
Identification of Cancer Driver Genes by Integrating Multiomics Data with Graph Neural Networks
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.1.1. Multiomics Data
2.1.2. Network Data
2.1.3. Assignment of Positive Labels and Negative Labels for Genes
- Genes associated with the expression level of cancer driver genes.
- Genes related to existing cancer pathways.
- Cancer genes in the OMIM dataset [23] (https://omim.org//downloads accessed on 1 May 2021).
- Known cancer driver genes in the DriverDBV3 dataset (http://driverdb.tms.cmu.edu.tw/ accessed on 1 May 2021).
- Cancer driver genes in the NCG dataset [24] (http://ncg.kcl.ac.uk/ accessed on 1 May 2021).
2.2. Feature Generation
2.2.1. Epigenetic Feature
2.2.2. Transcriptome Features
Expression Fold Change
Significance of Differential Expression
Gene Outlier Feature
2.2.3. Genomic Features
Gene Mutation Frequency
Variant Allele Fraction
MutSigCV Derived Attribute
- DNA replication time;
- Noncoding mutation rate;
- Local GC content;
- HiC compartment;
- Local gene density;
- Wgs mean depth;
- Wgs percent 20x;
- Capture on target rate;
- Capture mean depth;
- Capture pct200;
- Capture mean percentGC.
Mutant Base Conversion
Copy Number Variation Rate
2.2.4. Biological Network Derived Features
Bipartite Graph Attributes
BioGrid Network Features
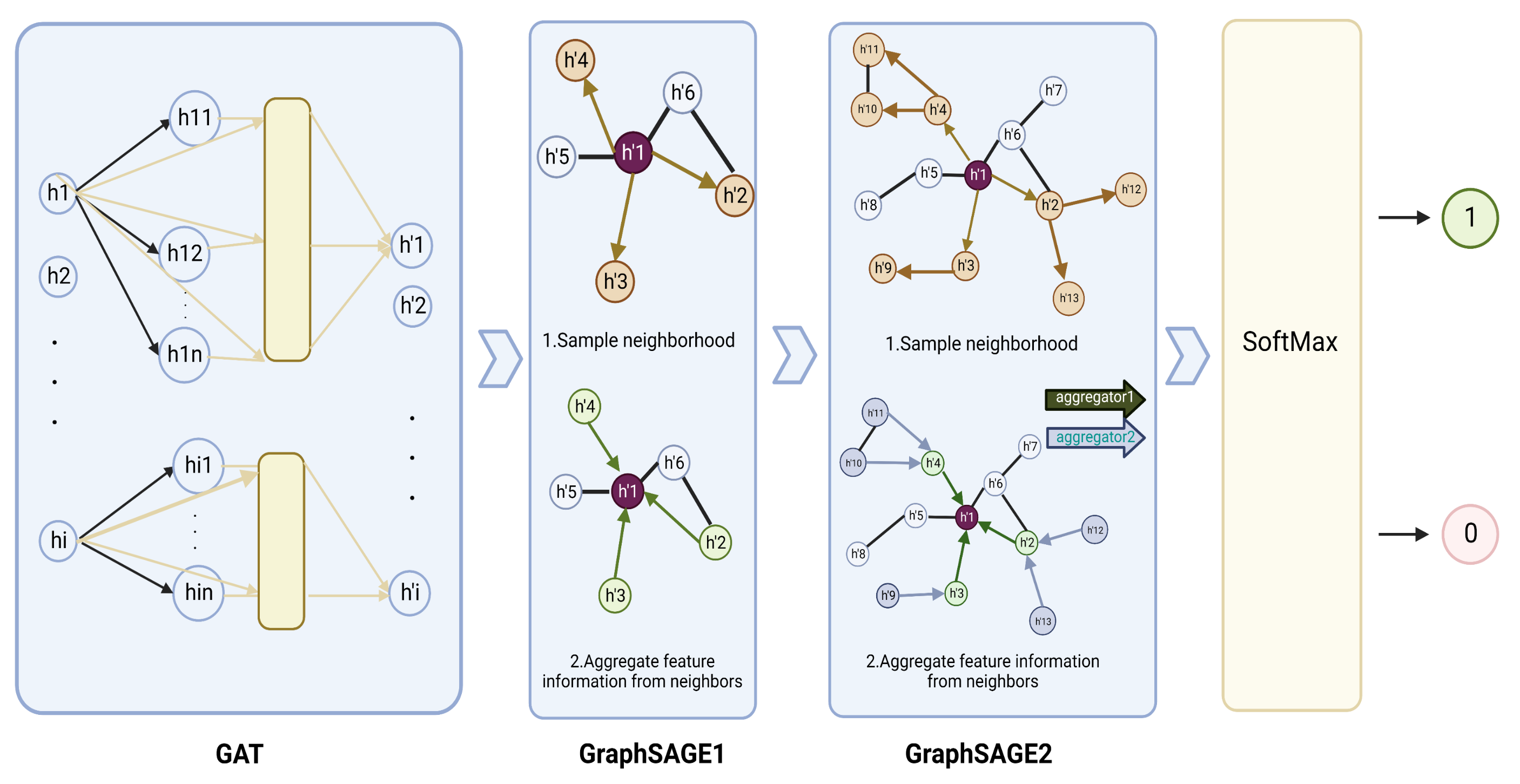
2.3. Model Construction
2.3.1. GraphSAGE Layer
2.3.2. GAT Layer
2.4. Model Training
3. Results
3.1. Evaluation Metrics
3.2. Performance of GGraphSAGE by Comparing with SOTA Methods
3.3. Performance of GGraphSAGE by Comparing with Classical Machine Learning Models
3.4. Ablation Experiments
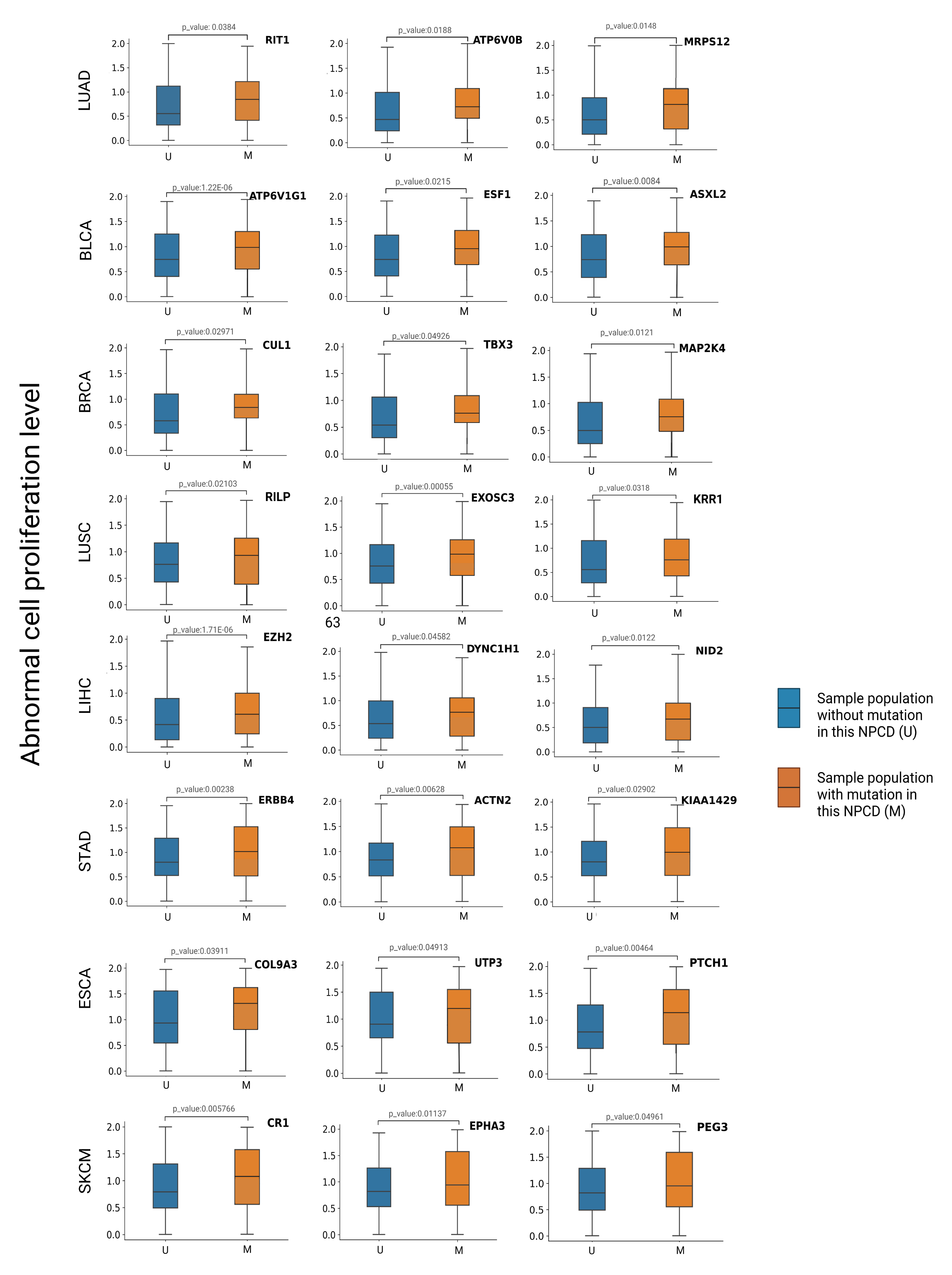
3.5. Association between Newly Predicted Genes and Cell Proliferation
3.6. Newly Predicted Cancer Driver Genes and Their Verified Functions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Gene ID | Literature Description | p-Value | Citation |
|---|---|---|---|---|
| STAD | ERBB4 | Members of the ErbB subfamily of receptor tyrosine kinases are important regulators of normal breast physiology, and abnormalities in their signaling have been associated with breast tumorigenesis. | 0.00238 | [41,45] |
| ACTN2 | ACTN2-overexpression in human hepatocellular carcinoma cells showed enhanced cell motility and invasive ability, suggesting possible functions in late metastatic stages, such as extravasation and lung colonization. | 0.00628 | [35] | |
