
Integrating Multiple Analytical Datasets to Compare Metabolite Profiles of Mouse Colonic-Cecal Contents and Feces

Abstract
:1. Introduction
2. Results and Discussion
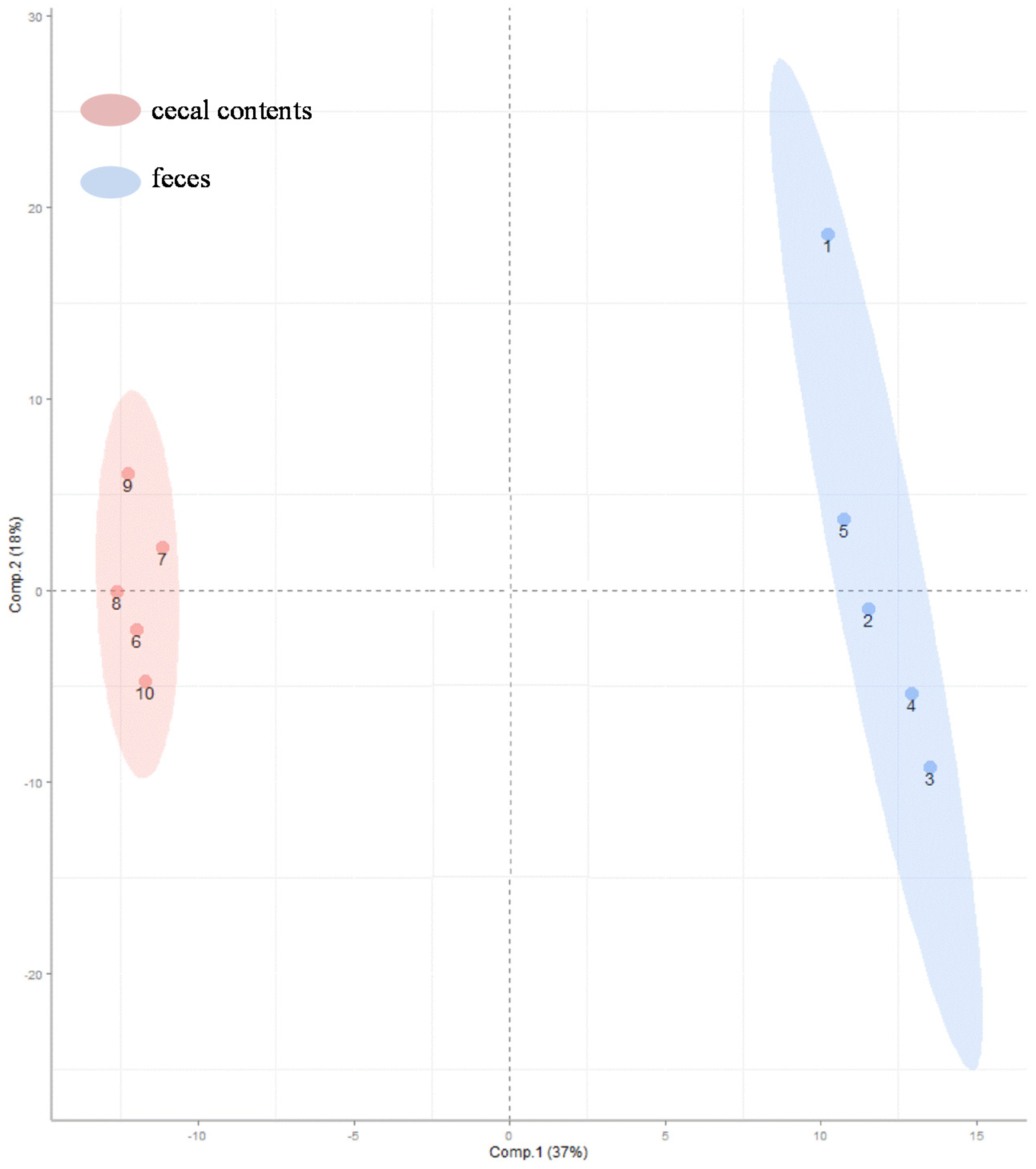
2.1. Metabolomic Comparisons of Sample Types

2.2. Metabolites Unique To Colonic-Cecal Contents or Feces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Colon-cecal contents | Feces |
|---|---|
| Unique metabolites | |
| 1-Hexadecanoyl-2-octadecadienoyl-sn-glycero-3-phosphocholine | 1H-Indole-3-carboxylic acid |
| L-Alanyl-L-norleucine | 1-Myristoyl-sn-glycero-3-phosphocholine |
| Leu-Val | 1-Octadecanoyl-sn-glycero-3-phosphocholine |
| N-Palmitoylsphingosine | Betaine |
| Palmityl-L-carnitine | L-Carnitine |
| Pregnan-20-one, 17-(acetyloxy)-3-hydroxy-6-methyl-, (3b,5b,6a)-(A) | Nicotinamide adenine dinucleotide (NAD) |
| 3a,12b-Dihydroxy-5b-cholanoic acid-(A) | Oxyquinoline |
| Deoxythymidine monophosphate (dTMP) | Pregnan-20-one, 17-(acetyloxy)-3-hydroxy-6-methyl-, (3b,5b,6a)-(B) |
| 13-hydroxy-9Z,11E-octadecadienoic acid | |
| 3a,12b-Dihydroxy-5b-cholanoic acid-(B) | |
| UDP-N-acetyl-D-galactosamine | |
| Respective biochemical pathways | |
| Ether lipid metabolism | Primary bile acid biosynthesis |
| Glycerophospholipid metabolism | Tryptophan metabolism |
| Metabolic pathways | Biosynthesis of secondary metabolites |
| Choline metabolism in cancer | Ether lipid metabolism |
| Valine, leucine and isoleucine degradation / biosynthesis | Glycerophospholipid metabolism |
| Sphingolipid metabolism | Metabolic pathways |
| Bile secretion | Choline metabolism in cancer |
| Fatty acid degradation | Glycine, serine and threonine metabolism |
| Steroid hormone biosynthesis | ABC transporters |
| Secondary bile acid biosynthesis | Bile secretion |
| Pyrimidine metabolism | Fatty acid degradation |
| Folate biosynthesis | |
| Nicotinate and nicotinamide metabolism | |
| Quinolines | |
| Phenylalanine, tyrosine and tryptophan biosynthesis | |
| Steroid hormone biosynthesis | |
| Biosynthesis of unsaturated fatty acids | |
| Linoleic acid metabolism | |
| Secondary bile acid biosynthesis | |
| Mucin type O-Glycan biosynthesis | |
| Amino sugar and nucleotide sugar metabolism | |
| Mucin type O-Glycan biosynthesis | |
2.3. Metabolite Patterns and Networks



