
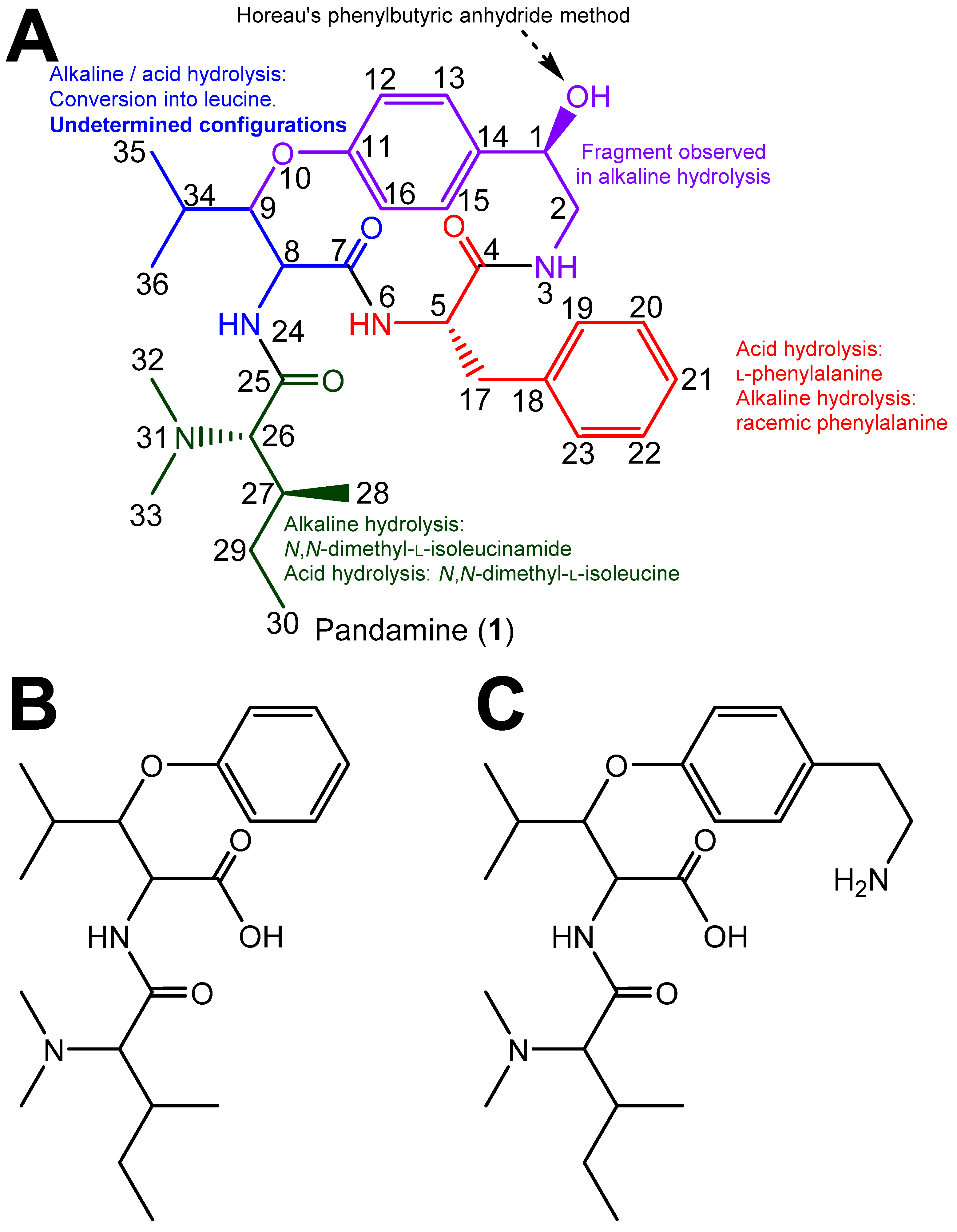
Clarifying the Configuration of Pandamine by an Extensive Spectroscopic Reinvestigation of the Authentic 1964 Sample
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
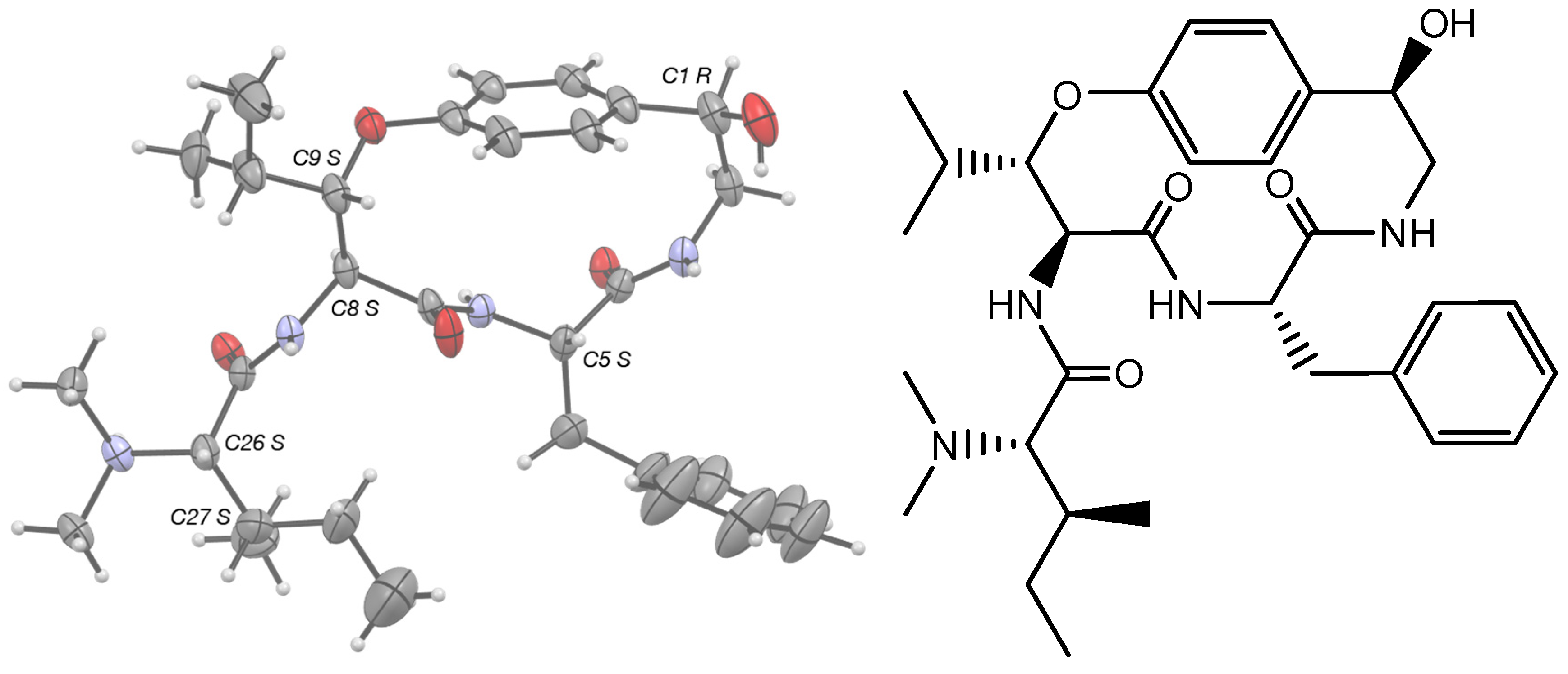
2.2. X-ray Crystallographic Analysis of Pandamine
2.3. Antimicrobial Activity Assay by Microdilutions
2.4. Cytotoxicity Assessment
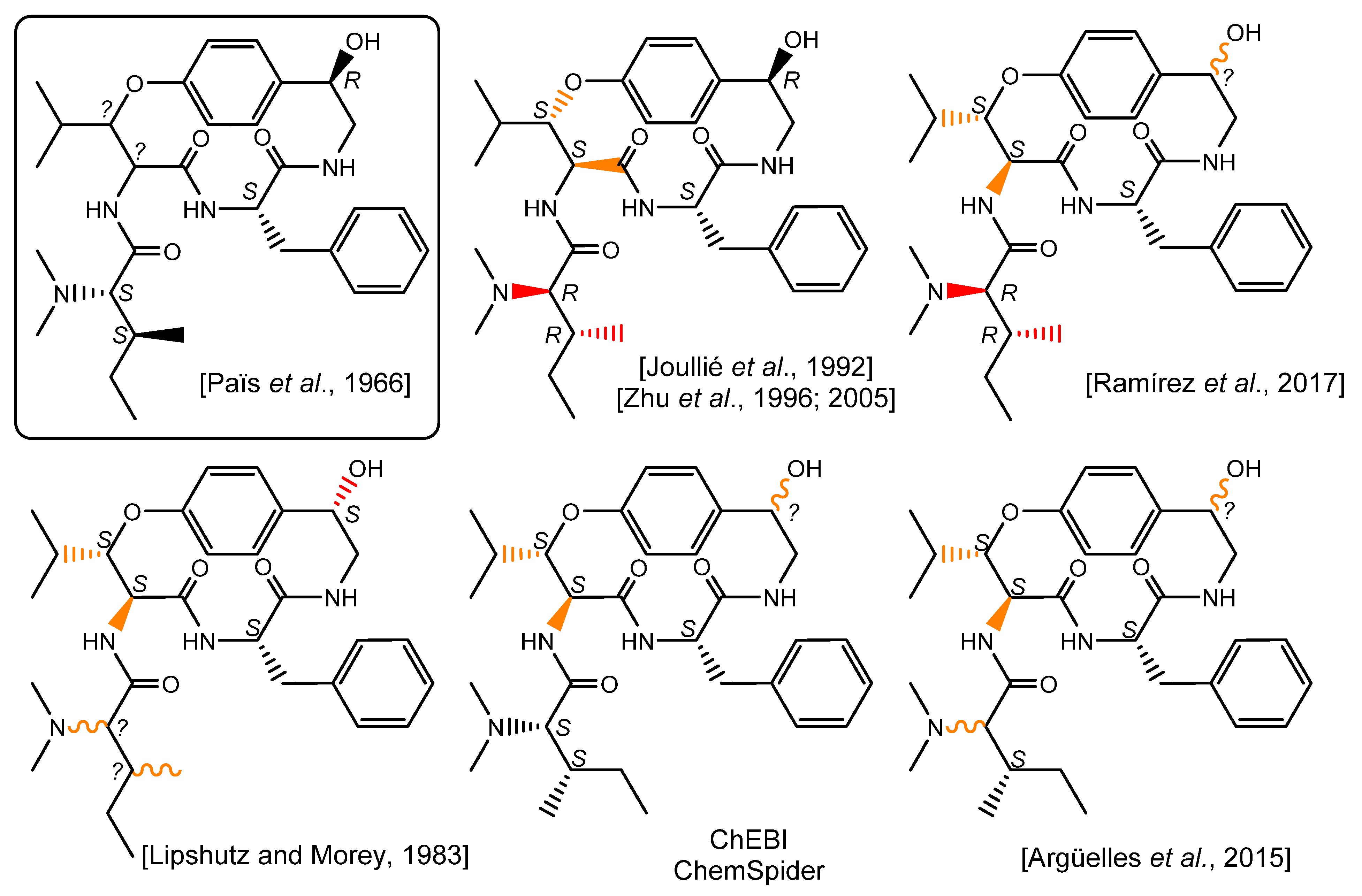
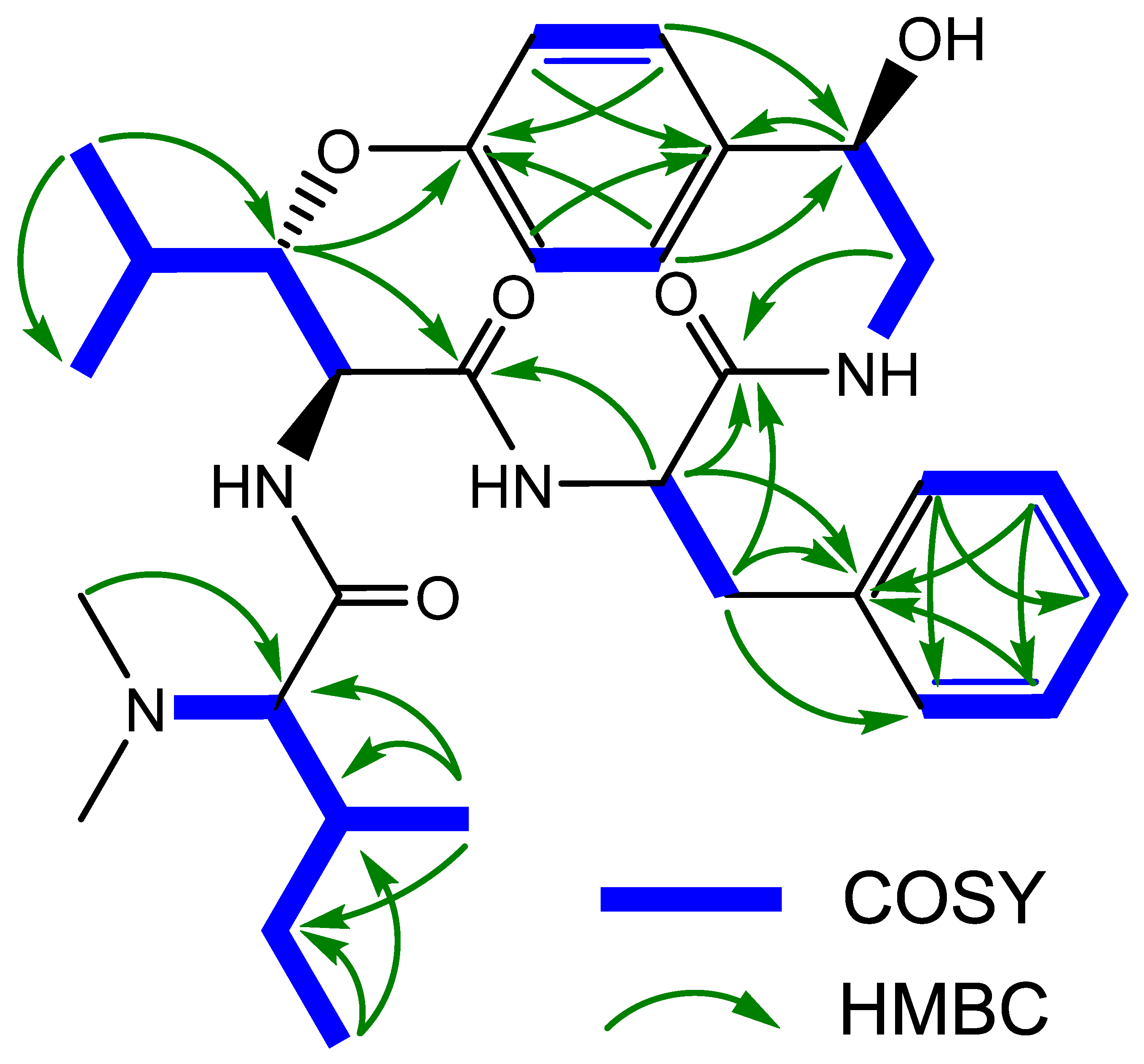
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Païs, M.; Lusinchi, X.; Goutarel, R.; Monseur, X. Alcaloides peptidiques. II. Structure de la pandamine alcaloide du panda oleosa pierre (pandacees). communication preliminaire. Bull. Soc. Chim. Fr. 1964, 4, 817–821. [Google Scholar]
