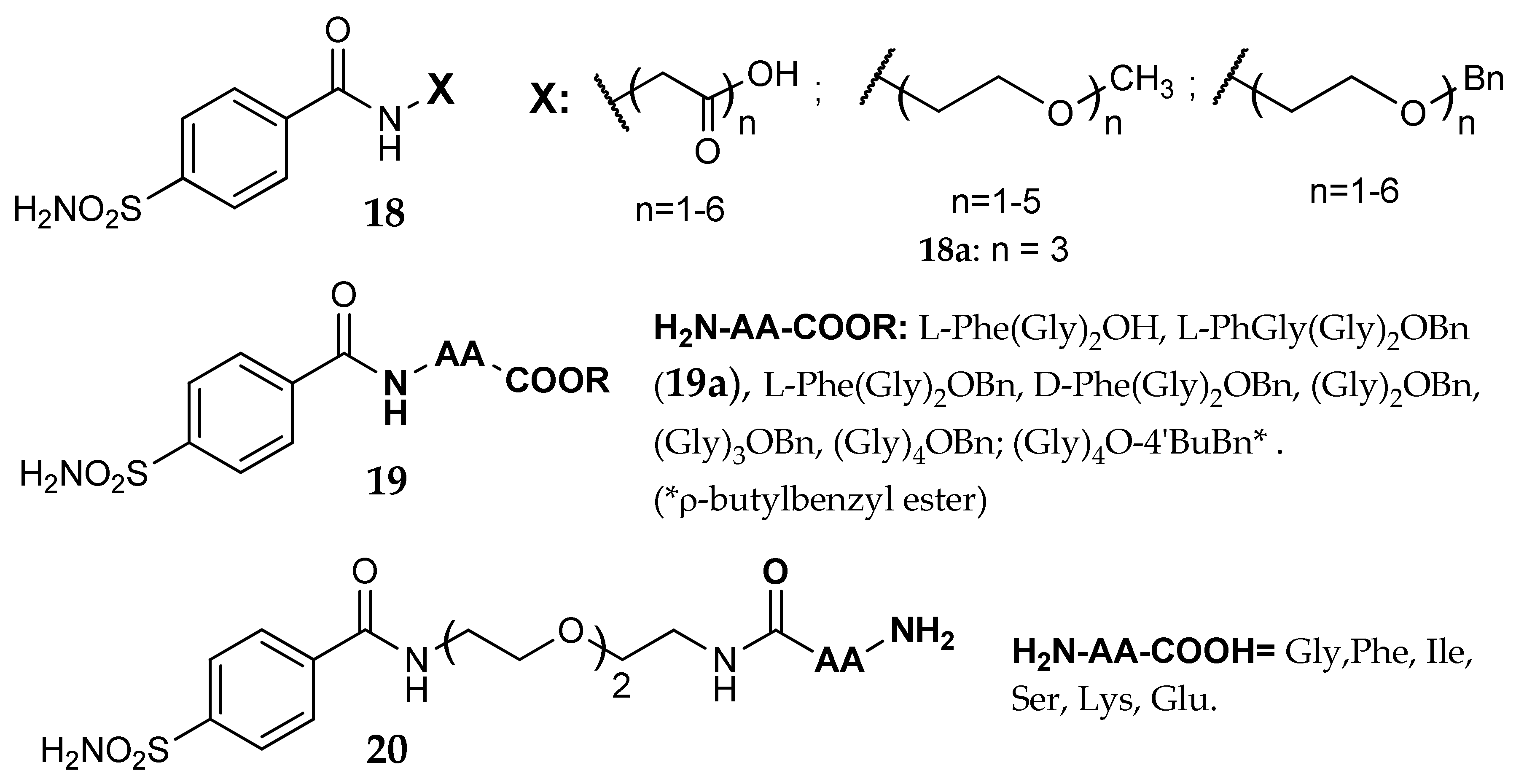
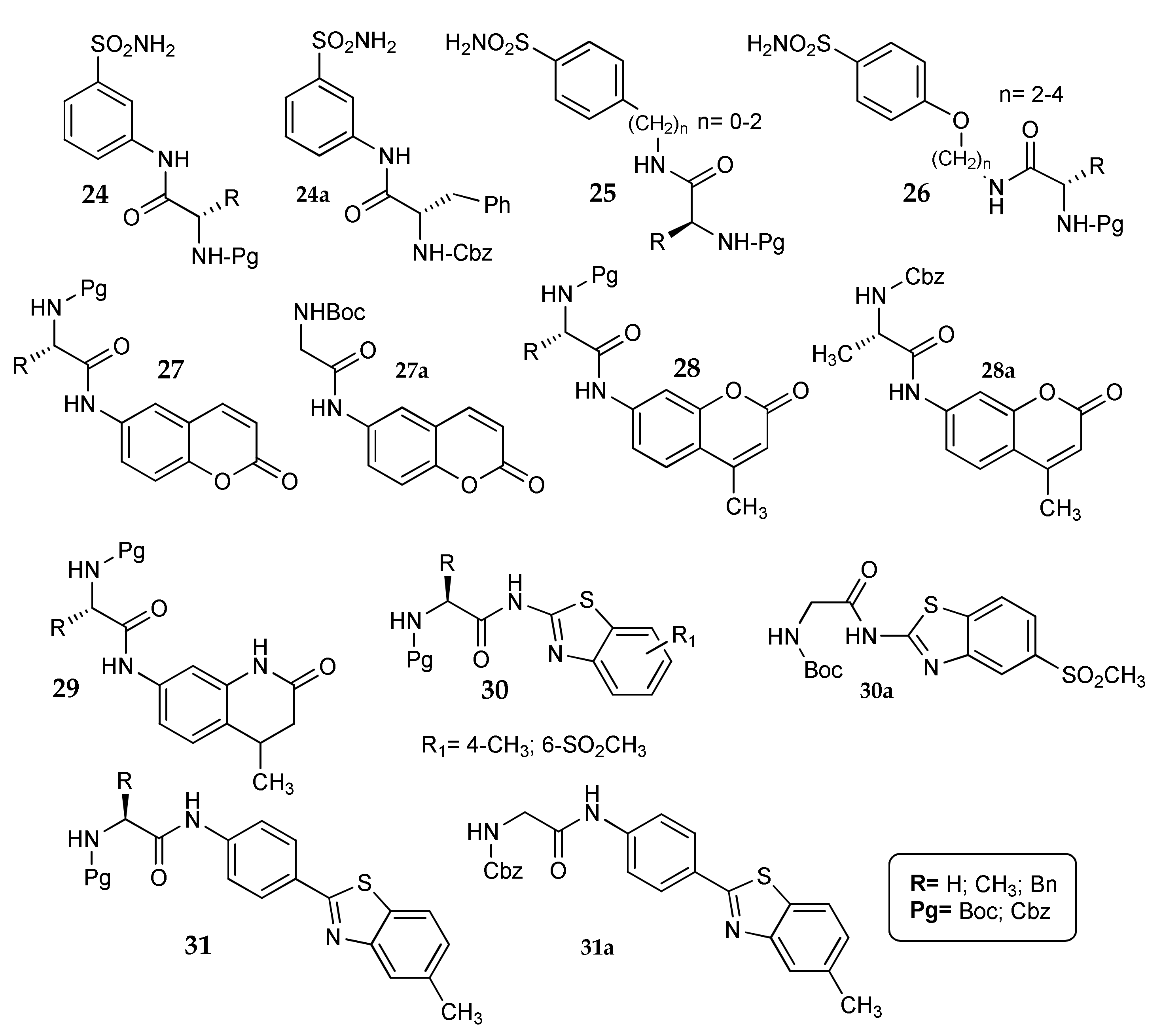
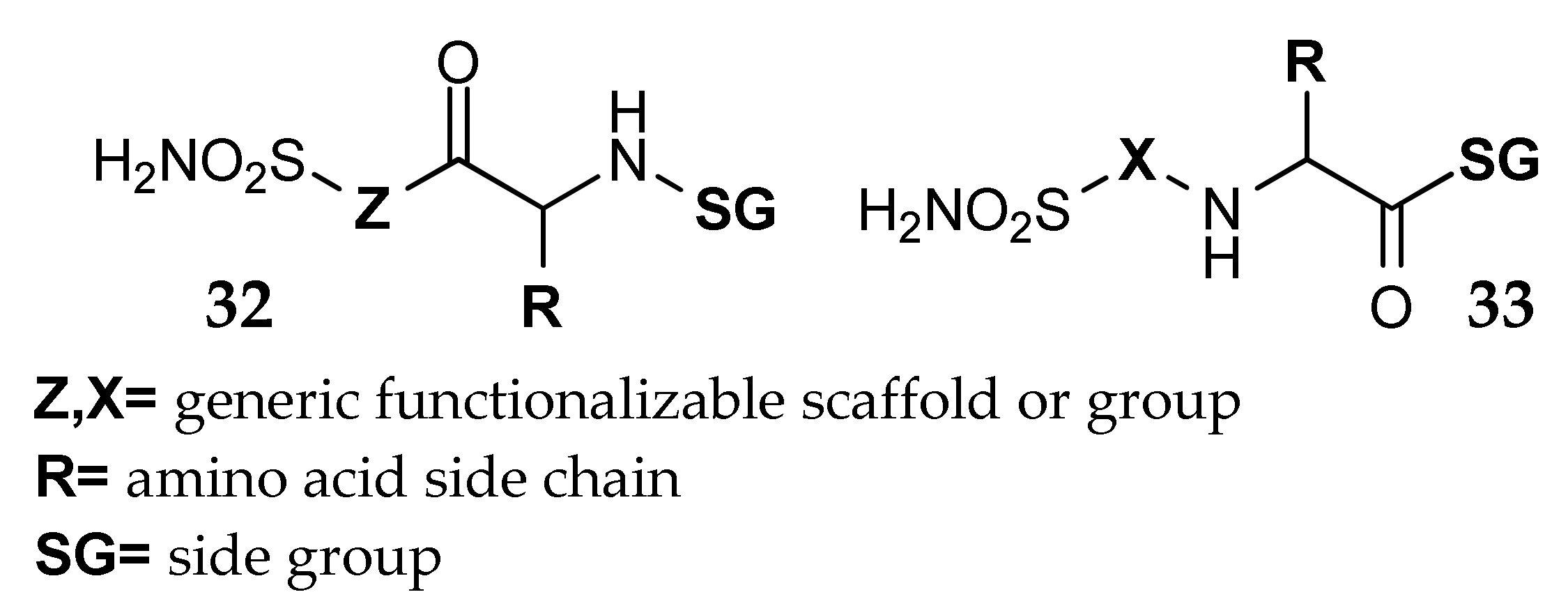
- Küçükbay, F.Z.; Küçükbay, H.; Tanc, M.; Supuran, C.T. Synthesis and carbonic anhydrase I, II, IV and XII inhibitory properties of N-protected amino acid-sulfonamide conjugates. J. Enzym. Inhib. Med. Chem. 2016, 31, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Küçükbay, F.Z.; Küçükbay, H.; Tanc, M.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory properties of amino acid–coumarin/quinolinone conjugates incorporating glycine, alanine and phenylalanine moieties. J. Enzym. Inhib. Med. Chem. 2016, 31, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Küçükbay, F.Z.; Buğday, N.; Küçükbay, H.; Tanc, M.; Supuran, C.T. Synthesis, characterization and carbonic anhydrase inhibitory activity of novel benzothiazole derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; Supuran, C.T.; McKenna, R. Non-classical inhibition of carbonic anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. [Google Scholar] [CrossRef] [PubMed]
- Žalubovskis, R. In a search for selective inhibitors of carbonic anhydrases: Coumarin and its bioisosteres—Synthesis and derivatization. Chem. Heterocycl. Compd. 2015, 51, 607–612. [Google Scholar] [CrossRef]
- Kolb, H.C.; Walsh, J.C.; Kasi, D.; Mocharla, V.; Wang, B.; Gangadharmath, U.B.; Duclos, B.A.; Chen, K.; Zhang, W.; Chen, G.; et al. Development of Triazole Derivatives as Molecular Imaging Probes for Carbonic Anhydrase-IX Using Click Chemistry. PCT International Application EP20080745260, 7 April 2008. 188p. [Google Scholar]
