
Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy

Abstract
:1. Introduction
2. General Iodine and Thyroid Metabolism
2.1. Iodine Absorption and Excretion
2.2. Thyroid Metabolism
3. Physiologic Metabolism during Pregnancy
3.1. Iodine
3.2. Maternal and Fetal Thyroid
3.2.1. Maternal Thyroid Metabolism
3.2.2. Fetal Thyroid
3.3. Thyroid Metabolites Placental Transport
3.3.1. TH Placental Transport
3.3.2. TSH
3.3.3. TRH
3.3.4. TRAb/TPOAb
3.4. Deiodinases
4. Maternal Pathologies during Pregnancy
4.1. Gestational Transient Hyperthyroidism (GTH)
4.2. Gestational Transient Hyperthyroidism (GTH)
4.3. Distinguishing GHT from Graves’ Disease
4.4. Thyroid Storm
4.5. Maternal Hypothyroidism
5. Fetal Consequences of Pathological Thyroid Metabolism
5.1. Fetal Anomalies Caused by Hyper or Hypothyroidism
5.1.1. Congenital Heart Defects
5.1.2. Impairments to Brain Development
5.1.3. Birthweight
5.2. Fetal Hyperthyroidism
5.3. Fetal Hypothyroidism
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma, J.; Hatch-Mcchesney, A.; Lieberman, H.R. Iodine and Iodine Deficiency: A Comprehensive Review of a Re-Emerging Issue. Nutrients 2022, 14, 3474. [Google Scholar] [CrossRef]
- Zimmermann, M.B. The effects of iodine deficiency in pregnancy and infancy. Paediatr. Perinat. Epidemiol. 2012, 26, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ittermann, T.; Albrecht, D.; Arohonka, P.; Bilek, R.; De Castro, J.J.; Dahl, L.; Filipsson Nystrom, H.; Gaberscek, S.; Garcia-Fuentes, E.; Gheorghiu, M.L.; et al. Standardized Map of Iodine Status in Europe. Thyroid 2020, 30, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- De Escobar, G.M.; Obregón, M.J.; Del Rey, F.E. Role of thyroid hormone during early brain development. Eur. J. Endocrinol. 2004, 151 (Suppl. S3), U25–U37. [Google Scholar] [CrossRef] [PubMed]
- Calvo, R.M.; Jauniaux, E.; Gulbis, B.; Asunción, M.; Gervy, C.; Contempré, B.; De Escobar, G.M. Fetal Tissues Are Exposed to Biologically Relevant Free Thyroxine Concentrations during Early Phases of Development. J. Clin. Endocrinol. Metab. 2002, 87, 1768–1777. [Google Scholar] [CrossRef]
- Thorpe-Beeston, J.G.; Nicolaides, K.H.; Felton, C.V.; Butler, J.; McGregor, A.M. Maturation of the secretion of thyroid hormone and thyroid-stimulating hormone in the fetus. N. Engl. J. Med. 1991, 324, 532–536. [Google Scholar] [CrossRef]
- Haldimann, M.; Alt, A.; Blanc, A.; Blondeau, K. Iodine content of food groups. J. Food Compos. Anal. 2005, 18, 461–471. [Google Scholar] [CrossRef]
- De Miranda Milagres, R.C.R.; De Souza, E.C.G.; Do Carmo Gouveia Peluzio, M.; Do Carmo Castro Franceschini, S.; Duarte, M.S.L. Food Iodine Content Table compiled from international databases. Rev. Nutr. 2020, 33, e190222. [Google Scholar] [CrossRef]
- Pearce, E.N.; Pino, S.; He, X.; Bazrafshan, H.R.; Lee, S.L.; Braverman, L.E. Sources of dietary iodine: Bread, cows’ milk, and infant formula in the Boston area. J. Clin. Endocrinol. Metab. 2004, 89, 3421–3424. [Google Scholar] [CrossRef]
- Katagiri, R.; Asakura, K.; Uechi, K.; Masayasu, S.; Sasaki, S. Iodine Excretion in 24-hour Urine Collection and Its Dietary Determinants in Healthy Japanese Adults. J. Epidemiol. 2016, 26, 613. [Google Scholar] [CrossRef]
- Tadesse, S.; Hymete, A.; Lieberman, M.; Gebreyesus, S.H.; Ashenef, A. Iodine status, household salt iodine content, knowledge and practice assessment among pregnant women in Butajira, South Central Ethiopia. PLoS ONE 2022, 17, e0277208. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Du, Y.; Meng, F.; Liu, L.; Li, M.; Liu, P.; Sun, D. How to Decide the Iodine Content in Salt for a Country-China as an Example. Nutrients 2022, 14, 4606. [Google Scholar] [CrossRef] [PubMed]
- Clar, C.; Wu, T.; Liu, G.; Li, P. Iodized salt for iodine deficiency disorders. A systematic review. Endocrinol. Metab. Clin. North Am. 2002, 31, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Esche, J.; Thamm, M.; Remer, T. Contribution of iodized salt to total iodine and total salt intake in Germany. Eur. J. Nutr. 2020, 59, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.R.; Ovadia, Y.S.; Anteby, E.Y.; Fytlovich, S.; Aharoni, D.; Zamir, D.; Gefel, D.; Shenhav, S. Low intake of iodized salt and iodine containing supplements among pregnant women with apparently insufficient iodine status-time to change policy? Isr. J. Health Policy Res. 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- De La Vieja, A.; Santisteban, P. Role of iodide metabolism in physiology and cancer. Endocr. Relat. Cancer 2018, 25, R225–R245. [Google Scholar] [CrossRef]
- Portulano, C.; Paroder-Belenitsky, M.; Carrasco, N. The Na+/I− Symporter (NIS): Mechanism and Medical Impact. Endocr. Rev. 2014, 35, 106–149. [Google Scholar] [CrossRef]
- Shcheynikov, N.; Yang, D.; Wang, Y.; Zeng, W.; Karniski, L.P.; So, I.; Wall, S.M.; Muallem, S. The Slc26a4 transporter functions as an electroneutral Cl−/I−/HCO3− exchanger: Role of Slc26a4 and Slc26a6 in I− and HCO3− secretion and in regulation of CFTR in the parotid duct. J. Physiol. 2008, 586, 3813–3824. [Google Scholar] [CrossRef]
- Perez-Cornejo, P.; Gokhale, A.; Duran, C.; Cui, Y.; Xiao, Q.; Hartzell, H.C.; Faundez, V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc. Natl. Acad. Sci. USA 2012, 109, 10376–10381. [Google Scholar] [CrossRef]
- Yoshida, A.; Hisatome, I.; Taniguchi, S.; Sasaki, N.; Yamamoto, Y.; Miake, J.; Fukui, H.; Shimizu, H.; Okamura, T.; Okura, T.; et al. Mechanism of iodide/chloride exchange by pendrin. Endocrinology 2004, 145, 4301–4308. [Google Scholar] [CrossRef]
- Altorjay, Á.; Dohán, O.; Szilágyi, A.; Paroder, M.; Wapnir, I.L.; Carrasco, N. Expression of the Na+/l-symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer 2007, 7, 5. [Google Scholar] [CrossRef]
- Mazzone, A.; Bernard, C.E.; Strege, P.R.; Beyder, A.; Galietta, L.J.V.; Pasricha, P.J.; Rae, J.L.; Parkman, H.P.; Linden, D.R.; Szurszewski, J.H.; et al. Altered expression of ano1 variants in human diabetic gastroparesis. J. Biol. Chem. 2011, 286, 13393–13403. [Google Scholar] [CrossRef] [PubMed]