| KIAA1429 | M6 RNA methylation has a huge impact on RNA production/ metabolism and is involved in the pathogenesis of many diseases, including cancer. M6 is modified by m-mounted 6 methyltransferases (METTL3/14, WTAP, RBM15/15B and KIAA1429, referred to as “writers”), reduced by (FTO and ALKBH5, referred to as “erasers”) and recognized by m-mount6 binding proteins (YTHDF1/2/3, IGF2BP1 and HNRNPA2B1, called “readers”). | 0.02902 | [46] | |
| BLCA | ESF1 | Transcription of the E1A gene of highly oncogenic adenovirus 12 (Ad12) starts at two initiation sites (TS1 and TS2). We have previously shown that the distal ends of the E2F and ATF motifs of TS1 are synergistically involved in E1A self-stimulation in the TS1 promoter region. Here, we report the identification of a second E2F-like target region (E2DFII) upstream of the E1A stimulatory factor 1 binding site (ESF-1), which is important for 13S-mediated self-activation of TS2. | 0.0215 | [47] |
| ATP6V1G1 | Low mRNA and protein expression of ATP6V1s members were found to be significantly associated with clinical cancer stage, lymph node metastasis status, and patient gender in KIRC patients. In addition, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1C2, ATP6V1D, ATP6V1E1, ATP6V1E1, ATP6V1E1, ATP6V1E1, ATP6V1E1, ATP6V1G1, and ATP6V1H had lower mRNA expression but shorter OS. | [48] | ||
| ASXL2 | Upregulation of ASXL2 was associated with advanced clinical staging. Patients exhibiting high levels of ASXL2 expression had poorer overall survival, while those with low ASXL2 expression levels survived longer. A multifactorial Cox regression analysis showed that ASXL2 expression could be considered as an independent prognostic factor for CRC. Inhibition or overexpression of ASXL2 significantly affected the proliferation of CRC cells. | 0.0084 | [41,49] | |
| BRCA | CUL1 | CUL1 significantly promoted breast cancer cell migration, invasion, and in vitro tube formation, as well as angiogenesis and metastasis in vivo. Mechanistically, global transcriptional changes in MDA-MB-231 cells were determined using human gene expression profiling, and autocrine expression of cytokines CXCL8 and IL11 were identified as target genes of CUL1 in breast cancer cell migration, invasion, metastasis, and angiogenesis. | 0.02971 | [34,41] |
| BRCA | TBX3 | TBX3 has no known function in adult tissues but is frequently overexpressed in a wide range of epithelial- and mesenchymal-derived cancers. This overexpression greatly affects several features of cancer, including senescent bypass, apoptosis and deactivation, promotion of proliferation, tumor formation, angiogenesis, invasive and metastatic capacity, and cancer stem cell expansion. | 0.04926 | [41,50] |
