
Prediction of a Large-Scale Database of Collision Cross-Section and Retention Time Using Machine Learning to Reduce False Positive Annotations in Untargeted Metabolomics
 , , , and
, , , and
Abstract
: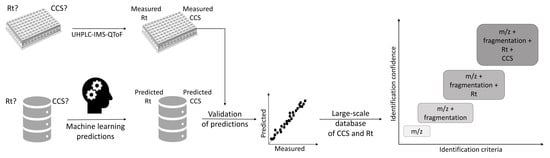
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standards
2.2. LC-MS Conditions
2.3. CCS Prediction
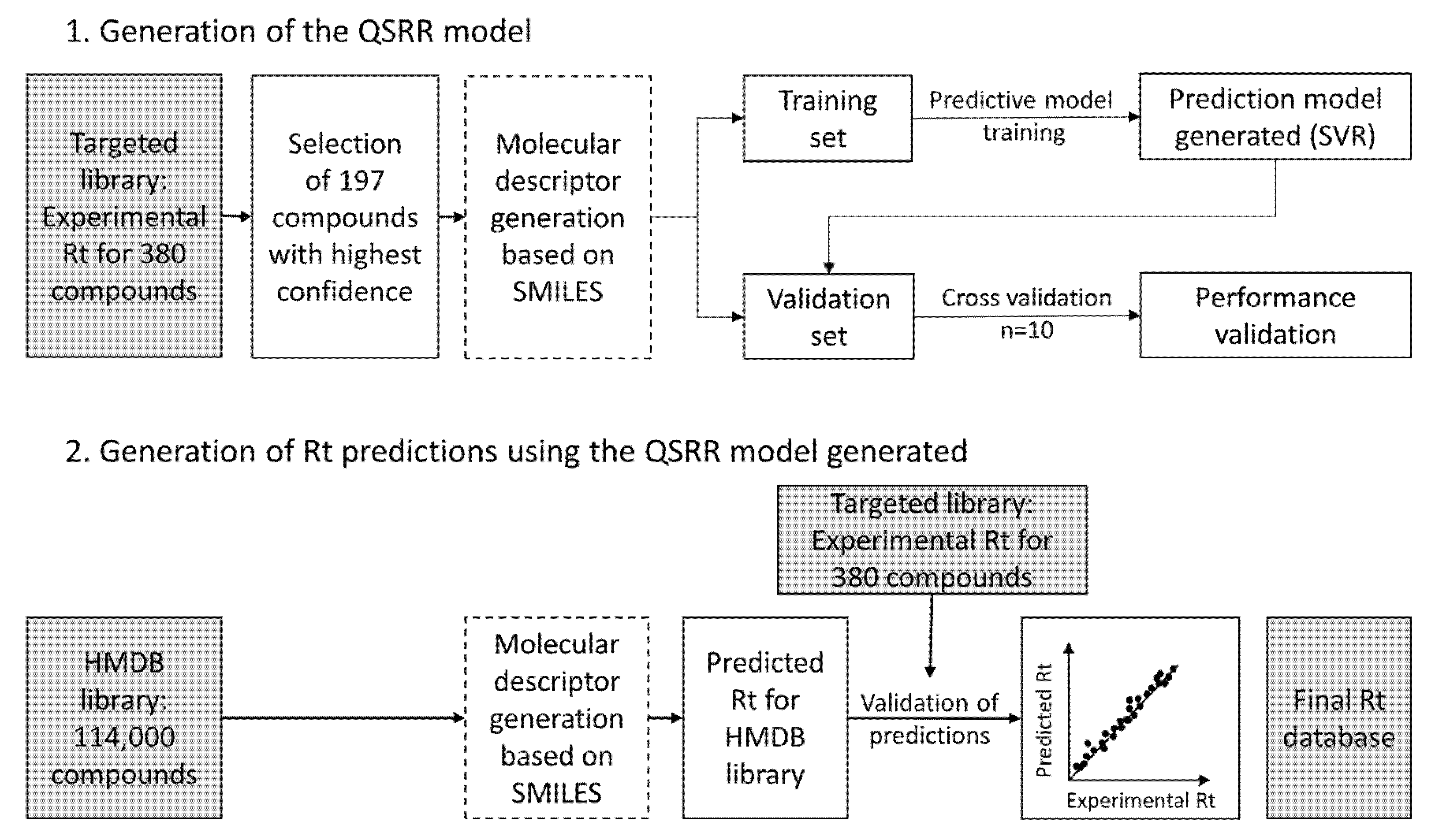
2.4. Rt Prediction of Small Molecules
2.5. Reduction of the Occurrence of False Positive Annotations in Untargeted Metabolomics: Application to Toxicology Data
3. Results
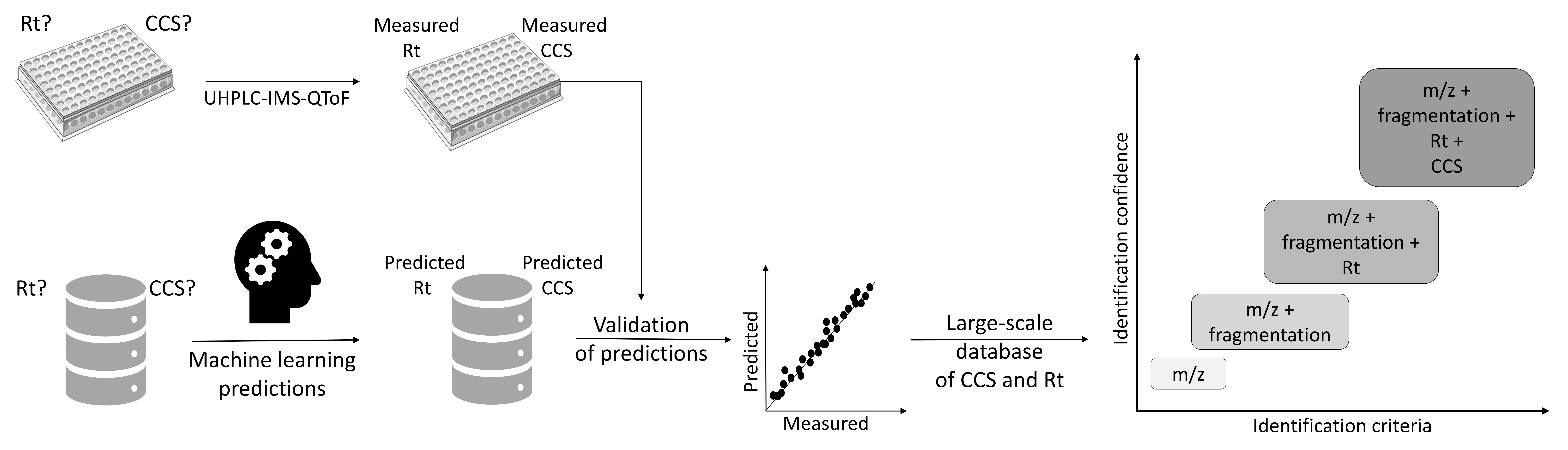
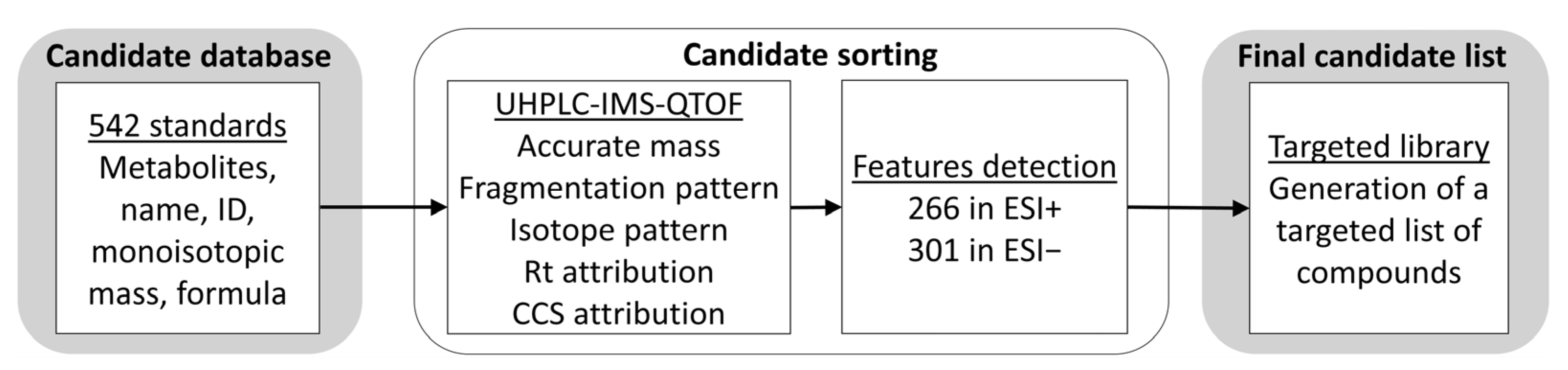
3.1. Analysis of Standard Compounds and Generation of an In-House Database
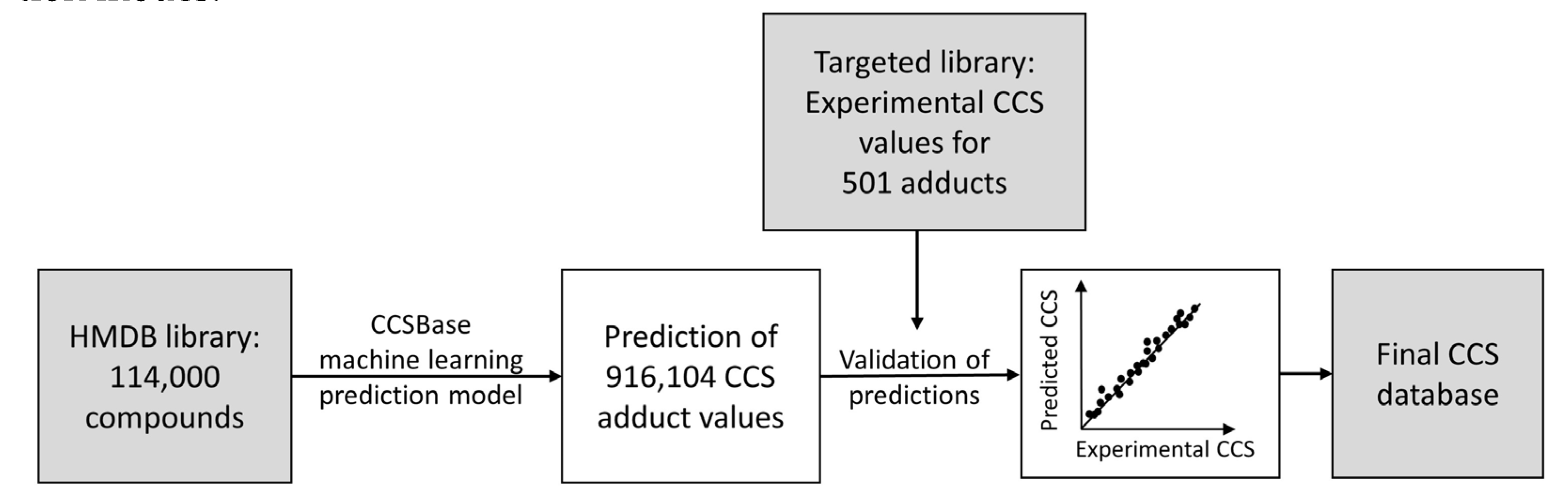
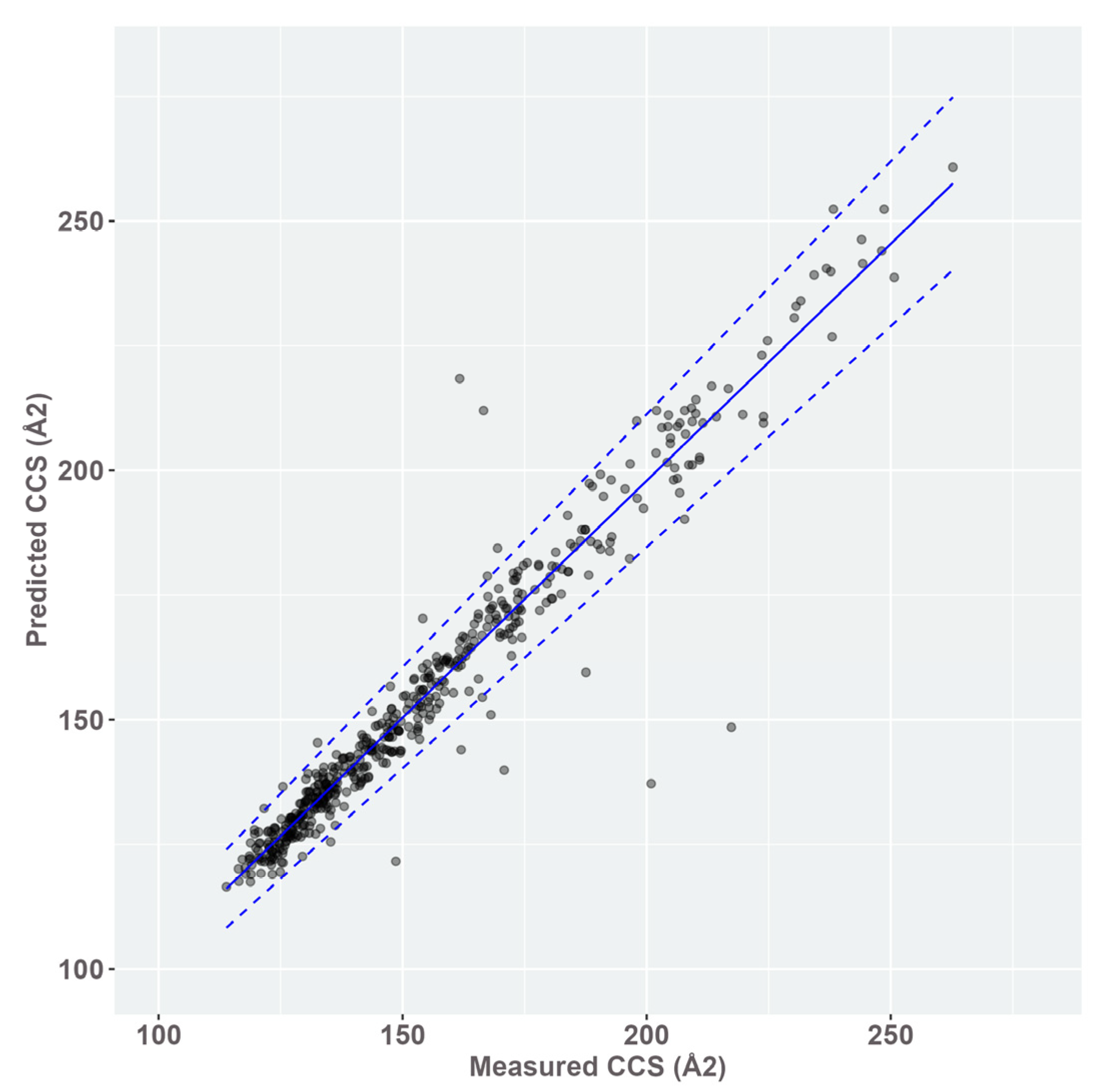
3.2. CCS Prediction and Creation of a CCS Database
3.3. Rt Prediction and Creation of an Rt Database
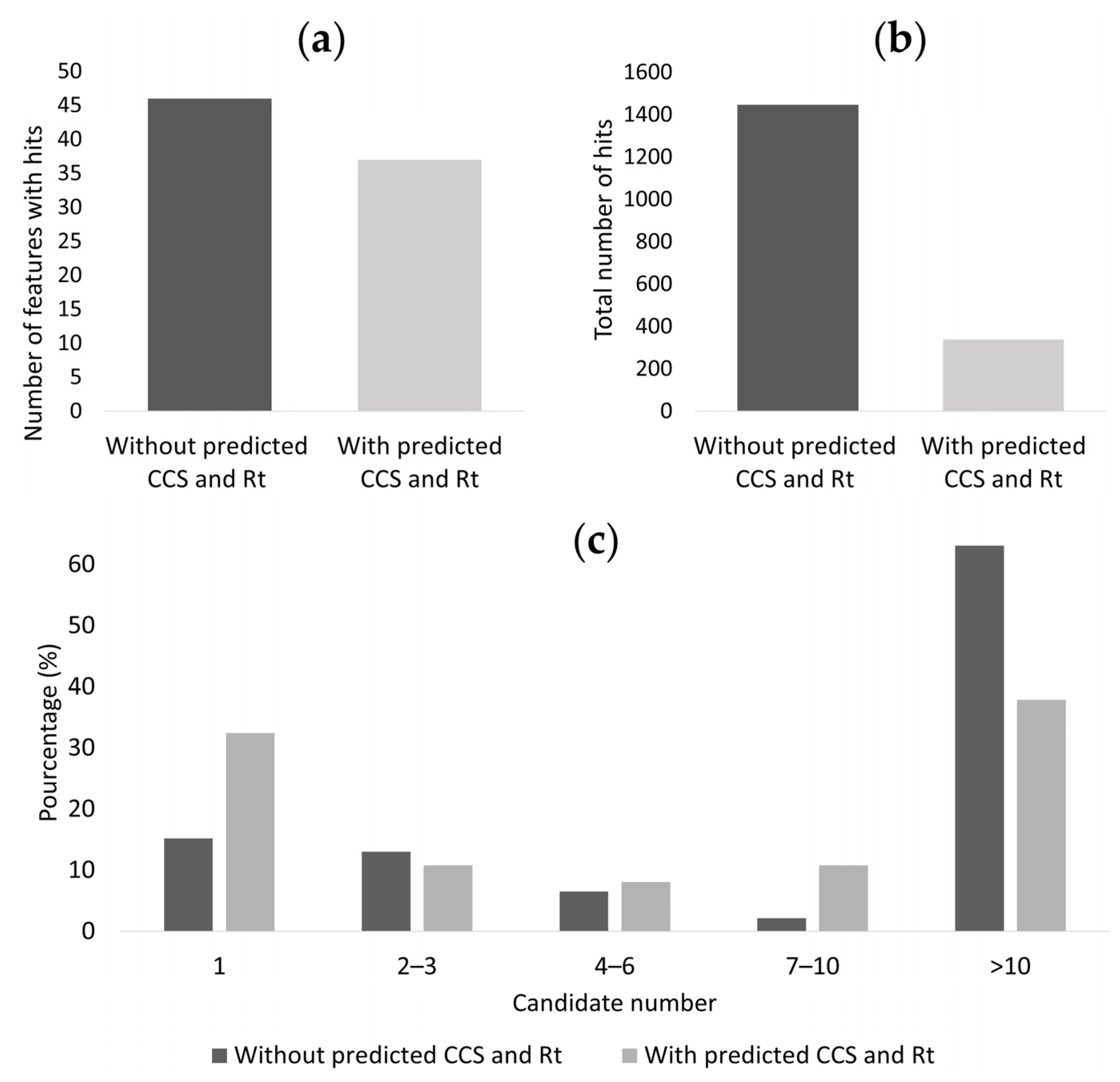
3.4. Reduction of the Occurrence of False Positive Annotations in Untargeted Metabolomics: Application to Toxicology Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCS | collision cross-section |
| ESI | electrospray ionization |
| HMDB | human metabolome database |
| HRMS | high-resolution mass spectrometry |
| IMS | ion mobility spectrometry |
| LC | liquid chromatography |
| m/z | mass to charge ratio |
| MS | mass spectrometry |
| MSI | metabolomics standards initiative |
| MSMLS | mass spectrometry metabolite library of standards (Sigma-Aldrich) |
| QSRR | quantitative structure retention relationships |
| QTOf | quadrupole time-of-flight |
| Rt | retention time |
| SD | standard deviation |
| SMILES | simplified molecular input line entry system |
| SVR | support vector regression |
| UHPLC | ultra high-performance liquid chromatography |
References