- Peeters, S.G.J.A.; Dubois, L.; Lieuwes, N.G.; Laan, D.; Mooijer, M.; Schuit, R.C.; Vullo, D.; Supuran, C.T.; Eriksson, J.; Windhorst, A.D.; et al. [18F]VM4-037 MicroPET Imaging and Biodistribution of Two In Vivo CAIX-Expressing Tumor Models. Mol. Imaging Biol. 2015, 17, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Kolb, H.C.; Walsh, J.C.; Mocharla, V.P.; Zhu, Z.; Haka, M.; Alpaugh, R.K.; Chen, D.Y.T.; Yu, J.Q. Biodistribution and radiation dosimetry of the carbonic anhydrase IX imaging agent [18F]VM4-037 determined from PET/CT scans in healthy volunteers. Mol. Imaging Biol. 2014, 16, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Rutkauskas, K.; Zubrienė, A.; Tumosienė, I.; Kantminienė, K.; Kažemėkaitė, M.; Smirnov, A.; Kazokaitė, J.; Morkūnaitė, V.; Čapkauskaitė, E.; Manakova, E.; et al. 4-Amino-substituted Benzenesulfonamides as Inhibitors of Human Carbonic Anhydrases. Molecules 2014, 19, 17356–17380. [Google Scholar] [CrossRef] [PubMed]
- Vaškevičienė, I.; Paketurytė, V.; Zubrienė, A.; Kantminienė, K.; Mickevičius, V.; Matulis, D. N-Sulfamoylphenyl- and N-sulfamoylphenyl-N-thiazolyl-β-alanines and their derivatives as inhibitors of human carbonic anhydrases. Bioorg. Chem. 2017, 75, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Ceruso, M.; Antel, S.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Inhibition studies of new ureido-substituted sulfonamides incorporating a GABA moiety against human carbonic anhydrase isoforms I-XIV. Bioorg. Med. Chem. 2014, 22, 6768–6775. [Google Scholar] [CrossRef] [PubMed]