| MAP2K4 | Genetic variants (copy number variants and single nucleotide polymorphisms) and acquired somatic copy number aberrations (CNAs) were associated with approximately 40% of gene expression, with the landscape dominated by cis and trans CNAs. By depicting genes with expression aberrations driven by CNA in cis, we identified putative cancer genes, including deletions in PPP2R2A, MTAP, and MAP2K4. | 0.0121 | [41,51] | |
| ESCA | COL9A3 | USP3 promotes GC progression and metastasis by deubiquitinating C- OL9A3 and COL6A5. These findings identify a mechanism for the USP3-mediated deubiquitination of enzymatic activity in GC metastasis and suggest a potential therapeutic target for GC management. | 0.03911 | [52] |
| PTCH1 | Dysregulation of the Hh signaling pathway is associated with developmental abnormalities and cancers (including Gorlin syndrome) and sporadic cancers (e.g., basal cell carcinoma, medulloblastoma, pancreatic, breast, colon, ovarian, and small-cell lung cancers). Abnormal activation of the Hh signaling pathway is caused by mutations in related genes (ligand nondependent signaling) or by overexpression of Hh signaling molecules (ligand-dependent signaling—autocrine or paracrine). | 0.00464 | [41,53] | |
| UTP3 | UTP3, the small subunit process group homolog (UTP3), and prostaglandin E synthase 3 (PTGES3). Thus, pathway functions in dynamic module 3 (ubiquitin-mediated protein hydrolysis and ribosomes) and several seed genes (PPP1R12A, UTP3, and PTGES3) may be associated with OS progression and could serve as potential therapeutic targets in OS. | 0.04913 | [54] | |
| LIHC | EZH2 | Recent findings on the role of EZH2 in cancer genesis, progression, metastasis, metabolism, drug resistance and immune regulation. In addition, we highlight the progress of targeted EZH2 therapies in experimental and clinical studies. | [41,55] | |
| DYNC1H1 | The deleterious mutations were found to affect the function of DYNC1H1 leading to the formation of associated cancers. | 0.04582 | [36] | |
| NID2 | NID2, SPARC, and MFAP2 were upregulated in gastric tumor tissues and significantly associated with poor overall survival. Thus, by using these 3 upregulated DEGs, the predictive value of the risk score model used for gastric cancer prognosis could be improved. | 0.0122 | [56] | |
| LUSC | RILP | Methylation of RILP in lung cancer promotes tumor cell proliferation and invasion. | 0.02103 | [37] |
| LUSC | EXOSC3 | In inflamed mucosa, EXOSC3 and CNOT4-mediated RNA stabilization, including that of MYD88, may trigger cancer development and could serve as potential predictive markers and innovative therapies to control cancer progression. | 0.00055 | [57] |