- Roessner, U.; Bowne, J. What Is Metabolomics All About? BioTechniques 2009, 46, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics Enables Precision Medicine: “A White Paper, Community Perspective”. Metabolomics Off. J. Metabolomic Soc. 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonova, O.P.; Maslov, D.L.; Balashova, E.E.; Lokhov, P.G. Current State and Future Perspectives on Personalized Metabolomics. Metabolites 2023, 13, 67. [Google Scholar] [CrossRef]
- Ma, X. Recent Advances in Mass Spectrometry-Based Structural Elucidation Techniques. Molecules 2022, 27, 6466. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, E.; Lenski, M.; Bruno, C.; Thibert, V.; Contreras, P.; Privat, K.; Ameline, A.; Fabresse, N. High-Resolution Mass Spectrometry: Theoretical and Technological Aspects. Toxicol. Anal. Clin. 2022, 34, 3–18. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The Apogee of the Omics Trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, S.; Benton, H.P.; Casazza, K.; Cooper, S.J.; Cui, X.; Du, X.; Engler, J.; Kabarowski, J.H.; Li, S.; Pathmasiri, W.; et al. Training in Metabolomics Research. II. Processing and Statistical Analysis of Metabolomics Data, Metabolite Identification, Pathway Analysis, Applications of Metabolomics and Its Future. J. Mass Spectrom. JMS 2016, 51, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Nash, W.J.; Dunn, W.B. From Mass to Metabolite in Human Untargeted Metabolomics: Recent Advances in Annotation of Metabolites Applying Liquid Chromatography-Mass Spectrometry Data. TrAC Trends Anal. Chem. 2019, 120, 115324. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed Minimum Reporting Standards for Chemical Analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics Off. J. Metabolomic Soc. 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Zhu, Z.