- Païs, M.; Jarreau, F.-X.; Lusinchi, X.; Goutarel, R. Alcaloïdes Peptidiques, III. Pandamine et Pandaminine, Alcaloïdes Du Panda oleosa Pierre (Pandacées). Ann. Chim. 1966, 1, 83–105. [Google Scholar]
- Warnhoff, E.W. Peptide Alkaloids. In Fortschritte der Chemie Organischer Naturstoffe; Springer: Vienna, Austria, 1970; pp. 162–203. [Google Scholar]
- Gournelis, D.C.; Laskaris, G.G.; Verpoorte, R. Cyclopeptide Alkaloids. In Progress in the Chemistry of Organic Natural Products; Springer: Vienna, Austria, 1998; pp. 1–179. [Google Scholar]
- Tan, N.-H.; Zhou, J. Plant Cyclopeptides. Chem. Rev. 2006, 106, 840–895. [Google Scholar] [CrossRef]
- Tuenter, E.; Exarchou, V.; Apers, S.; Pieters, L. Cyclopeptide Alkaloids. Phytochem. Rev. 2017, 16, 623–637. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Morey, M.C. An Approach to the Cyclopeptide Alkaloids (Phencyclopeptines) via Heterocyclic Diamide/Dipeptide Equivalents. Preparation and N-Alkylation Studies of 2, 4 (5)-Disubstituted Imidazoles. J. Org. Chem. 1983, 48, 3745–3750. [Google Scholar] [CrossRef]
- Heffner, R.J.; Jiang, J.; Joullié, M.M. Total Synthesis of (-)-Nummularine F. J. Am. Chem. Soc. 1992, 114, 10181–10189. [Google Scholar] [CrossRef]
- Zhu, J.; Laïb, T.; Chastanet, J.; Beugelmans, R. A Novel Strategy towards the Total Synthesis of Cyclopeptide Alkaloids. Angew. Chem. Int. Ed. 1996, 35, 2517–2519. [Google Scholar] [CrossRef]
- Cristau, P.; Temal-Laïb, T.; Bois-Choussy, M.; Martin, M.-T.; Vors, J.-P.; Zhu, J. Total Synthesis of Mauritines A, B, C, and F: Cyclopeptide Alkaloids with a 14-Membered Paracyclophane Unit. Chem. Eur. J. 2005, 11, 2668–2679. [Google Scholar] [CrossRef]