| KRR1 | Tumor-associated antigens KRR1 and ZRF1 and their cognate autoantibodies may be considered as potential molecular markers for breast cancer. | 0.0318 | [58] | |
| LUAD | ATP6V0B | miRNA-15a, which could regulate ATPase, H(+) transporting, lysosomal21 kDa, V0 subunit b(ATP6V0B), and miRNA-155, were found to be the most significant. | 0.0188 | [59] |
| RIT1 | The results provide a genome-wide map of oncogenic RIT1 functional interactions and identify components of the RAS pathway, spindle assembly checkpoint, and Hippo/YAP1 network as candidate therapeutic targets for RIT1 mutant lung cancer. | 0.0384 | [41,60] | |
| MRPS12 | MRPS12 functions as a potential oncogene in ovarian cancer and is a promising prognostic candidate. | 0.0148 | [61] | |
| SKCM | CR1 | The HER2 CR1 domain can be antagonized by the clinically used HER2 antibody pertuzumab. Pertuzumab significantly reduced tumorigenicity in TNBC cells expressing circ-HER2/HER2-103. | 0.005766 | [62] |
| EPHA3 | EphA3, originally isolated from leukemia and melanoma cells, is currently one of the most promising therapeutic targets, with multiple tumor-promoting effects in multiple cancer types. | 0.01137 | [63] | |
| PEG3 | Paternal expression gene 3 (PEG3) was the only imprinted gene associated with prognosis of colon cancer patients at the mRNA level, and a high expression of PEG3 mRNA was associated with a poor prognosis. | 0.04961 | [64] |
References
- Liu, F.; Gai, X.; Wu, Y.; Zhang, B.; Wu, X.; Cheng, R.; Tang, B.; Shang, K.; Zhao, N.; Deng, W.; et al. Oncogenic β -catenin stimulation of AKT2–CAD-mediated pyrimidine synthesis is targetable vulnerability in liver cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2202157119. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M. Patterns of somatic mutation in human cancer genomes. EJC Suppl. 2008, 9, 6. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. [Special Issue Review] Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Wensheng, Z.; Flemington, E.K.; Kun, Z. Driver gene mutations based clustering of tumors: Methods and applications. Bioinformatics 2018, 34, i404–i411. [Google Scholar]
- Mao, Y.; Chen, H.; Liang, H.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. CanDrA: Cancer-Specific Driver Missense Mutation Annotation with Optimized Features. PLoS ONE 2013, 8, e77945. [Google Scholar] [CrossRef] [Green Version]
- Bashashati, A.; Haffari, G.; Ding, J.; Ha, G.; Lui, K.; Rosner, J.; Huntsman, D.G.; Caldas, C.; Aparicio, S.A.; Shah, S.P. DriverNet: Uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012, 13, R124. [Google Scholar] [CrossRef] [Green Version]