-J. Overview of Tandem Mass Spectral and Metabolite Databases for Metabolite Identification in Metabolomics. Methods Mol. Biol. Clifton NJ 2020, 2104, 139–148. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A.; Haddad, P.R. Retention Index Prediction Using Quantitative Structure-Retention Relationships for Improving Structure Identification in Nontargeted Metabolomics. Anal. Chem. 2018, 90, 9434–9440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shen, X.; Tu, J.; Zhu, Z.-J. Large-Scale Prediction of Collision Cross-Section Values for Metabolites in Ion Mobility-Mass Spectrometry. Anal. Chem. 2016, 88, 11084–11091. [Google Scholar] [CrossRef]
- Blaženović, I.; Shen, T.; Mehta, S.S.; Kind, T.; Ji, J.; Piparo, M.; Cacciola, F.; Mondello, L.; Fiehn, O. Increasing Compound Identification Rates in Untargeted Lipidomics Research with Liquid Chromatography Drift Time-Ion Mobility Mass Spectrometry. Anal. Chem. 2018, 90, 10758–10764. [Google Scholar] [CrossRef]
- Hinnenkamp, V.; Klein, J.; Meckelmann, S.W.; Balsaa, P.; Schmidt, T.C.; Schmitz, O.J. Comparison of CCS Values Determined by Traveling Wave Ion Mobility Mass Spectrometry and Drift Tube Ion Mobility Mass Spectrometry. Anal. Chem. 2018, 90, 12042–12050. [Google Scholar] [CrossRef]
- Zhang, X.; Kew, K.; Reisdorph, R.; Sartain, M.; Powell, R.; Armstrong, M.; Quinn, K.; Cruickshank-Quinn, C.; Walmsley, S.; Bokatzian, S.; et al. Performance of a High-Pressure Liquid Chromatography-Ion Mobility-Mass Spectrometry System for Metabolic Profiling. Anal. Chem. 2017, 89, 6384–6391. [Google Scholar] [CrossRef]
- Zheng, X.; Aly, N.A.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A Structural Examination and Collision Cross Section Database for over 500 Metabolites and Xenobiotics Using Drift Tube Ion Mobility Spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef] [Green Version]