- Pablo Nicola, J.; Basquin, C.; Portulano, C.; Reyna-Neyra, A.; Paroder, M.; Carrasco, N. The Na/I symporter mediates active iodide uptake in the intestine. Am. J. Physiol. Cell Physiol. 2009, 296, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Fahlke, C. Ion permeation and selectivity in ClC-type chloride channels. Am. J. Physiol. Renal Physiol. 2001, 280, F748–F757. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Andersson, M. Assessment of iodine nutrition in populations: Past, present, and future. Nutr. Rev. 2012, 70, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C.; Dutton, C.M.; Castro, M.R.; Bergert, E.R.; Goellner, J.R.; Heufelder, A.E.; Morris, J.C. Expression of the sodium iodide symporter in human kidney. Kidney Int. 2001, 59, 1013–1023. [Google Scholar] [CrossRef]
- Wapnir, I.L.; Van De Rijn, M.; Nowels, K.; Amenta, P.S.; Walton, K.; Montgomery, K.; Greco, R.S.; Dohán, O.; Carrasco, N. Immunohistochemical Profile of the Sodium/Iodide Symporter in Thyroid, Breast, and Other Carcinomas Using High Density Tissue Microarrays and Conventional Sections. J. Clin. Endocrinol. Metab. 2003, 88, 1880–1888. [Google Scholar] [CrossRef]
- Xu, J.; Barone, S.; Li, H.; Holiday, S.; Zahedi, K.; Soleimani, M. Slc26a11, a chloride transporter, localizes with the vacuolar H+ -ATPase of A-intercalated cells of the kidney. Kidney Int. 2011, 80, 926–937. [Google Scholar] [CrossRef]
- Svenningsen, P.; Nielsen, M.R.; Marcussen, N.; Walter, S.; Jensen, B.L. TMEM16A is a Ca2+-activated Cl− channel expressed in the renal collecting duct. Acta Physiol. 2014, 212, 166–174. [Google Scholar] [CrossRef]
- Pearce, E.N.; Caldwell, K.L. Urinary iodine, thyroid function, and thyroglobulin as biomarkers of iodine status. Am. J. Clin. Nutr. 2016, 104 (Suppl. S3), 898S–901S. [Google Scholar] [CrossRef]
- Remer, T.; Fonteyn, N.; Alexy, U.; Berkemeyer, S. Longitudinal examination of 24-h urinary iodine excretion in schoolchildren as a sensitive, hydration status-independent research tool for studying iodine status. Am. J. Clin. Nutr. 2006, 83, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Karumbunathan, V.; Zimmermann, M.B. Global iodine status in 2011 and trends over the past decade. J. Nutr. 2012, 142, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Zhong, G.; Ma, X. Urinary iodine concentration (UIC) could be a promising biomarker for predicting goiter among school-age children: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0174095. [Google Scholar] [CrossRef]
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Ris-Stalpers, C. Physiology and pathophysiology of the DUOXes. Antioxid. Redox Signal. 2006, 8, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Aycan, Z.; Cangul, H.; Muzza, M.; Bas, V.N.; Fugazzola, L.; Chatterjee, V.K.; Persani, L.; Schoenmakers, N. Digenic DUOX1 and DUOX2 Mutations in Cases with Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3085. [Google Scholar] [CrossRef] [PubMed]
- Pilo, A.; Iervasi, G.; Vitek, F.; Ferdeghini, M.; Cazzuola, F.; Bianchi, R. Thyroidal and peripheral production of 3,5,3′-triiodothyronine in humans by multicompartmental analysis. Am. J. Physiol. 1990, 258, E715–E726. [Google Scholar] [CrossRef]
- Janssen, S.T.; Janssen, O.E. Directional thyroid hormone distribution via the blood stream to target sites. Mol. Cell. Endocrinol. 2017, 458, 16–21. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355. [Google Scholar] [CrossRef]
- De Felice, M.; Di Lauro, R. Thyroid development and its disorders: Genetics and molecular mechanisms. Endocr. Rev. 2004, 25, 722–746. [Google Scholar] [CrossRef]
- Ohmori, M.; Endo, T.; Harii, N.; Onaya, T. A Novel Thyroid Transcription Factor Is Essential for Thyrotropin-Induced Up-Regulation of Na+/I− Symporter Gene Expression. Mol. Endocrinol. 1998, 12, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Zannini, M.; Levy, O.; Carrasco, N.; di Lauro, R. The Paired-Domain Transcription Factor Pax8 Binds to the Upstream Enhancer of the Rat Sodium/Iodide Symporter Gene and Participates in Both Thyroid-Specific and Cyclic-AMP-Dependent Transcription. Mol. Cell. Biol. 1999, 19, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.L.; Espinoza, C.R.; Loos, U. Characterization of a Thyroid-Specific and Cyclic Adenosine Monophosphate-Responsive Enhancer Far Upstream from the Human Sodium Iodide Symporter Gene. Thyroid 2004, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Kogai, T.; Kanamoto, Y.; Hershman, J.M.; Brent, G.A. A Thyroid-Specific Far-Upstream Enhancer in the Human Sodium/Iodide Symporter Gene Requires Pax-8 Binding and Cyclic Adenosine 3′,5′-Monophosphate Response Element-Like Sequence Binding Proteins for Full Activity and Is Differentially Regulated in Normal and Thyroid Cancer Cells. Mol. Endocrinol. 2002, 16, 2266–2282. [Google Scholar] [CrossRef]
- Fernández, L.P.; López-Márquez, A.; Martínez, Á.M.; Gómez-López, G.; Santisteban, P. New Insights into FoxE1 Functions: Identification of Direct FoxE1 Targets in Thyroid Cells. PLoS ONE 2013, 8, e62849. [Google Scholar] [CrossRef]
- Kang, H.S.; Kumar, D.; Liao, G.; Lichti-Kaiser, K.; Gerrish, K.; Liao, X.H.; Refetoff, S.; Jothi, R.; Jetten, A.M. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J. Clin. Investig. 2022, 127, 4326–4337. [Google Scholar] [CrossRef]
- Giuliani, C.; Bucci, I.; Napolitano, G. The role of the transcription factor Nuclear Factor-kappa B in thyroid autoimmunity and cancer. Front. Endocrinol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Nicola, J.P.; Nazar, M.; Mascanfroni, I.D.; Pellizas, C.G.; Masini-Repiso, A.M. NF-κB p65 Subunit Mediates Lipopolysaccharide-Induced Na+/I− Symporter Gene Expression by Involving Functional Interaction with the Paired Domain Transcription Factor Pax8. Mol. Endocrinol. 2010, 24, 1846–1862. [Google Scholar] [CrossRef]
- Elefant, É. Placental immunoglobulin transfer. Bull. Acad. Natl. Med. 2012, 196, 1601–1612. [Google Scholar] [CrossRef]
- Zimmermann, M.B. Iodine deficiency in pregnancy and the effects of maternal iodine supplementation on the offspring: A review. Am. J. Clin. Nutr. 2009, 89, 668S–672S. [Google Scholar] [CrossRef]