- Carter, H. Computational Assessment of Somatic Missense Mutations Detected in Tumor Sequencing Studies with Cancer-Specific High-Throughput Annotation of Somatic Mutations (CHASM). Ph.D. Thesis, The Johns Hopkins University, Baltimore, MD, USA, 2012. [Google Scholar]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615–621. [Google Scholar]
- Yin, C.; Cao, Y.; Sun, P.; Zhang, H.; Li, Z.; Xu, Y.; Sun, H. Molecular Subtyping of Cancer Based on Robust Graph Neural Network and Multi-Omics Data Integration. Front. Genet. 2022, 13, 884028. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Gong, L.; Cheng, Q. Exploiting Edge Features in Graph Neural Networks. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018. [Google Scholar]
- Xu, D.; Ruan, C.; Korpeoglu, E.; Kumar, S.; Achan, K. Inductive representation learning on temporal graphs. arXiv 2020, arXiv:2002.07962. [Google Scholar]
- Zhu, J.; Yan, Y.; Zhao, L.; Heimann, M.; Akoglu, L.; Koutra, D. Beyond Homophily in Graph Neural Networks: Current Limitations and Effective Designs. Adv. Neural Inf. Process. Syst. 2020, 33, 7793–7804. [Google Scholar]
- Zhu, J.; Jin, J.; Loveland, D.; Schaub, M.T.; Koutra, D. On the Relationship between Heterophily and Robustness of Graph Neural Networks. arXiv 2021, arXiv:2106.07767. [Google Scholar]
- Velickovic, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. Stat 2017, 1050, 20. [Google Scholar]
- Liu, J.; Ong, G.P.; Chen, X. GraphSAGE-Based Traffic Speed Forecasting for Segment Network with Sparse Data. IEEE Trans. Intell. Transp. Syst. 2022, 23, 1755–1766. [Google Scholar] [CrossRef]
- Weinstein, J.; Collisson, E.; Mills, G.; Shaw, K.; Ozenberger, B.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.; Network, C. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [Green Version]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; Mckusick, V.A. Online Mendelian Inheritance In Man (OMIM). Hum. Mutat. 2000, 15, 57–61. [Google Scholar] [CrossRef]
- D’Antonio, M.; Pendino, V.; Sinha, S.; Ciccarelli, F.D. Network of Cancer Genes (NCG 3.0): Integration and analysis of genetic and network properties of cancer genes. Nucleic Acids Res. 2012, 40, D978–D983. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imambi, S.; Prakash, K.B.; Kanagachidambaresan, G.R. PyTorch. Program. Tensorflow Solut. Edge Comput. Appl. 2021, 87–104. Available online: https://www.semanticscholar.org/paper/PyTorch-Imambi-Prakash/d668f12be54174141e6197fad737006b7b0c0571 (accessed on 1 May 2021).