- Righetti, L.; Bergmann, A.; Galaverna, G.; Rolfsson, O.; Paglia, G.; Dall’Asta, C. Ion Mobility-Derived Collision Cross Section Database: Application to Mycotoxin Analysis. Anal. Chim. Acta 2018, 1014, 50–57. [Google Scholar] [CrossRef]
- Picache, J.A.; Rose, B.S.; Balinski, A.; Leaptrot, K.L.; Sherrod, S.D.; May, J.C.; McLean, J.A. Collision Cross Section Compendium to Annotate and Predict Multi-Omic Compound Identities. Chem. Sci. 2019, 10, 983–993. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; Le Bizec, B.; Monteau, F.; García-Campaña, A.M.; Dervilly-Pinel, G. Collision Cross Section (CCS) Database: An Additional Measure to Characterize Steroids. Anal. Chem. 2018, 90, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Luo, M.; Chen, X.; Yin, Y.; Xiong, X.; Wang, R.; Zhu, Z.-J. Ion Mobility Collision Cross-Section Atlas for Known and Unknown Metabolite Annotation in Untargeted Metabolomics. Nat. Commun. 2020, 11, 4334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tu, J.; Zhu, Z.-J. Advancing the Large-Scale CCS Database for Metabolomics and Lipidomics at the Machine-Learning Era. Curr. Opin. Chem. Biol. 2018, 42, 34–41. [Google Scholar] [CrossRef]
- Rainey, M.A.; Watson, C.A.; Asef, C.K.; Foster, M.R.; Baker, E.S.; Fernández, F.M. CCS Predictor 2.0: An Open-Source Jupyter Notebook Tool for Filtering Out False Positives in Metabolomics. Anal. Chem. 2022, 94, 17456–17466. [Google Scholar] [CrossRef] [PubMed]
- Plante, P.-L.; Francovic-Fontaine, É.; May, J.C.; McLean, J.A.; Baker, E.S.; Laviolette, F.; Marchand, M.; Corbeil, J. Predicting Ion Mobility Collision Cross-Sections Using a Deep Neural Network: DeepCCS. Anal. Chem. 2019, 91, 5191–5199. [Google Scholar] [CrossRef]