- Caldwell, K.L.; Jones, R.; Hollowell, J.G. Urinary iodine concentration: United States National Health And Nutrition Examination Survey 2001–2002. Thyroid 2005, 15, 692–699. [Google Scholar] [CrossRef]
- Brander, L.; Als, C.; Buess, H.; Haldimann, F.; Harder, M.; Hänggi, W.; Herrmann, U.; Lauber, K.; Niederer, U.; Zürcher, T.; et al. Urinary iodine concentration during pregnancy in an area of unstable dietary iodine intake in Switzerland. J. Endocrinol. Investig. 2003, 26, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Stilwell, G.; Reynolds, P.J.; Parameswaran, V.; Blizzard, L.; Greenaway, T.M.; Burgess, J.R. The Influence of Gestational Stage on Urinary Iodine Excretion in Pregnancy. J. Clin. Endocrinol. Metab. 2008, 93, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Cai, Y.; Ji, C.; Zhao, C.; Tian, C.; Pang, B.; Shi, M.; Li, X.; Liu, Y.; Sun, D. Evaluation of iodine nutritional status during pregnancy by estimated 24-h urinary iodine excretion: Population variation range and individual accuracy. Public Health Nutr. 2022, 25, 237–247. [Google Scholar] [CrossRef] [PubMed]
- De Groot, L.; Abalovich, M.; Alexander, E.K.; Amino, N.; Barbour, L.; Cobin, R.H.; Eastman, C.J.; Lazarus, J.H.; Luton, D.; Mandel, S.J.; et al. Management of Thyroid Dysfunction during Pregnancy and Postpartum: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2543–2565. [Google Scholar] [CrossRef]
- Alexander, E.K.; Pearce, E.N.; Brent, G.A.; Brown, R.S.; Chen, H.; Dosiou, C.; Grobman, W.A.; Laurberg, P.; Lazarus, J.H.; Mandel, S.J.; et al. 2017 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease during Pregnancy and the Postpartum. Thyroid 2017, 27, 315–389. [Google Scholar] [CrossRef]
- Lazarus, J.; Brown, R.S.; Daumerie, C.; Hubalewska-Dydejczyk, A.; Negro, R.; Vaidya, B. 2014 European thyroid association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur. Thyroid J. 2014, 3, 76–94. [Google Scholar] [CrossRef]
- Andersson, M.; De Benoist, B.; Delange, F.; Zupan, J. Prevention and control of iodine deficiency in pregnant and lactating women and in children less than 2-years-old: Conclusions and recommendations of the Technical Consultation. Public Health Nutr. 2007, 10, 1606–1611. [Google Scholar] [CrossRef]
- Miles, E.A.; Vahlberg, T.; Calder, P.C.; Houttu, N.; Pajunen, L.; Koivuniemi, E.; Mokkala, K.; Laitinen, K. Iodine status in pregnant women and infants in Finland. Eur. J. Nutr. 2022, 61, 2919–2927. [Google Scholar] [CrossRef]
- Bidart, J.-M.; Lacroix, L.; Evain-Brion, D.; Caillou, B.; Lazar, V.; Frydman, R.; Bellet, D.; Filetti, S.; Schlumberger, M. Expression of Na+/I− symporter and Pendred syndrome genes in trophoblast cells. J. Clin. Endocrinol. Metab. 2000, 85, 4367–4372. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Manley, S.W.; Morris, J.C.; Powell, K.A.; Bergert, E.R.; Mortimer, R.H. Sodium iodide symporter (NIS) gene expression in human placenta. Placenta 2001, 22, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Degrelle, S.A.; Guibourdenche, J.; Galland, F.; Bidart, J.M.; Fournier, T.; Evain-Brion, D. Iodide transporters expression in early human invasive trophoblast. Placenta 2013, 34, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bidart, J.-M.; Mian, C.; Lazar, V.; Russo, D.; Filetti, S.; Caillou, B.; Schlumberger, M. Expression of Pendrin and the Pendred Syndrome (PDS) Gene in Human Thyroid Tissues*. J. Clin. Endocrinol. Metab. 2000, 85, 2028–2033. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.W.; Li, H.; Mortimer, R.H. The BeWo choriocarcinoma cell line as a model of iodide transport by placenta. Placenta 2005, 26, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Nwabuobi, C.; Arlier, S.; Schatz, F.; Guzeloglu-Kayisli, O.; Lockwood, C.J.; Kayisli, U.A. hCG: Biological Functions and Clinical Applications. Int. J. Mol. Sci. 2017, 18, 2037. [Google Scholar] [CrossRef]
- Morgan, F.J.; Birken, S.; Canfield, R.E. The amino acid sequence of human chorionic gonadotropin. The alpha subunit and beta subunit. J. Biol. Chem. 1975, 250, 5247–5258. [Google Scholar] [CrossRef]
- Kraiem, Z.; Sadeh, O.; Blithe, D.L.; Nisula, B.C. Human chorionic gonadotropin stimulates thyroid hormone secretion, iodide uptake, organification, and adenosine 3′,5′-monophosphate formation in cultured human thyrocytes. J. Clin. Endocrinol. Metab. 1994, 79, 595–599. [Google Scholar] [CrossRef]
- Yoshimura, M.; Hershman, J.M. Thyrotropic action of human chorionic gonadotropin. Thyroid 1995, 5, 425–434. [Google Scholar] [CrossRef]
- Pekonen, F.; Alfthan, H.; Stenman, U.H.; Ylikorkala, O. Human chorionic gonadotropin (hCG) and thyroid function in early human pregnancy: Circadian variation and evidence for intrinsic thyrotropic activity of hCG. J. Clin. Endocrinol. Metab. 1988, 66, 853–856. [Google Scholar] [CrossRef]
- Arturi, F.; Presta, I.; Scarpelli, D.; Bidart, J.M.; Schlumberger, M.; Filetti, S.; Russo, D. Stimulation of iodide uptake by human chorionic gonadotropin in FRTL-5 cells: Effects on sodium/iodide symporter gene and protein expression. Eur. J. Endocrinol. 2002, 147, 655–661. [Google Scholar] [CrossRef]
- Li, H.; Richard, K.; McKinnon, B.; Mortimer, R.H. Effect of Iodide on Human Choriogonadotropin, Sodium-Iodide Symporter Expression, and Iodide Uptake in BeWo Choriocarcinoma Cells. J. Clin. Endocrinol. Metab. 2007, 92, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.; O’Herlihy, C.; Smyth, P.P.A. Regulation of Iodide Uptake in Placental Primary Cultures. Eur. Thyroid J. 2013, 2, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schröder-Van Der Elst, J.P.; Van Der Heide, D.; Kastelijn, J.; Rousset, B.; Obregón, M.J. The expression of the sodium/iodide symporter is up-regulated in the thyroid of fetuses of iodine-deficient rats. Endocrinology 2001, 142, 3736–3741. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.; O’Herlihy, C.; Smyth, P.P.A. The placenta as a compensatory iodine storage organ. Thyroid 2011, 21, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Neven, K.Y.; Marien, C.B.D.; Janssen, B.G.; Roels, H.A.; Waegeneers, N.; Nawrot, T.S.; Ruttens, A. Variability of iodine concentrations in the human placenta. Sci. Rep. 2020, 10, 161. [Google Scholar] [CrossRef]