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef]
- Wong, W.C.; Kim, D.; Carter, H.; Diekhans, M.; Ryan, M.C.; Karchin, R. CHASM and SNVBox: Toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics 2011, 27, 2147–2148. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, Y.L. Random Forests; Karelian Research Centre Russian Academy of Sciences: Petrozavodsk, Russia, 1997. [Google Scholar]
- Abeywickrama, T.; Cheema, M.A.; Taniar, D. k-Nearest Neighbors on Road Networks: A Journey in Experimentation and In-Memory Implementation. arXiv 2016, arXiv:1601.01549. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V.N. Support-vector networks. Mach. Learn. 2004, 20, 273–297. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Liu, X.; Fan, X.; Hong, Y.; Wang, Y.; Huang, Y.L.; Xie, M.Q. Prioritizing disease genes with an improved dual label propagation framework. BMC Bioinform. 2018, 19, 47. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.F.; Zhang, Z.; Zhang, M.; Chen, Y.S.; Song, J.; Hou, P.F.; Yong, H.M.; Zheng, J.N.; Bai, J. CUL1 promotes breast cancer metastasis through regulating EZH2-induced the autocrine expression of the cytokines CXCL8 and IL11. Cell Death Dis. 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, L.H.; Lam, C.Y.; To, J.C.; Chiu, C.H.; Keng, V.W. Sleeping Beauty insertional mutagenesis screen identifies the pro-metastatic roles of CNPY2 and ACTN2 in hepatocellular carcinoma tumor progression. Biochem. Biophys. Res. Commun. 2021, 541, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Sucularli, C.; Arslantas, M. Computational prediction and analysis of deleterious cancer associated missense mutations in DYNC1H1. Mol. Cell. Probes 2017, 34, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhuo, Y.; Yin, Y.; Qiu, L.; Li, X.; Lai, F. Methylation of RILP in lung cancer promotes tumor cell proliferation and invasion. Mol. Cell. Biochem. 2021, 476, 853–861. [Google Scholar] [CrossRef]
- Porta-Pardo, E.; Garcia-Alonso, L.; Hrabe, T.; Dopazo, J.; Godzik, A. A Pan-Cancer Catalogue of Cancer Driver Protein Interaction Interfaces. PLoS Comput. Biol. 2015, 11, e1004518. [Google Scholar] [CrossRef]
- Bertrand, D.; Chng, K.R.; Sherbaf, F.G.; Kiesel, A.; Chia, B.K.; Sia, Y.Y.; Huang, S.K.; Hoon, D.S.; Liu, E.T.; Hillmer, A.; et al. Patient-specific driver gene prediction and risk assessment through integrated network analysis of cancer omics profiles. Nucleic Acids Res. 2015, 43, e44. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Masand, N.; Patil, V.M. Understanding Molecular Process and Chemotherapeutics for the Management of Breast Cancer. Curr. Chem. Biol. 2021, 15, 69–84. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Craene, B.D.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Kilpinen, S. Role of ErbB4 in breast cancer. J. Mammary Gland. Biol. Neoplasia 2008, 13, 259–268. [Google Scholar]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Pützer, B.M.; Gnauck, J.; Kirch, H.C.; Brockmann, D.; Esche, H. A cis-acting element 7 bp upstream of the ESF-1-binding motif is involved in E1A 13S autoregulation of the adenovirus 12 TS2 promoter. J. Gen. Virol. 