- Zhou, Z.; Xiong, X.; Zhu, Z.-J. MetCCS Predictor: A Web Server for Predicting Collision Cross-Section Values of Metabolites in Ion Mobility-Mass Spectrometry Based Metabolomics. Bioinforma. Oxf. Engl. 2017, 33, 2235–2237. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Tu, J.; Xiong, X.; Shen, X.; Zhu, Z.-J. LipidCCS: Prediction of Collision Cross-Section Values for Lipids with High Precision To Support Ion Mobility-Mass Spectrometry-Based Lipidomics. Anal. Chem. 2017, 89, 9559–9566. [Google Scholar] [CrossRef]
- Grisoni, F.; Ballabio, D.; Todeschini, R.; Consonni, V. Molecular Descriptors for Structure-Activity Applications: A Hands-On Approach. Methods Mol. Biol. Clifton NJ 2018, 1800, 3–53. [Google Scholar] [CrossRef]
- Ross, D.H.; Cho, J.H.; Xu, L. Breaking Down Structural Diversity for Comprehensive Prediction of Ion-Neutral Collision Cross Sections. Anal. Chem. 2020, 92, 4548–4557. [Google Scholar] [CrossRef]
- Rainville, P.D.; Wilson, I.D.; Nicholson, J.K.; Isaac, G.; Mullin, L.; Langridge, J.I.; Plumb, R.S. Ion Mobility Spectrometry Combined with Ultra Performance Liquid Chromatography/Mass Spectrometry for Metabolic Phenotyping of Urine: Effects of Column Length, Gradient Duration and Ion Mobility Spectrometry on Metabolite Detection. Anal. Chim. Acta 2017, 982, 1–8. [Google Scholar] [CrossRef]
- Stanstrup, J.; Neumann, S.; Vrhovšek, U. PredRet: Prediction of Retention Time by Direct Mapping between Multiple Chromatographic Systems. Anal. Chem. 2015, 87, 9421–9428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchi, F.; Bertozzi, S.M.; Ottonello, G.; Ruda, G.F.; Colombano, G.; Fiorelli, C.; Martucci, C.; Bertorelli, R.; Scarpelli, R.; Cavalli, A.; et al. Kernel-Based, Partial Least Squares Quantitative Structure-Retention Relationship Model for UPLC Retention Time Prediction: A Useful Tool for Metabolite Identification. Anal. Chem. 2016, 88, 