- Brent, G.A. Maternal thyroid function: Interpretation of thyroid function tests in pregnancy. Clin. Obstet. Gynecol. 1997, 40, 3–15. [Google Scholar] [CrossRef]
- Lazarus, J.H. Thyroid function in pregnancy. Br. Med. Bull. 2011, 97, 137–148. [Google Scholar] [CrossRef]
- Lee, R.H.; Spencer, C.A.; Mestman, J.H.; Miller, E.A.; Petrovic, I.; Braverman, L.E.; Goodwin, T.M. Free T4 immunoassays are flawed during pregnancy. Am. J. Obstet. Gynecol. 2009, 200, 260.e1–260.e6. [Google Scholar] [CrossRef]
- Knøsgaard, L.; Andersen, S.; Hansen, A.B.; Vestergaard, P.; Andersen, S.L. Classification of maternal thyroid function in early pregnancy using repeated blood samples. Eur. Thyroid J. 2022, 11, e210055. [Google Scholar] [CrossRef]
- Ain, K.B.; Mori, Y.; Refetoff, S. Reduced clearance rate of thyroxine-binding globulin (TBG) with increased sialylation: A mechanism for estrogen-induced elevation of serum TBG concentration. J. Clin. Endocrinol. Metab. 1987, 65, 689–696. [Google Scholar] [CrossRef]
- Pradhan, R.; Agarwal, A.; Lombardi, C.P.; Raffaelli, M. Applied Embryology of the Thyroid and Parathyroid Glands. Surg. Thyroid Parathyr. Gland. 2021, 1, 15–25.e4. [Google Scholar] [CrossRef]
- Arrangoiz, R.; Cordera, F.; Caba, D.; Muñoz, M.; Moreno, E.; de León, E.L.; Arrangoiz, R.; Cordera, F.; Caba, D.; Muñoz, M.; et al. Comprehensive Review of Thyroid Embryology, Anatomy, Histology, and Physiology for Surgeons. Int. J. Otolaryngol. Head Neck Surg. 2018, 7, 160–188. [Google Scholar] [CrossRef]
- Ozguner, G.; Sulak, O. Size and location of thyroid gland in the fetal period. Surg. Radiol. Anat. 2014, 36, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Dom, G.; Dmitriev, P.; Lambot, M.A.; Van Vliet, G.; Glinoer, D.; Libert, F.; Lefort, A.; Dumont, J.E.; Maenhaut, C. Transcriptomic Signature of Human Embryonic Thyroid Reveals Transition From Differentiation to Functional Maturation. Front. Cell Dev. Biol. 2021, 9, 1481. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Fagman, H. Development of the thyroid gland. Development 2017, 144, 2123–2140. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Mishra, S.; Kaul, J.M. Development of Parafollicular Cells and their Relationship with Developing Thyroid Follicles in Human Foetuses. J. Clin. Diagn. Res. 2017, 11, AC01. [Google Scholar] [CrossRef]
- Trueba, S.S.; Augé, J.; Mattei, G.; Etchevers, H.; Martinovic, J.; Czernichow, P.; Vekemans, M.; Polak, M.; Attié-Bitach, T. PAX8, TITF1, and FOXE1 gene expression patterns during human development: New insights into human thyroid development and thyroid dysgenesis-associated malformations. J. Clin. Endocrinol. Metab. 2005, 90, 455–462. [Google Scholar] [CrossRef]
- Chen, Z.; Peeters, R.P.; Leeuwenburgh, S.; Broekhuizen, M.; Neuman, R.I.; Hitzerd, E.; Tan, L.; Jongejan, R.M.S.; de Rijke, Y.B.; Reiss, I.K.M.; et al. Asymmetrical Transport of Thyroxine Across Human Term Placenta. Thyroid, 2023; ahead of print. [Google Scholar] [CrossRef]
- Landers, K.; Richard, K. Traversing barriers—How thyroid hormones pass placental, blood-brain and blood-cerebrospinal fluid barriers. Mol. Cell. Endocrinol. 2017, 458, 22–28. [Google Scholar] [CrossRef]
- Costa, A.; Arisio, R.; Benedetto, C.; Bertino, E.; Fabris, C.; Giraudi, G.; Marozio, L.; Maulà, V.; Pagliano, M.; Testori, O.; et al. Thyroid hormones in tissues from human embryos and fetuses. J. Endocrinol. Investig. 1991, 14, 559–568. [Google Scholar] [CrossRef]
- Contempré, B.; Jauniaux, E.; Calvo, R.; Jurkovic, D.; Campbell, S.; De Escobar, G.M. Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J. Clin. Endocrinol. Metab. 1993, 77, 1719–1722. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Manley, S.W.; Mortimer, R.H. Membrane transport of thyroid hormone in the human choriocarcinoma cell line, JAR. Mol. Cell. Endocrinol. 1992, 87, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, L.F.F.; Muñoz, Y.S.; Pustovrh, M.C. Thyroid hormones: Metabolism and transportation in the fetoplacental unit. Mol. Reprod. Dev. 2022, 89, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Meima, M.E.; Peeters, R.P.; Visser, W.E. Thyroid Hormone Transporters in Pregnancy and Fetal Development. Int. J. Mol. Sci. 2022, 23, 15113. [Google Scholar] [CrossRef] [PubMed]
- Roost, M.S.; Van Iperen, L.; Ariyurek, Y.; Buermans, H.P.; Arindrarto, W.; Devalla, H.D.; Passier, R.; Mummery, C.L.; Carlotti, F.; De Koning, E.J.P.; et al. KeyGenes, a Tool to Probe Tissue Differentiation Using a Human Fetal Transcriptional Atlas. Stem Cell Rep. 2015, 4, 1112–1124. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Jose, A.; Poojari, V.G.; Shetty, S.; Prabhu RV, K.; Rao, M. Role and Clinical Significance of Monocarboxylate Transporter 8 (MCT8) During Pregnancy. Reprod. Sci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; De Souza, E.C.L.; Meima, M.E.; Peeters, R.P.; Edward Visser, W.; Visser, T.J. Outward-Open Model of Thyroid Hormone Transporter Monocarboxylate Transporter 8 Provides Novel Structural and Functional Insights. Endocrinology 2017, 158, 3292–3306. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Docter, R.; Moerings, E.P.C.M.; Verrey, F.; Krenning, E.P.; Hennemann, G.; Visser, T.J. Thyroid Hormone Transport by the Heterodimeric Human System L Amino Acid Transporter. Endocrinology 2001, 142, 4339–4348. [Google Scholar] [CrossRef]
- Zevenbergen, C.; Meima, M.E.; De Souza, E.C.L.; Peeters, R.P.; Kinne, A.; Krause, G.; Edward Visser, W.; Visser, T.J. Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology 2015, 156, 4345–4355. [Google Scholar] [CrossRef]
- Kinne, A.; Wittner, M.; Wirth, E.K.; Hinz, K.M.; Schülein, R.; Köhrle, J.; Krause, G. Involvement of the L-Type Amino Acid Transporter Lat2 in the Transport of 3,3’-Diiodothyronine across the Plasma Membrane. Eur. Thyroid J. 2015, 4, 42–50. [Google Scholar] [CrossRef]