3. Experimental Section
3.1. Animals, Diets, and Treatment
3.2. Sample Extraction
3.3. GC-TOF Analysis
3.4. LC-Q-TOF Data Collection and Analysis
3.5. Data Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Akin, H.; Tözün, N. Diet, microbiota, and colorectal cancer. J. Clin. Gastroenterol. 2014, 48, S67–S69. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Cardona, F.; Tinahones, F.J.; Queipo-Ortuño, M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Jankevics, A.; Burgess, K.E.; Breitling, R.; Barrett, M.P. IDEOM: an Excel interface for analysis of LC-MS-based metabolomics data. Bioinformatics 2012, 28, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. Novartis Found Symp. 2002, 247, 91–101. [Google Scholar]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut microbial activity, implications for health and disease: the potential role of metabolite analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef]
- Zeng, H.; Lazarova, D.L.; Bordonaro, M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J. Gastrointest. Oncol. 2014, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Geypens, B.; Luypaerts, A.; Hiele, M.; Ghoos, Y.; Rutgeerts, P. Digestibility of cooked and raw egg protein in humans as assessed by stable isotope techniques. J. Nutr. 1998, 128, 1716–1722. [Google Scholar] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC. Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Nordgaard, I.; Mortensen, P.B. Digestive processes in the human colon. Nutrition 1995, 11, 37–45. [Google Scholar] [PubMed]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.E. Colonic bacterial metabolites and human health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Setoguchi, T.; Katsuki, T. Effect of cecum and appendix on 7 alpha-dehydroxylation and 7 beta-epimerization of chenodeoxycholic acid in the rabbit. J. Lipid Res. 1980, 21, 215–222. [Google Scholar] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: the neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Bergkvist, A.; Rusnakova, V.; Sindelka, R.; Garda, J.M.; Sjögreen, B.; Lindh, D.; Forootan, A.; Kubista, M. Gene expression profiling—Clusters of possibilities. Methods 2010, 50, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, R. Chemometrics with R: Multivariate Data Analysis in the Natural Sciences and Life Sciences. Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar]
- Okada, T.; Afendi, F.M.; Altaf-Ul-Amin, M.; Takahashi, H.; Nakamura, K.; Kanaya, S. Metabolomics of medicinal plants: the importance of multivariate analysis of analytical chemistry data. Curr. Comput. Aided Drug Des. 2010, 6, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, B.S. Role of the gut microbiota in human nutrition and metabolism. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 4), 9–17. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, do Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Weckwerth, W.; Wenzel, K.; Fiehn, O. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 2004, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Extending the breadth of metabolite profiling by gas chromatography coupled to mass spectrometry. Trends Analyt. Chem. 2008, 27, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R.H. A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M. Setup and annotation of metabolomic experiments by integrating biological and mass spectrometric metadata. In Data intergration in the life Sciences, Proceedings of Second International Workshop, DILS 2005, San Diego, CA, USA, 20–22 July 2005; Springer: Berlin Heidelberg, Germany, 2005; pp. 224–239. [Google Scholar]
- Scholz, M.; Fiehn, O. SetupX—a public study design database for metabolomic projects. Pac. Symp. Biocomput. 2007, 169–180. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar]
- Klaus, B.; Strimmer, K. fdrtool: Estimation of (local) false discovery rates and higher criticism, R package version 1.2.10; 2012. Avaible online: https://cran.r-project.org/web/packages/fdrtool/index.html (acessed on 8 September 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Grapov, D.; Jackson, M.I.; Fahrmann, J.; Fiehn, O.; Combs, G.F. Integrating Multiple Analytical Datasets to Compare Metabolite Profiles of Mouse Colonic-Cecal Contents and Feces. Metabolites 2015, 5, 489-501. https://doi.org/10.3390/metabo5030489
Zeng H, Grapov D, Jackson MI, Fahrmann J, Fiehn O, Combs GF. Integrating Multiple Analytical Datasets to Compare Metabolite Profiles of Mouse Colonic-Cecal Contents and Feces. Metabolites. 2015; 5(3):489-501. https://doi.org/10.3390/metabo5030489
Chicago/Turabian StyleZeng, Huawei, Dmitry Grapov, Matthew I. Jackson, Johannes Fahrmann, Oliver Fiehn, and Gerald F. Combs. 2015. "Integrating Multiple Analytical Datasets to Compare Metabolite Profiles of Mouse Colonic-Cecal Contents and Feces" Metabolites 5, no. 3: 489-501. https://doi.org/10.3390/metabo5030489