9510–9517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creek, D.J.; Jankevics, A.; Breitling, R.; Watson, D.G.; Barrett, M.P.; Burgess, K.E.V. Toward Global Metabolomics Analysis with Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry: Improved Metabolite Identification by Retention Time Prediction. Anal. Chem. 2011, 83, 8703–8710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonini, P.; Kind, T.; Tsugawa, H.; Barupal, D.K.; Fiehn, O. Retip: Retention Time Prediction for Compound Annotation in Untargeted Metabolomics. Anal. Chem. 2020, 92, 7515–7522. [Google Scholar] [CrossRef]
- Liapikos, T.; Zisi, C.; Kodra, D.; Kademoglou, K.; Diamantidou, D.; Begou, O.; Pappa-Louisi, A.; Theodoridis, G. Quantitative Structure Retention Relationship (QSRR) Modelling for Analytes’ Retention Prediction in LC-HRMS by Applying Different Machine Learning Algorithms and Evaluating Their Performance. J. Chromatogr. B 2022, 1191, 123132. [Google Scholar] [CrossRef]
- Park, S.H.; De Pra, M.; Haddad, P.R.; Grosse, S.; Pohl, C.A.; Steiner, F. Localised Quantitative Structure-Retention Relationship Modelling for Rapid Method Development in Reversed-Phase High Performance Liquid Chromatography. J. Chromatogr. A 2020, 1609, 460508. [Google Scholar] [CrossRef]
- Goryński, K.; Bojko, B.; Nowaczyk, A.; Buciński, A.; Pawliszyn, J.; Kaliszan, R. Quantitative Structure-Retention Relationships Models for Prediction of High Performance Liquid Chromatography Retention Time of Small Molecules: Endogenous Metabolites and Banned Compounds. Anal. Chim. Acta 2013, 797, 13–19. [Google Scholar] [CrossRef]
- Naylor, B.C.; Catrow, J.L.; Maschek, J.A.; Cox, J.E. QSRR Automator: A Tool for Automating Retention Time Prediction in Lipidomics and Metabolomics. Metabolites 2020, 10, 237. [Google Scholar] [CrossRef]