- Hinz, K.M.; Neef, D.; Rutz, C.; Furkert, J.; Köhrle, J.; Schülein, R.; Krause, G. Molecular features of the L-type amino acid transporter 2 determine different import and export profiles for thyroid hormones and amino acids. Mol. Cell. Endocrinol. 2017, 443, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wirth, E.K.; Roth, S.; Blechschmidt, C.; Hölter, S.M.; Becker, L.; Racz, I.; Zimmer, A.; Klopstock, T.; Gailus-Durner, V.; Fuchs, H.; et al. Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J. Neurosci. 2009, 29, 9439–9449. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Adachi, H.; Nishio, T.; Unno, M.; Tokui, T.; Okabe, M.; Onogawa, T.; Suzuki, T.; Asano, N.; Tanemoto, M.; et al. Identification of Thyroid Hormone Transporters in Humans: Different Molecules Are Involved in a Tissue-Specific Manner. Endocrinology 2001, 142, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.H.; Jansen, J.; Jachtenberg, J.W.; Visser, W.E.; Kester, M.H.A.; Visser, T.J. Effective Cellular Uptake and Efflux of Thyroid Hormone by Human Monocarboxylate Transporter 10. Mol. Endocrinol. 2008, 22, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.H.; Grueters, P.A.; Biebermann, H.; Krude, H.; Von Moers, A.; Reeser, M.; Barrett, T.G.; Mancilla, E.E.; Svensson, J.; Kester, M.H.A.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Chan, S.Y.; Martín-Santos, A.; Loubière, L.S.; González, A.M.; Stieger, B.; Logan, A.; Mccabe, C.J.; Franklyn, J.A.; Kilby, M.D. The expression of thyroid hormone transporters in the human fetal cerebral cortex during early development and in N-Tera-2 neurodifferentiation. J. Physiol. 2011, 589, 2827–2845. [Google Scholar] [CrossRef]
- Heuer, H.; Maier, M.K.; Iden, S.; Mittag, J.; Friesema, E.C.H.; Visser, T.J.; Bauer, K. The Monocarboxylate Transporter 8 Linked to Human Psychomotor Retardation Is Highly Expressed in Thyroid Hormone-Sensitive Neuron Populations. Endocrinology 2005, 146, 1701–1706. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Visser, T.J.; Borgers, A.J.; Kalsbeek, A.; Swaab, D.F.; Fliers, E.; Alkemade, A. Thyroid hormone transporters and deiodinases in the developing human hypothalamus. Eur. J. Endocrinol. 2012, 167, 379–386. [Google Scholar] [CrossRef]
- Heinrichs, C.; De Zegher, F.; Vansnick, F.; Vokaer, A.; Christophe, C.; Frankenne, F. Fetal hypopituitarism: Perinatal endocrine and morphological studies in two cases. Acta Paediatr. 1994, 83, 448–451. [Google Scholar] [CrossRef]
- Bajoria, R.; Fisk, N.M. Permeability of Human Placenta and Fetal Membranes to Thyrotropin-Stimulating Hormone in Vitro. Pediatr. Res. 1998, 43, 621–628. [Google Scholar] [CrossRef]
- Yoshida, K.; Sakurada, T.; Takahashi, T.; Furuhashi, N.; Kaise, K.; Yoshinaga, K. Measurement of tsh in human amniotic fluid: Diagnosis of fetal thyroid abnormality in utero. Clin. Endocrinol. 1986, 25, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Guibourdenche, J.; Noël, M.; Chevenne, D.; Vuillard, E.; Voluménie, J.L.; Polak, M.; Boissinot, C.; Porquet, D.; Luton, D. Biochemical investigation of foetal and neonatal thyroid function using the ACS-180SE analyser: Clinical application. Ann. Clin. Biochem. 2001, 38, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Polak, M. Human fetal thyroid function. Endocr. Dev. 2014, 26, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Sousa, N. Maternal hormonal milieu influence on fetal brain development. Brain Behav. 2018, 8, e00920. [Google Scholar] [CrossRef]
- Theodoropoulos, T.; Braverman, L.E.; Vagenakis, A.G. Thyrotropin-Releasing Hormone is not Required for Thyrotropin Secretion in the Perinatal Rat. J. Clin. Investig. 1979, 63, 588–594. [Google Scholar] [CrossRef]
- Ansari, M.A.; Demello, D.E.; Polk, D.H.; Devaskar, U.P. Thyrotropin-releasing hormone accelerates fetal mouse lung ultrastructural maturation via stimulation of extra thyroidal pathway. Pediatr. Res. 1997, 42, 709–714. [Google Scholar] [CrossRef]
- Yang, C.; He, B.; Zhang, H.; Wang, X.; Zhang, Q.; Dai, W. IgG Fc Affinity Ligands and Their Applications in Antibody-Involved Drug Delivery: A Brief Review. Pharmaceutics 2023, 15, 187. [Google Scholar] [CrossRef]
- Gitlin, D.; Kumate, J.; Urrusti, J.; Morales, C. The selectivity of the human placenta in the transfer of plasma proteins from mother to fetus. J. Clin. Investig. 1964, 43, 1938–1951. [Google Scholar] [CrossRef]
- Volkov, M.; Brinkhaus, M.; van Schie, K.A.; Bondt, A.; Kissel, T.; van der Kooi, E.J.; Bentlage, A.E.H.; Koeleman, C.A.M.; de Taeye, S.W.; Derksen, N.I.; et al. IgG Fab Glycans Hinder FcRn-Mediated Placental Transport. J. Immunol. 2023, 210, 158–167. [Google Scholar] [CrossRef]
- Firan, M.; Bawdon, R.; Radu, C.; Ober, R.J.; Eaken, D.; Antohe, F.; Ghetie, V.; Ward, E.S. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int. Immunol. 2001, 13, 993–1002. [Google Scholar] [CrossRef]
- Szlauer, R.; Ellinger, I.; Haider, S.; Saleh, L.; Busch, B.L.; Knöfler, M.; Fuchs, R. Functional expression of the human neonatal Fc-receptor, hFcRn, in isolated cultured human syncytiotrophoblasts. Placenta 2009, 30, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, B.K. Human placental Fc gamma-binding proteins in the maternofetal transfer of IgG. APMIS. Suppl. 1996, 64, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bonagura, V.R.; Morrison, S.L.; Bjorkman, P.J. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 1995, 34, 14649–14657. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Chen, M.Y.; Gastinel, L.N.; Bjorkman, P.J. Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity 1994, 1, 303–315. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004, 101, 11076–11081. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Kuhn, P.; Nicolaides, K.H.; Schneider, H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am. J. Reprod. Immunol. 1996, 36, 248–255. [Google Scholar] [CrossRef]
- Lozano, N.A.; Lozano, A.; Marini, V.; Saranz, R.J.; Blumberg, R.S.; Baker, K.; Agresta, M.F.; Ponzio, M.F. Expression of FcRn receptor in placental tissue and its relationship with IgG levels in term and pre-term newborns HHS Public Access. Am. J. Reprod. Immunol. 2018, 80, 12972. [Google Scholar] [CrossRef]