- Horeau, A. Determination of the Configuration of Secondary Alcohols by Partial Resolution. In Stereochemistry: Fundamentals and Methods; Kagan, H.B., Ed.; Thieme: Stuttgart, Germany, 1978; Volume 3, pp. 51–94. [Google Scholar]
- König, W.A.; Gehrcke, B.; Weseloh, G. Determination of the Absolute Configuration of Secondary Alcohols with Horeau’s Method Including Enantioselective Gas Chromatography. Chirality 1994, 6, 141–147. [Google Scholar] [CrossRef]
- Da Silva, U.F.; Cardoso, C.D.; Zanatta, N.; Morel, A.F.; Icheln, D.; Gehrcke, B. Determination of the Stereochemistry of the N, N-Dimethyl Amino Acid and the α-Amino Acid Residue of Peptide Alkaloids by Chiral Gas Chromatography. Phytochem. Anal. 1996, 7, 20–23. [Google Scholar] [CrossRef]
- Païs, M.; Jarreau, F.-X.; Sierra, M.G.; Mascaretti, O.A.; Ruveda, E.A.; Chang, C.-J.; Hagaman, E.W.; Wenkert, E. Carbon-13 NMR Analysis of Cyclic Peptide Alkaloids. Phytochemistry 1979, 18, 1869–1872. [Google Scholar] [CrossRef]
- Ramírez, M.; Ochoa-Terán, A.; Somanathan, R.; Aguirre, G. Synthesis of Cis-Enamide Macrocycles via Ring-Closing Metathesis. Org. Chem. 2017, 4, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Argüelles, A.J.; Cordell, G.A.; Maruenda, H. Molecular Docking and Binding Mode Analysis of Plant Alkaloids as in Vitro and in Silico Inhibitors of Trypanothione Reductase from Trypanosoma cruzi. Nat. Prod. Commun. 2016, 11, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DNP. Available online: http://Dnp.Chemnetbase.Com/ (accessed on 30 October 2022).