- Gritti, F. Perspective on the Future Approaches to Predict Retention in Liquid Chromatography. Anal. Chem. 2021, 93, 5653–5664. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Dusautoir, R.; Zarcone, G.; Verriele, M.; Garçon, G.; Fronval, I.; Beauval, N.; Allorge, D.; Riffault, V.; Locoge, N.; Lo-Guidice, J.-M.; et al. Comparison of the Chemical Composition of Aerosols from Heated Tobacco Products, Electronic Cigarettes and Tobacco Cigarettes and Their Toxic Impacts on the Human Bronchial Epithelial BEAS-2B Cells. J. Hazard. Mater. 2021, 401, 123417. [Google Scholar] [CrossRef]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass Appeal: Metabolite Identification in Mass Spectrometry-Focused Untargeted Metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- Bittremieux, W.; Wang, M.; Dorrestein, P.C. The Critical Role That Spectral Libraries Play in Capturing the Metabolomics Community Knowledge. Metabolomics Off. J. Metabolomic Soc. 2022, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J.; González-Ruiz, V.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Guillarme, D.; Schappler, J.; Picard, D.; Boccard, J.; Rudaz, S. A Scoring Approach for Multi-Platform Acquisition in Metabolomics. J. Chromatogr. A 2019, 1592, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Guijas, C.; Billings, E.; Montenegro-Burke, J.R.; Uritboonthai, W.; Aisporna, A.E.; Chen, E.; Benton, H.P.; Siuzdak, G. The METLIN Small Molecule Dataset for Machine Learning-Based Retention Time Prediction. Nat. Commun. 2019, 10, 5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A Lipidome Atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Mollerup, C.B.; Mardal, M.; Dalsgaard, P.W.; Linnet, K.; Barron, L.P. Prediction of Collision Cross Section and Retention Time for Broad Scope Screening in Gradient Reversed-Phase Liquid