- Ciobanu, A.M.; Dumitru, A.E.; Gica, N.; Botezatu, R.; Peltecu, G.; Panaitescu, A.M. Benefits and Risks of IgG Transplacental Transfer. Diagnostics 2020, 10, 583. [Google Scholar] [CrossRef]
- Seror, J.; Amand, G.; Guibourdenche, J.; Ceccaldi, P.F.; Luton, D. Anti-TPO antibodies diffusion through the placental barrier during pregnancy. PLoS ONE 2014, 9, e0084647. [Google Scholar] [CrossRef]
- Köhrle, J. The deiodinase family: Selenoenzymes regulating thyroid hormone availability and action. Cell. Mol. Life Sci. 2000, 57, 1853–1863. [Google Scholar] [CrossRef]
- Salvatore, D.; Bartha, T.; Harney, J.W.; Larsen, P.R. Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology 1996, 137, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Local activation and inactivation of thyroid hormones: The deiodinase family. Mol. Cell. Endocrinol. 1999, 151, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kachilele, S.; Hobbs, E.; Bulmer, J.N.; Boelaert, K.; McCabe, C.J.; Driver, P.M.; Bradwell, A.R.; Kester, M.; Visser, T.J.; et al. Placental iodothyronine deiodinase expression in normal and growth-restricted human pregnancies. J. Clin. Endocrinol. Metab. 2003, 88, 4488–4495. [Google Scholar] [CrossRef] [PubMed]
- Koopdonk-Kool, J.M.; de Vijlder, J.J.; Veenboer, G.J.; Ris-Stalpers, C.; Kok, J.H.; Vulsma, T.; Boer, K.; Visser, T.J. Type II and type III deiodinase activity in human placenta as a function of gestational age. J. Clin. Endocrinol. Metab. 1996, 81, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Vulsma, T.; Gons, M.H.; de Vijlder, J.J.M. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N. Engl. J. Med. 1989, 321, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Glinoer, D.; de Nayer, P.; Bourdoux, P.; Lemone, M.; Robyn, C.; van Steirteghem, A.; Kinthaert, J.; Lejeune, B. Regulation of Maternal Thyroid during Pregnancy. J. Clin. Endocrinol. Metab. 1990, 71, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.P.; Khoo, D.H.C.; Eng, P.H.K.; Tan, H.K.; Yo, S.L.; Jacob, E. Prevalence of gestational thyrotoxicosis in Asian women evaluated in the 8th to 14th weeks of pregnancy: Correlations with total and free beta human chorionic gonadotrophin. Clin. Endocrinol. 2001, 55, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Virmani, S.; Srinivas, S.B.; Bhat, R.; Rao, R.; Kudva, R. Transient Thyrotoxicosis in Molar Pregnancy. J. Clin. Diagn. Res. 2017, 11, QD01. [Google Scholar] [CrossRef]
- Glinoer, D. The regulation of thyroid function in pregnancy: Pathways of endocrine adaptation from physiology to pathology. Endocr. Rev. 1997, 18, 404–433. [Google Scholar] [CrossRef]
- Nguyen, C.T.; Sasso, E.B.; Barton, L.; Mestman, J.H. Graves’ hyperthyroidism in pregnancy: A clinical review. Clin. Diabetes Endocrinol. 2018, 4, 1–9. [Google Scholar] [CrossRef]
- De Bruin, R.; Van Dalen, S.L.; Franx, S.J.; Simons, S.H.P.; Flint, R.B.; Van Den Bosch, G.E. Risk for neonatal hypoglycaemia and bradycardia after beta-blocker use during pregnancy or lactation: A systematic review and meta-analysis protocol. BMJ Open 2022, 12, e055292. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, A.; Stan, M. Thyrotropin Receptor Antibodies—An Overview. Ophthal. Plast. Reconstr. Surg. 2018, 34, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S.; Laurberg, P. Hyperthyroidism in pregnancy. Lancet Diabetes Endocrinol. 2013, 1, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Amino, N.; Tanizawa, O.; Mori, H.; Iwatani, Y.; Yamada, T.; Kurachi, K.; Kumahara, Y.; Miyai, K. Aggravation of thyrotoxicosis in early pregnancy and after delivery in Graves’ disease. J. Clin. Endocrinol. Metab. 1982, 55, 108–112. [Google Scholar] [CrossRef]
- Tagami, T.; Hagiwara, H.; Kimura, T.; Usui, T.; Shimatsu, A.; Naruse, M. The incidence of gestational hyperthyroidism and postpartum thyroiditis in treated patients with Graves’ disease. Thyroid 2007, 17, 767–772. [Google Scholar] [CrossRef]
- Davis, L.E.; Lucas, M.J.; Hankins, G.D.V.; Roark, M.L.; Cunningham, F.G. Thyrotoxicosis complicating pregnancy. Am. J. Obstet. Gynecol. 1989, 160, 63–70. [Google Scholar] [CrossRef]
- Laurberg, P.; Bournaud, C.; Karmisholt, J.; Orgiazzi, J. Management of Graves’ hyperthyroidism in pregnancy: Focus on both maternal and foetal thyroid function, and caution against surgical thyroidectomy in pregnancy. Eur. J. Endocrinol. 2009, 160, 1–8. [Google Scholar] [CrossRef]
- Guo, N.; Xue, M.; Liang, Z. Advances in the differential diagnosis of transient hyperthyroidism in pregnancy and Graves’ disease. Arch. Gynecol. Obstet. 2022. [Google Scholar] [CrossRef]
- Yoshihara, A.; Noh, J.Y.; Mukasa, K.; Suzuki, M.; Ohye, H.; Matsumoto, M.; Kunii, Y.; Watanabe, N.; Suzuki, N.; Kameda, T.; et al. Serum human chorionic gonadotropin levels and thyroid hormone levels in gestational transient thyrotoxicosis: Is the serum hCG level useful for differentiating between active Graves’ disease and GTT? Endocr. J. 2015, 62, 557–560. [Google Scholar] [CrossRef]
- Xue, M.; Shi, Q.L.; Tan, K.N.; Wu, Y.; Zhou, R. The role of color doppler ultrasonography, thyroid function and auto antibody for the screening of Graves’ disease in pregnancy. Zhonghua Nei Ke Za Zhi 2016, 55, 470–473. [Google Scholar] [CrossRef]
- Hiraiwa, T.; Tsujimoto, N.; Tanimoto, K.; Terasaki, J.; Amino, N.; Hanafusa, T. Use of color Doppler ultrasonography to measure thyroid blood flow and differentiate graves’ disease from painless thyroiditis. Eur. Thyroid J. 2013, 2, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Metsman, J.H. Hyperthyroidism in pregnancy. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 267–288. [Google Scholar] [CrossRef]
- Patel, K.N.; Yip, L.; Lubitz, C.C.; Grubbs, E.G.; Miller, B.S.; Shen, W.; Angelos, P.; Chen, H.; Doherty, G.M.; Fahey, T.J.; et al. The American Association of Endocrine Surgeons Guidelines for the Definitive Surgical Management of Thyroid Disease in Adults. Ann. Surg. 2020, 271, E21–E93. [Google Scholar] [CrossRef] [PubMed]