1997, 78, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wang, L.; Sha, J.; Lou, G.; Lu, N.; Hang, B.; Mao, J.H.; Zou, X. Expression and transcriptional regulation of human ATP6V1A gene in gastric cancers. Sci. Rep. 2017, 7, 3015. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Yang, L.; Wang, Y.; Zhong, M.; Yu, M.; Chen, B. Elevated Expression of ASXL2 is Associated with Poor Prognosis in Colorectal Cancer by Enhancing Tumorigenesis and Inducing Cell Proliferation. Cancer Manag. Res. 2020, 12, 10221. [Google Scholar] [CrossRef]
- Khan, S.F.; Damerell, V.; Omar, R.; Du Toit, M.; Khan, M.; Maranyane, H.M.; Mlaza, M.; Bleloch, J.; Bellis, C.; Sahm, B.D.; et al. The roles and regulation of TBX3 in development and disease. Gene 2020, 726, 144223. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Wu, X.; Wang, H.; Zhu, D.; Chai, Y.; Wang, J.; Dai, W.; Xiao, Y.; Tang, W.; Li, J.; Hong, L.; et al. USP3 promotes gastric cancer progression and metastasis by deubiquitination-dependent COL9A3/COL6A5 stabilisation. Cell Death Dis. 2021, 13, 10. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Sontag, J.M.; Nunbhakdi-Craig, V.; Mitterhuber, M.; Ogris, E.; Sontag, E. Regulation of protein phosphatase 2A methylation by LCMT1 and PME-1 plays a critical role in differentiation of neuroblastoma cells. J. Neurochem. 2010, 115, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Wang, W.; Tong, Y.; Zhang, J. Genome-scale analysis identified NID2, SPARC, and MFAP2 as prognosis markers of overall survival in gastric cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e929558-1. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Noguchi, M.; Kurai, T.; Ichihashi, Y.; Ise, K.; Wang, L.; Ishida, Y.; Tanino, M.; Hirano, S.; Asaka, M.; et al. Aberrant expression of MYD88 via RNA-controlling CNOT4 and EXOSC3 in colonic mucosa impacts generation of colonic cancer. Cancer Sci. 2021, 112, 5100. [Google Scholar] [CrossRef]
- Dyachenko, L.; Havrysh, K.; Lytovchenko, A.; Dosenko, I.; Antoniuk, S.; Filonenko, V.; Kiyamova, R. Autoantibody response to ZRF1 and KRR1 SEREX antigens in patients with breast tumors of different histological types and grades. Dis. Markers 2016, 2016, 5128720. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Teng, L.; Liu, W.; Cao, Y.; Ding, D.; Wang, W.; Chen, H.; Li, C.; An, R. Identification of biological targets of therapeutic intervention for clear cell renal cell carcinoma based on bioinformatics approach. Cancer Cell Int. 2016, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Vichas, A.; Riley, A.K.; Nkinsi, N.T.; Kamlapurkar, S.; Parrish, P.C.; Lo, A.; Duke, F.; Chen, J.; Fung, I.; Watson, J.; et al. Integrative oncogene-dependency mapping identifies RIT1 vulnerabilities and synergies in lung cancer. Nat. Commun. 2021, 12, 4789. [Google Scholar] [CrossRef]
- Qiu, X.; Guo, D.; Du, J.; Bai, Y.; Wang, F. A novel biomarker, MRPS12 functions as a potential oncogene in ovarian cancer and is a promising prognostic candidate. Medicine 2021, 100, e24898. [Google Scholar] [CrossRef]