- Rigaku, O.D. CrysAlis PRO. Rigaku Oxford Diffraction; Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. CheckCIF Validation ALERTS: What They Mean and How to Respond. Acta Crystallogr. Sect. E 2020, 76, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lagarias, J.C.; Yokoyama, W.H.; Bordner, J.; Shih, W.C.; Klein, M.P.; Rapoport, H. Cyclopeptide Alkaloids. Conformational Analysis of the Dihydro-p-Phencyclopeptine Nucleus. J. Am. Chem. Soc. 1983, 105, 1031–1040. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Takai, M.; Kawai, K.; Ogihara, Y.; Iitaka, Y.; Shibata, S. X-Ray Analysis of Tri-N-Methylfrangulanine Methiodide. J. Chem. Soc. Chem. Commun. 1974, 16, 653a. [Google Scholar] [CrossRef]
- Takai, M.; Ogihara, Y.; Iitaka, Y.; Shibata, S. Peptides in Higher Plants. I. The Conformation of Frangulanine. Chem. Pharm. Bull. 1975, 23, 2556–2559. [Google Scholar] [CrossRef] [Green Version]
- Flack, H.D. On Enantiomorph-Polarity Estimation. Acta Crystallogr. Sect. A 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Hooft, R.W.; Straver, L.H.; Spek, A.L. Determination of Absolute Structure Using Bayesian Statistics on Bijvoet Differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of Intensity Quotients and Differences in Absolute Structure Refinement. Acta Crystallogr. Sect. B 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira Ribeiro, S.; Fontaine, V.; Mathieu, V.; Zhiri, A.; Baudoux, D.; Stévigny, C.; Souard, F. Antibacterial and Cytotoxic Activities of Ten Commercially Available Essential Oils. Antibiotics 2020, 9, 717. [Google Scholar] [CrossRef] [PubMed]
- Ravon, F.; Menchi, E.; Lambot, C.; Al Kattar, S.; Chraibi, S.; Remmelink, M.; Fontaine, V.; Wauthoz, N. In Vitro and in Vivo Local Tolerability of a Synergistic Anti-Tuberculosis Drug Combination Intended for Pulmonary Delivery. J. Appl. Toxicol. 2023, 43, 298–311. [Google Scholar] [CrossRef]
- Beniddir, M.A.; Genta-Jouve, G.; Lewin, G. Resolving the (19 R) Absolute Configuration of Lanciferine, a Monoterpene Indole Alkaloid from Alstonia Boulindaensis. J. Nat. Prod. 2018, 81, 1075–1078. [Google Scholar] [CrossRef]
- Kouamé, T.; Bernadat, G.; Turpin, V.; Litaudon, M.; Okpekon, A.T.; Gallard, J.-F.; Leblanc, K.; Rharrabti, S.; Champy, P.; Poupon, E.; et al. Structure Reassignment of Melonine and Quantum-Chemical Calculations-Based Assessment of Biosynthetic Scenarios Leading to Its Revised and Original Structures. Org. Lett. 2021, 23, 5964–5968. [Google Scholar] [CrossRef]
- Jagora, A.; Gallard, J.-F.; Beniddir, M.A.; Le Pogam, P. A Reappraisal of the Structure of Lyaline as the First Naturally Occurring Nacycline Monoterpene Indole Alkaloid. J. Nat. Prod. 2021, 84, 2617–2622. [Google Scholar] [CrossRef]
- Sierra, M.G.; Mascaretti, O.A.; Diaz, F.J.; Rúveda, E.A.; Chang, C.-J.; Hagaman, E.W.; Wenkert, E. The Stereochemistry of the β-Hydroxyleucine Unit of Frangulanine. J. Chem. Soc. Chem. Commun. 1972, 915–916. [Google Scholar] [CrossRef]