Chromatography-Ion Mobility-High Resolution Accurate Mass Spectrometry. J. Chromatogr. A 2018, 1542, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Celma, A.; Bade, R.; Sancho, J.V.; Hernandez, F.; Humphries, M.; Bijlsma, L. Prediction of Retention Time and Collision Cross Section (CCSH+, CCSH–, and CCSNa+) of Emerging Contaminants Using Multiple Adaptive Regression Splines. J. Chem. Inf. Model. 2022, 62, 5425–5434. [Google Scholar] [CrossRef]
- Ross, D.H.; Cho, J.H.; Zhang, R.; Hines, K.M.; Xu, L. LiPydomics: A Python Package for Comprehensive Prediction of Lipid Collision Cross Sections and Retention Times and Analysis of Ion Mobility-Mass Spectrometry-Based Lipidomics Data. Anal. Chem. 2020, 92, 14967–14975. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenski, M.; Maallem, S.; Zarcone, G.; Garçon, G.; Lo-Guidice, J.-M.; Anthérieu, S.; Allorge, D. Prediction of a Large-Scale Database of Collision Cross-Section and Retention Time Using Machine Learning to Reduce False Positive Annotations in Untargeted Metabolomics. Metabolites 2023, 13, 282. https://doi.org/10.3390/metabo13020282
Lenski M, Maallem S, Zarcone G, Garçon G, Lo-Guidice J-M, Anthérieu S, Allorge D. Prediction of a Large-Scale Database of Collision Cross-Section and Retention Time Using Machine Learning to Reduce False Positive Annotations in Untargeted Metabolomics. Metabolites. 2023; 13(2):282. https://doi.org/10.3390/metabo13020282
Chicago/Turabian StyleLenski, Marie, Saïd Maallem, Gianni Zarcone, Guillaume Garçon, Jean-Marc Lo-Guidice, Sébastien Anthérieu, and Delphine Allorge. 2023. "Prediction of a Large-Scale Database of Collision Cross-Section and Retention Time Using Machine Learning to Reduce False Positive Annotations in Untargeted Metabolomics" Metabolites 13, no. 2: 282. https://doi.org/10.3390/metabo13020282