- de Mul, N.; Damstra, J.; Nieveen van Dijkum, E.J.M.; Fischli, S.; Kalkman, C.J.; Schellekens, W.J.M.; Immink, R.V. Risk of perioperative thyroid storm in hyperthyroid patients: A systematic review. Br. J. Anaesth. 2021, 127, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Abalovich, M.; Gutierrez, S.; Alcaraz, G.; Maccallini, G.; Garcia, A.; Levalle, O. Overt and subclinical hypothyroidism complicating pregnancy. Thyroid 2002, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.M.; Middleton, P.; Cossich, M.C.; Crowther, C.A.; Bain, E. Interventions for clinical and subclinical hypothyroidism pre-pregnancy and during pregnancy. Cochrane Database Syst. Rev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Dhillon-Smith, R.K.; Tobias, A.; Smith, P.P.; Middleton, L.J.; Sunner, K.K.; Baker, K.; Farrell-Carver, S.; Bender-Atik, R.; Agrawal, R.; Bhatia, K.; et al. The Prevalence of Thyroid Dysfunction and Autoimmunity in Women with History of Miscarriage or Subfertility. J. Clin. Endocrinol. Metab. 2020, 105, 2667–2677. [Google Scholar] [CrossRef]
- Toloza, F.J.K.; Derakhshan, A.; Männistö, T.; Bliddal, S.; Popova, P.V.; Carty, D.M.; Chen, L.; Taylor, P.; Mosso, L.; Oken, E.; et al. Association between maternal thyroid function and risk of gestational hypertension and pre-eclampsia: A systematic review and individual-participant data meta-analysis. Lancet Diabetes Endocrinol. 2022, 10, 243–252. [Google Scholar] [CrossRef]
- Geng, X.; Chen, Y.; Li, S.; Wang, W.; Wu, W.; Sun, C.; Li, N.; Wang, L. Systematic review and meta-analysis on the influence of thyroid dysfunction in early pregnancy on pregnancy outcomes under ultrasound guidance. Ann. Palliat. Med. 2022, 11, 1001–1016. [Google Scholar] [CrossRef]
- Thangaratinam, S.; Tan, A.; Knox, E.; Kilby, M.D.; Franklyn, J.; Coomarasamy, A. Association between thyroid autoantibodies and miscarriage and preterm birth: Meta-analysis of evidence. BMJ 2011, 342, d2616. [Google Scholar] [CrossRef]
- Spinillo, A.; De Maggio, I.; Ruspini, B.; Bellingeri, C.; Cavagnoli, C.; Giannico, S.; Boschetti, A.; Magri, F.; Lovati, E.; Beneventi, F. Placental pathologic features in thyroid autoimmunity. Placenta 2021, 112, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ye, E.; Sun, M.; Lin, H.; Yu, L.; Lin, Z.; Peng, M.; Lin, D.; Lu, X. Association between third trimester maternal isolated hypothyroxinemia and adverse pregnancy outcomes. Endocr. J. 2023, EJ22-0528. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E.; Pinz, I.; Preda, M.; Norton, C.R.; Gridley, T.; Hernandez, A. DIO3 protects against thyrotoxicosis-derived cranio-encephalic and cardiac congenital abnormalities. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Peng, T.; Li, M.Q.; Xie, F.; Wu, J.N. Association between Maternal Thyroxine and Risk of Fetal Congenital Heart Defects: A Hospital-Based Cohort Study. Int. J. Endocrinol. 2022, 2022, 3859388. [Google Scholar] [CrossRef]
- Ochi, S.; Manabe, S.; Kikkawa, T.; Osumi, N. Thirty Years’ History since the Discovery of Pax6: From Central Nervous System Development to Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 6115. [Google Scholar] [CrossRef]
- Xu, Y.; Xi, J.; Wang, G.; Guo, Z.; Sun, Q.; Lu, C.; Ma, L.; Wu, Y.; Jia, W.; Zhu, S.; et al. PAUPAR and PAX6 sequentially regulate human embryonic stem cell cortical differentiation. Nucleic Acids Res. 2021, 49, 1935–1950. [Google Scholar] [CrossRef]
- Hevner, R.F. Intermediate progenitors and Tbr2 in cortical development. J. Anat. 2019, 235, 616–625. [Google Scholar] [CrossRef]
- Mohan, V.; Sinha, R.A.; Pathak, A.; Rastogi, L.; Kumar, P.; Pal, A.; Godbole, M.M. Maternal thyroid hormone deficiency affects the fetal neocorticogenesis by reducing the proliferating pool, rate of neurogenesis and indirect neurogenesis. Exp. Neurol. 2012, 237, 477–488. [Google Scholar] [CrossRef]
- Vancamp, P.; Le Blay, K.; Butruille, L.; Sébillot, A.; Boelen, A.; Demeneix, B.A.; Remaud, S. Developmental thyroid disruption permanently affects the neuroglial output in the murine subventricular zone. Stem Cell Rep. 2022, 17, 459. [Google Scholar] [CrossRef]
- Shimokawa, N.; Yousefi, B.; Morioka, S.; Yamaguchi, S.; Ohsawa, A.; Hayashi, H.; Azuma, A.; Mizuno, H.; Kasagi, M.; Masuda, H.; et al. Altered cerebellum development and dopamine distribution in a rat genetic model with congenital hypothyroidism. J. Neuroendocrinol. 2014, 26, 164–175. [Google Scholar] [CrossRef]
- Engel, S.M.; Villanger, G.D.; Herring, A.; Nethery, R.C.; Drover, S.S.M.; Zoeller, R.T.; Meltzer, H.M.; Zeiner, P.; Knudsen, G.P.; Reichborn-Kjennerud, T.; et al. Gestational thyroid hormone concentrations and risk of attention-deficit hyperactivity disorder in the Norwegian Mother, Father and Child Cohort Study. Paediatr. Perinat. Epidemiol. 2023, 37, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, Z.C.; Zhang, T.; Fan, P.; Ma, R.; Zhang, J.; Ouyang, F. Maternal Thyroid Dysfunction and Neuropsychological Development in Children. J. Clin. Endocrinol. Metab. 2023, 108, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, A.; Peeters, R.P.; Taylor, P.N.; Bliddal, S.; Carty, D.M.; Meems, M.; Vaidya, B.; Chen, L.; Knight, B.A.; Ghafoor, F.; et al. Association of maternal thyroid function with birth weight: A systematic review and individual-participant data meta-analysis HHS Public Access. Lancet Diabetes Endocrinol. 2020, 8, 501–510. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, Y.; Liu, Y.; Liu, C.; Ren, J.; Liu, X.; Korevaar, T.I.M.; Fan, J. Triglycerides Mediate the Relationship between Maternal Free Thyroxine and Birth Weight: A Prospective Cohort Study from China. Thyroid 2023. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhong, H. Correlation between Hypothyroidism During Pregnancy and Glucose and Lipid Metabolism in Pregnant Women and Its Influence on Pregnancy Outcome and Fetal Growth and Development. Front. Surg. 2022, 9, 863286. [Google Scholar] [CrossRef]
- Yang, M.; Sun, M.; Jiang, C.; Wu, Q.; Jiang, Y.; Xu, J.; Luo, Q. Thyroid hormones and carnitine in the second trimester negatively affect neonate birth weight: A prospective cohort study. Front. Endocrinol. 2023, 14, 1080969. [Google Scholar] [CrossRef]