- Maldaner, G.; Marangon, P.; Ilha, V.; Caro, M.S.B.; Burrow, R.A.; Dalcol, I.I.; Morel, A.F. Cyclopeptide Alkaloids from Scutia buxifolia Reiss. Phytochemistry 2011, 72, 804–809. [Google Scholar] [CrossRef]
- Park, M.H.; Suh, D.-Y.; Han, B.H. Absolute Configuration of a Cyclopeptide Alkaloid, Sanjoinine-G1, from Zizyphus vulgaris var. spinosus. Phytochemistry 1996, 43, 701–704. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recorded in DMSO-d6 | Recorded in TFA-d | |||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 4.89 (br s) | 70.6 | 5.27 (d, 3.9) | 75.7 |
| 1-OH | 5.21 (br s) | |||
| 2α | 3.96 (ddd, 13.7, 10.7, 4.4) | 47.3 | 4.19 (dd, 15.1, 4.0) | 49.4 |
| 2β | 2.73 (d, 13.7) | 3.07 (d, 15.1) | ||
| 3 | 7.27 (d, 10.7) | |||
| 4 | 169.0 | 173.2 | ||
| 5 | 4.27 (dt, 6.1, 8.3) | 53.0 | 4.51 (dd, 8.7, 6.6) | 57.8 |
| 6 | 7.47 (br s) | |||
| 7 | 168.5 | 173.0 | ||
| 8 | 4.51 (m) | 54.9 | 4.84 (d, 9.2) | 58.5 |
| 9 | 4.69 (dd, 9.0, 1.8) | 79.1 | 4.78 (d, 9.2) | 79.7 |
| 11 | 155.5 | 159.4 | ||
| 12 | 6.70 (dd, 8.3, 2.5) | 118.5 | 6.70 (dd, 8.6, 2.4) | 121.0 |
| 13 | 6.86 (dd, 8.3, 2.0) | 126.6 | 6.97 (dd, 8.6, 1.8) | 130.0 |
| 14 | 134.9 | 132.3 | ||
| 15 | 7.25 (dd, 8.8, 2.0) | 126.5 | 7.18 (dd, 8.4, 2.1) | 128.4 |
| 16 | 6.81 (dd, 8.8, 2.5) | 113.4 | 6.82 (dd, 8.8, 2.5) | 114.0 |
| 17 | 2.35 (dd, 13.8, 6.2) 2.63 (dd, 13.8, 8.3) | 39.0 | 2.74 (dd, 13.6, 8.4) 2.60 (dd, 13.6, 6.4) | 41.5 |
| 18 | 136.8 | 136.2 | ||
| 19 | 7.02 (m) | 128.8 | 6.91 (dd, 6.6, 1.7) | 130.9 |
| 20 | 7.16 (m) | 127.8 | 7.11 (m) | 130.8 |
| 21 | 7.11 (t, 7.3) | 126.0 | 7.09 (ov) | 129.7 |
| 22 | 7.16 (m) | 127.8 | 7.11 (m) | 130.8 |
| 23 | 7.02 (m) | 128.8 | 6.91 (dd, 6.6, 1.7) | 130.9 |
| 24 | ||||
| 25 | n.d. | 168.2 | ||
| 26 | 3.73 (br s) | 70.2 | 3.97 (d, 4.4) | 75.5 |
| 27 | 1.94 (m) | 33.0 | 2.11 (m) | 36.7 |
| 28 | 0.72 (d, 6.4) | 13.4 | 0.84 (d, 6.9) | 13.6 |
| 29 | 0.99 (ov.), 1.49 (m) | 25.3 | 0.92 (m), 1.38 (m) | 28.6 |
| 30 | 0.83 (t, 7.3) | 10.9 | 0.92 (t, 7.2) | 12.4 |
| 32 | 2.71 (s) | 41.2 | 3.01 (s) | 42.8 |
| 33 | 2.71 (s) | 41.2 | 3.05 (s) | 45.6 |
| 34 | 2.13 (m) | 27.9 | 1.97 (hept, 6.6) | 31.3 |
| 35 | 1.07 (d, 7.3) | 20.1 | 1.05 (d, 6.7) | 20.3 |
| 36 | 0.95 (d, 6.7) | 14.4 | 0.96 (d, 6.7) | 14.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Retailleau, P.; Numbi Wa Ilunga, E.; Fontaine, V.; Gallard, J.-F.; Le Pogam, P. Clarifying the Configuration of Pandamine by an Extensive Spectroscopic Reinvestigation of the Authentic 1964 Sample. Metabolites 2023, 13, 470. https://doi.org/10.3390/metabo13040470
Retailleau P, Numbi Wa Ilunga E, Fontaine V, Gallard J-F, Le Pogam P. Clarifying the Configuration of Pandamine by an Extensive Spectroscopic Reinvestigation of the Authentic 1964 Sample. Metabolites. 2023; 13(4):470. https://doi.org/10.3390/metabo13040470
Chicago/Turabian StyleRetailleau, Pascal, Evodie Numbi Wa Ilunga, Véronique Fontaine, Jean-François Gallard, and Pierre Le Pogam. 2023. "Clarifying the Configuration of Pandamine by an Extensive Spectroscopic Reinvestigation of the Authentic 1964 Sample" Metabolites 13, no. 4: 470. https://doi.org/10.3390/metabo13040470