- Li, J.; Ma, M.; Yang, X.; Zhang, M.; Luo, J.; Zhou, H.; Huang, N.; Xiao, F.; Lai, B.; Lv, W.; et al. Circular HER2 RNA positive triple negative breast cancer is sensitive to Pertuzumab. Mol. Cancer 2020, 19, 142. [Google Scholar] [CrossRef]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef]
- Zhou, T.; Lin, W.; Zhu, Q.; Renaud, H.; Liu, X.; Li, R.; Tang, C.; Ma, C.; Rao, T.; Tan, Z.; et al. The role of PEG3 in the occurrence and prognosis of colon cancer. OncoTargets Ther. 2019, 12, 6001–6012. [Google Scholar] [CrossRef] [Green Version]








| Tumor Type | Number of Tumor Samples | Number of Control Samples |
|---|---|---|
| BLCA | 408 | 19 |
| BRCA | 1095 | 19 |
| LUSC | 501 | 51 |
| LUAD | 515 | 59 |
| ESCA | 184 | 13 |
| LIHC | 371 | 59 |
| STAD | 238 | 33 |
| SKCM | 466 | 5 |
| Tumor Type | Driver Genes | Nondriver Genes |
|---|---|---|
| BLCA | 78 | 3586 |
| BRCA | 65 | 3646 |
| LUSC | 78 | 3586 |
| LUAD | 44 | 3690 |
| ESCA | 73 | 3659 |
| LIHC | 33 | 3636 |
| STAD | 35 | 3707 |
| SKCM | 14 | 3801 |
| Tumor Type | GAT (Input/Output Size) | GraphSAGE1 (Input/Output Size) | GraphSAGE2 (Input/Output Size) |
|---|---|---|---|
| BRCA | 36/64 | 64/128 | 128/2 |
| BLCA | 36/64 | 64/128 | 128/2 |
| ESCA | 36/64 | 64/32 | 32/2 |
| LUAD | 36/256 | 256/64 | 64/2 |
| LIHC | 36/256 | 256/128 | 128/2 |
| LUSC | 36/128 | 128/32 | 32/2 |
| STAD | 36/128 | 128/64 | 64/2 |
| SKCM | 36/64 | 64/256 | 256/2 |
| Tumor Type | Gene Name | M/U Group Size | Median of Abnormal Cell Proliferation Level (M/U) |
|---|---|---|---|
| BRCA | CUL1 | 50/1045 | 0.971/0.551 |
| TBX3 | 85/1010 | 0.896/0.548 | |
| MAP2K4 | 104/991 | 0.743/0.51 | |
| BLCA | ATP6V1G1 | 22/389 | 0.998/0.75 |
| ESF2 | 53/358 | 0.999/0.749 | |
| ASXL2 | 64/347 | 1.12/0.561 | |
| ESCA | UTP3 | 23/161 | 1.25/0.987 |
| COL9A3 | 19/165 | 1.3/0.99 | |
| PTCH1 | 32/152 | 1.131/0.748 | |
| LUAD | RIT1 | 86/481 | 0.801/0.562 |
| ATP6V0B | 30/537 | 0.731/0.433 | |
| MRPS12 | 30/537 | 0.799/0.561 | |
| LIHC | EZH2 | 79/292 | 0.688/0.49 |
| DYNC1H1 | 63/308 | 0.789/0.55 | |
| NID2 | 48/323 | 0.727/0.501 | |
| LUSC | RILP | 53/448 | 0.975/0.691 |
| EXOSC3 | 39/462 | 1.081/0.75 | |
| KRR1 | 45/456 | 1.191/0.744 | |
| STAD | ERBB4 | 35/402 | 1.011/0.771 |
| ACTN2 | 48/389 | 1.232/0.822 | |
| KIAA1429 | 28/156 | 1.088/0.752 | |
| SKCM | CR1 | 68/399 | 1.11/0.786 |
| PEG3 | 85/382 | 0.977/0.812 | |
| EPHA3 | 48/413 | 1.134/0.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Yin, C.; Li, Z.; Feng, K.; Cao, Y.; Gu, Y.; Sun, H. Identification of Cancer Driver Genes by Integrating Multiomics Data with Graph Neural Networks. Metabolites 2023, 13, 339. https://doi.org/10.3390/metabo13030339
Song H, Yin C, Li Z, Feng K, Cao Y, Gu Y, Sun H. Identification of Cancer Driver Genes by Integrating Multiomics Data with Graph Neural Networks. Metabolites. 2023; 13(3):339. https://doi.org/10.3390/metabo13030339
Chicago/Turabian StyleSong, Hongzhi, Chaoyi Yin, Zhuopeng Li, Ke Feng, Yangkun Cao, Yujie Gu, and Huiyan Sun. 2023. "Identification of Cancer Driver Genes by Integrating Multiomics Data with Graph Neural Networks" Metabolites 13, no. 3: 339. https://doi.org/10.3390/metabo13030339