- Zakarija, M.; McKenzie, J.M. Pregnancy-associated changes in the thyroid-stimulating antibody of Graves’ disease and the relationship to neonatal hyperthyroidism. J. Clin. Endocrinol. Metab. 1983, 57, 1036–1040. [Google Scholar] [CrossRef]
- Polak, M. Hyperthyroidism in early infancy: Pathogenesis, clinical features and diagnosis with a focus on neonatal hyperthyroidism. Thyroid 1998, 8, 1171–1177. [Google Scholar] [CrossRef]
- Hébrant, A.; Van Staveren, W.C.G.; Maenhaut, C.; Dumont, J.E.; Leclère, J. Genetic hyperthyroidism: Hyperthyroidism due to activating TSHR mutations. Eur. J. Endocrinol. 2011, 164, 1–9. [Google Scholar] [CrossRef]
- Paschke, R.; Niedziela, M.; Vaidya, B.; Persani, L.; Rapoport, B.; Leclere, J.; Paschke, R. 2012 European Thyroid Association Guidelines for the Management of Familial and Persistent Sporadic Non-Autoimmune Hyperthyroidism Caused by Thyroid-Stimulating Hormone Receptor Germline Mutations. Eur. Thyroid J. 2012, 1, 142–147. [Google Scholar] [CrossRef]
- Cho, W.K.; Ahn, M.B.; Jang, W.; Chae, H.; Kim, M.; Suh, B.K. Nonautoimmune congenital hyperthyroidism due to p.Asp633Glu mutation in the TSHR gene. Ann. Pediatr. Endocrinol. Metab. 2018, 23, 235. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, H.P.; Wonerow, P.; Von Petrykowski, W.; Henschen, M.; Scherbaum, W.A.; Paschke, R. Sporadic congenital hyperthyroidism due to a spontaneous germline mutation in the thyrotropin receptor gene. J. Clin. Endocrinol. Metab. 1997, 82, 3879–3884. [Google Scholar] [CrossRef]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, R.; Dias, P.; Gouveia, R.; Sousa, A.B.; Oliveira, G. Neonatal McCune-Albright syndrome with systemic involvement: A case report. J. Med. Case Rep. 2015, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Weinstein, L.S.; Moran, A.; Pescovitz, O.H.; Charest, N.J.; Boney, C.M.; Van Wyk, J.J.; Merino, M.J.; Feuillan, P.P.; Spiegel, A.M. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J. Pediatr. 1993, 123, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Nakayama, M.; Baba, T.; Uehara, Y.; Niikawa, N.; Ito, M.; Tsuji, Y. A case of neonatal McCune-Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediatr. Scand. 1991, 80, 984–987. [Google Scholar] [CrossRef]
- Gaspari, L.; Paris, F.; Nicolino, M.; Hameury, F.; Bonnaure, H.; Pienkowski, C.; Servant, N.; Kalfa, N.; Sultan, C. Fetal ovarian cysts: An early manifestation of McCune-Albright syndrome? Prenat. Diagn. 2012, 32, 859–863. [Google Scholar] [CrossRef]
- Polak, M.; Legac, I.; Vuillard, E.; Guibourdenche, J.; Castanet, M.; Luton, D. Congenital Hyperthyroidism: The Fetus as a Patient. Horm. Res. Paediatr. 2006, 65, 235–242. [Google Scholar] [CrossRef]
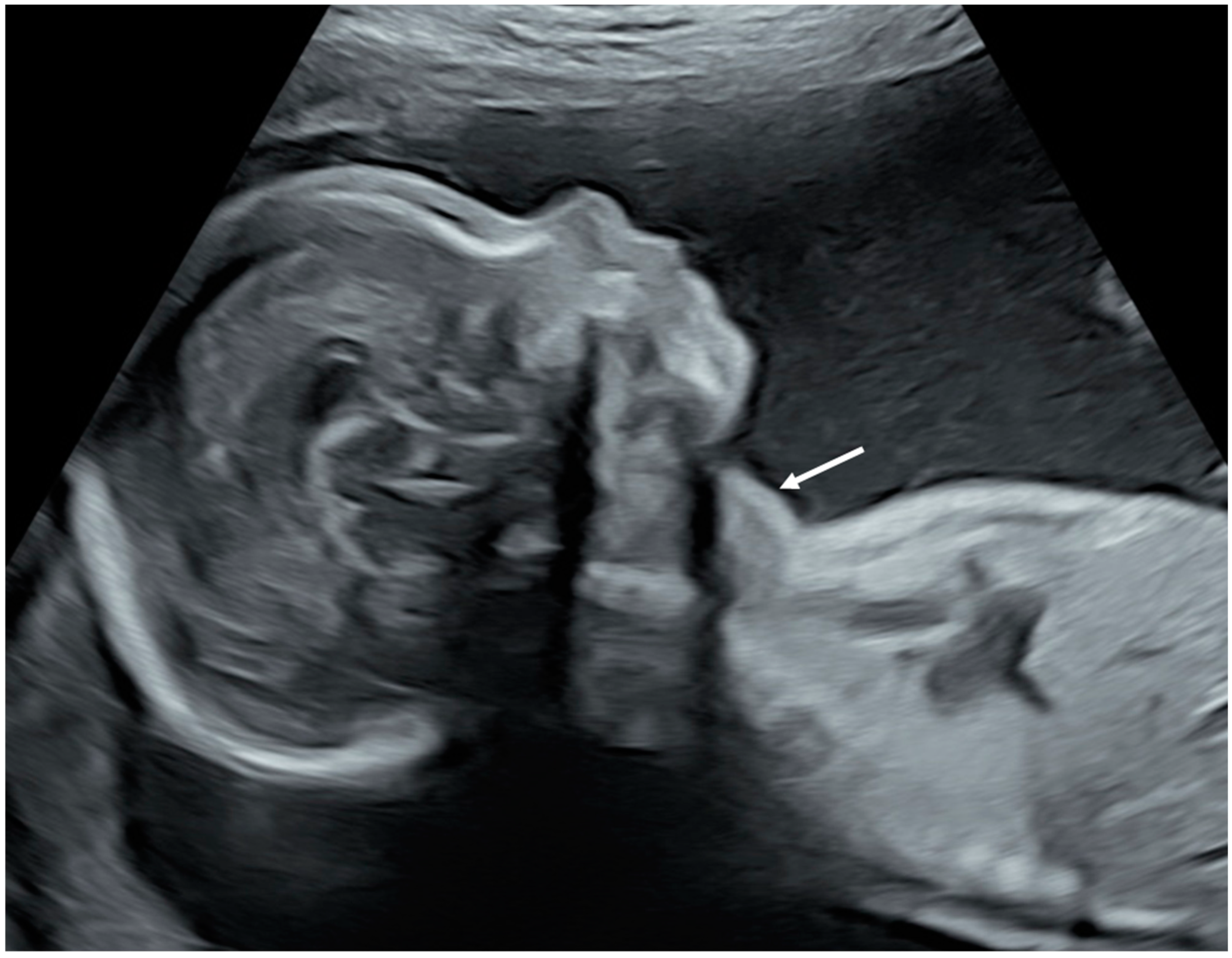
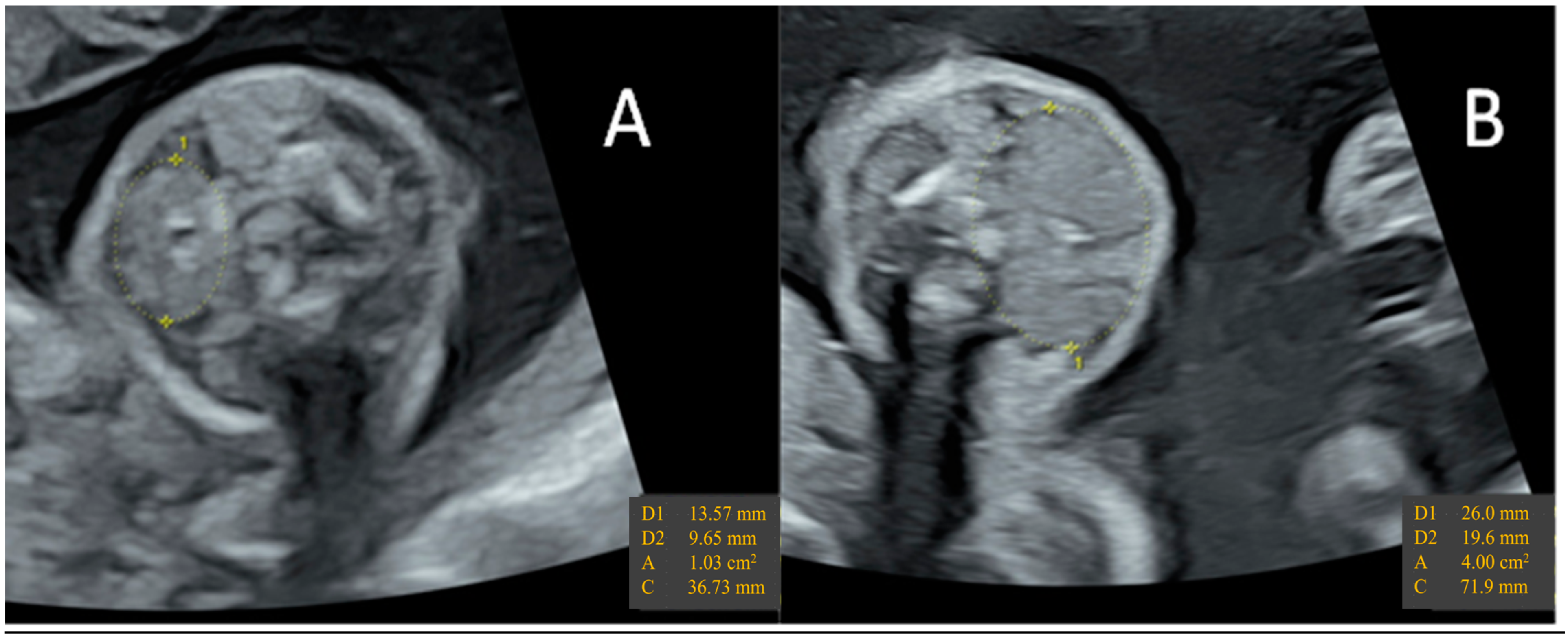
- Huel, C.; Guibourdenche, J.; Vuillard, E.; Ouahba, J.; Piketty, M.; Oury, J.F.; Luton, D. Use of ultrasound to distinguish between fetal hyperthyroidism and hypothyroidism on discovery of a goiter. Ultrasound Obstet. Gynecol. 2009, 33, 412–420. [Google Scholar] [CrossRef]
- Ranzini, A.C.; Ananth, C.V.; Smulian, J.C.; Kung, M.; Limbachia, A.; Vintzileos, A.M. Ultrasonography of the fetal thyroid: Nomograms based on biparietal diameter and gestational age. J. Ultrasound Med. 2001, 20, 613–617. [Google Scholar] [CrossRef]
- Luton, D.; Le Gac, I.; Vuillard, E.; Castanet, M.; Guibourdenche, J.; Noel, M.; Toubert, M.E.; Léger, J.; Boissinot, C.; Schlageter, M.H.; et al. Management of Graves’ Disease during Pregnancy: The Key Role of Fetal Thyroid Gland Monitoring. J. Clin. Endocrinol. Metab. 2005, 90, 6093–6098. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.V.; LaFranchi, S.H. Congenital hypothyroidism. Orphanet J. Rare Dis. 2010, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Bellisario, R.L.; Botero, D.; Fournier, L.; Abrams, C.A.; Cowger, M.L.; David, R.; Fort, P.; Richman, R.A. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J. Clin. Endocrinol. Metab. 1996, 81, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Gutch, M.; Philip, R.; Philip, R.; Toms, A.; Saran, S.; Gupta, K. Skeletal manifestations of juvenile hypothyroidism and the impact of treatment on skeletal system. Indian J. Endocrinol. Metab. 2013, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Vanderpas, J. Nutritional epidemiology and thyroid hormone metabolism. Annu. Rev. Nutr. 2006, 26, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Centre de Référence des Déficiences Intellectuelles de Causes Rares Filière DéfiScience Maladies Rares du Développement Cérébral et Déficience Intellectuelle Protocole National de Diagnostic et de Soins (PNDS) Syndrome de Allan Herndon-Dudley (SAHD) (MCT8 Thyroid Hormone Transporter) Argumentaire. Centre de Référence des Déficiences Intelectuelles de Causes Rares, Encéphalopathies, Centre de Compétence des Leucodystrophies et Leuco Encéphalopathies Rares, HAS. Protocole National de Diagnostic et de Soins: Syndrome de Allan Herndon-Dudley. 2019. Available online: https://www.has-sante.fr/upload/docs/application/pdf/2020-05/sahd_mct8_argumentaire.pdf (accessed on 12 March 2023).



{kind=link}
{kind=link}
| Placental Location | Evolution throughout Pregnancy | Substrates Uptake | Substrates Efflux | |||
|---|---|---|---|---|---|---|
| T1 | T2 | T3 | ||||
| LAT1 | SCT apical surface |  | T2 > rT3 > T3 > T4 | T2 | ||
| LAT2 | SCT |  | T2 > T3 | * | ||
| MCT8 | CT + SCT | * | T3, T4 > rT3 > T2 | T3, T4 | ||
| MCT10 | CT + SCT at T1 SCT at T3 | * | T3 > T4 | T3 | ||
| OATP1A2 | SCT basal surface CT and CT EV |  | T3 > rT3, T4 | * | ||
| OAPT4A1 | SCT apical surface |  | T3 > T4, rT3 | * | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mégier, C.; Dumery, G.; Luton, D. Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy. Metabolites 2023, 13, 633. https://doi.org/10.3390/metabo13050633
Mégier C, Dumery G, Luton D. Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy. Metabolites. 2023; 13(5):633. https://doi.org/10.3390/metabo13050633
Chicago/Turabian StyleMégier, Charles, Grégoire Dumery, and Dominique Luton. 2023. "Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy" Metabolites 13, no. 5: 633. https://doi.org/10.3390/metabo13050633