- Moeker, J.; Mahon, B.P.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; McKenna, R.; Poulsen, S.A. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J. Med. Chem. 2014, 57, 8635–8645. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lu, Y.T.; Sun, Y.; Shi, Z.H.; Li, N.G.; Tang, Y.P.; Duan, J.A. Recent opportunities in matrix metalloproteinase inhibitor drug design for cancer. Expert Opin. Drug Discov. 2018, 13, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Eckhardt, S.G. Development of Matrix Metalloproteinase Inhibitors in Cancer Therapy. JNCI J. Natl. Cancer Inst. 2001, 93, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Supuran, C.T. Carbonic anhydrase and matrix metalloproteinase inhibitors: Sulfonylated amino acid hydroxamates with MMP inhibitory properties act as efficient inhibitors of CA isozymes I, II, and IV, and N-hydroxysulfonamides inhibit both these zinc enzymes. J. Med. Chem. 2000, 43, 3677–3687. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Orlandini, E.; Nencetti, S.; Rossello, A.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase and matrix metalloproteinase inhibitors. Inhibition of human tumor-associated isozymes IX and cytosolic isozyme I and II with sulfonylated hydroxamates. Bioorg. Med. Chem. 2007, 15, 2298–2311. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, N.; Obaidi, O.A.; Senturk, M.; Astley, D.; Ekinci, D.; Supuran, C.T. Synthesis and biological activity of novel thiourea derivatives as carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2015, 30, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Akdemir, A.; Carradori, S.; Stefanucci, A.; Macedonio, G.; Ceruso, M.; Supuran, C.T. Exploring new Probenecid-based carbonic anhydrase inhibitors: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. 2015, 23, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Fidan, I.; Salmas, R.E.; Arslan, M.; Şentürk, M.; Durdagi, S.; Ekinci, D.; Şentürk, E.; Coşgun, S.; Supuran, C.T. Carbonic anhydrase inhibitors: Design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulfonamide derivatives. Bioorg. Med. Chem. 2015, 23, 7353–7358. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiaramonte, N.; Romanelli, M.N.; Teodori, E.; Supuran, C.T. Amino Acids as Building Blocks for Carbonic Anhydrase Inhibitors. Metabolites 2018, 8, 36. https://doi.org/10.3390/metabo8020036
Chiaramonte N, Romanelli MN, Teodori E, Supuran CT. Amino Acids as Building Blocks for Carbonic Anhydrase Inhibitors. Metabolites. 2018; 8(2):36. https://doi.org/10.3390/metabo8020036
Chicago/Turabian StyleChiaramonte, Niccolò, Maria Novella Romanelli, Elisabetta Teodori, and Claudiu T. Supuran. 2018. "Amino Acids as Building Blocks for Carbonic Anhydrase Inhibitors" Metabolites 8, no. 2: 36. https://doi.org/10.3390/metabo8020036