
Identification of the Transcription Factor ATF3 as a Direct and Indirect Regulator of the LDLR
 , , , and
, , , and 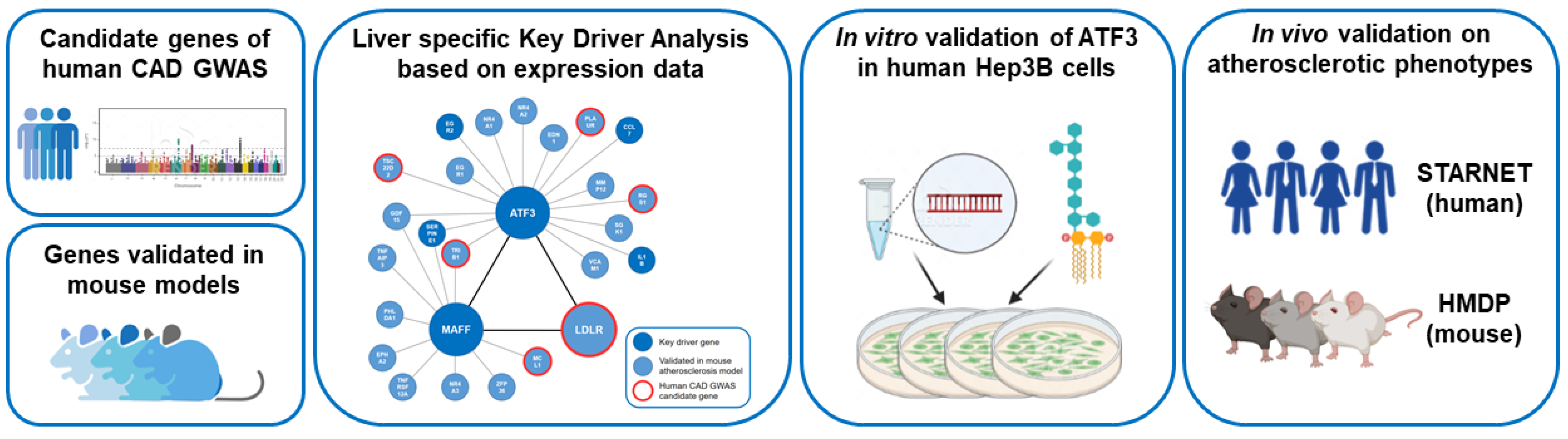{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
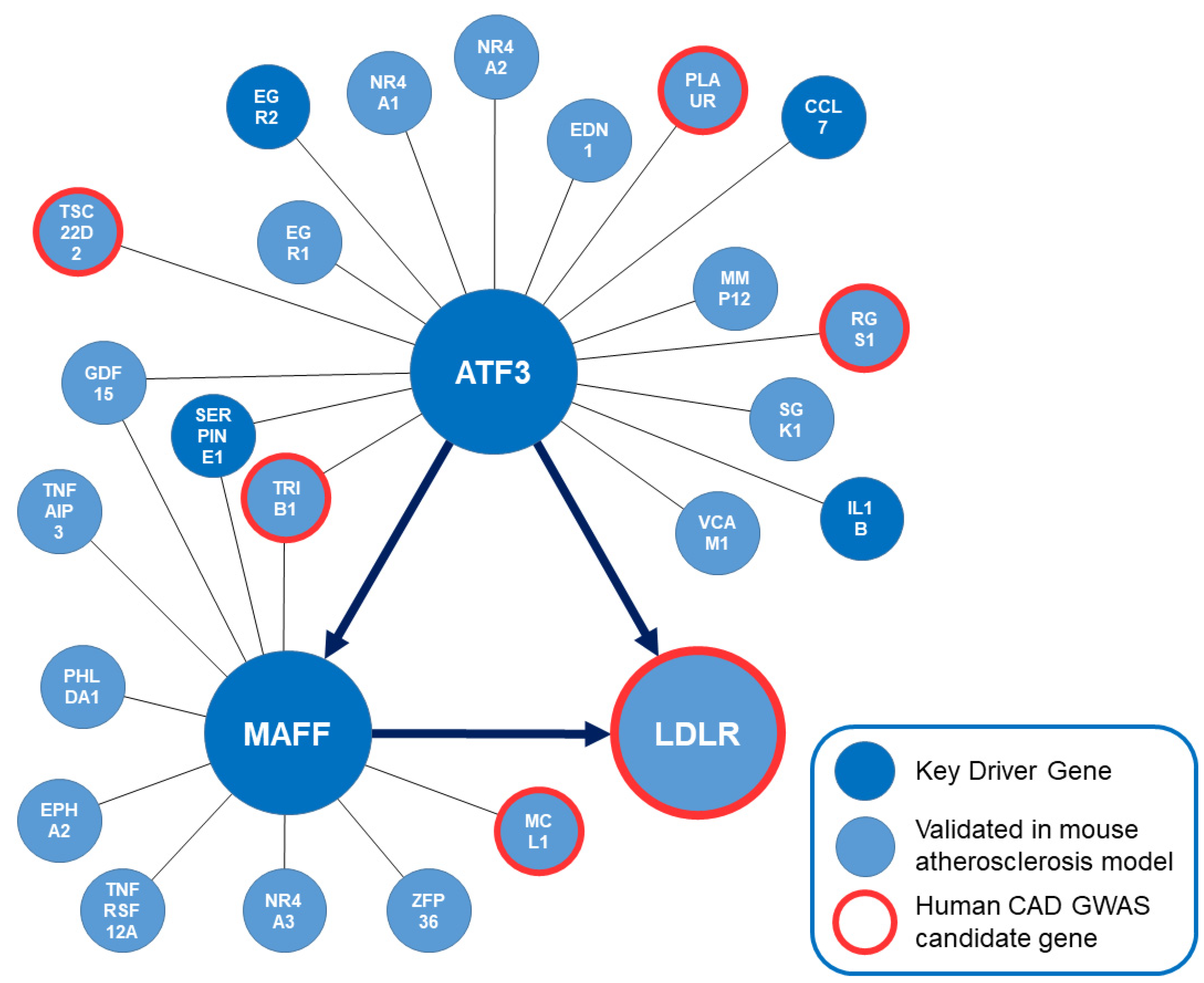
2.1. Identification of ATF3 As a Central Key Driver Gene of a Liver-Specific Regulatory Network
2.2. Identification of ATF3 As Direct and Indirect Regulator of LDLR
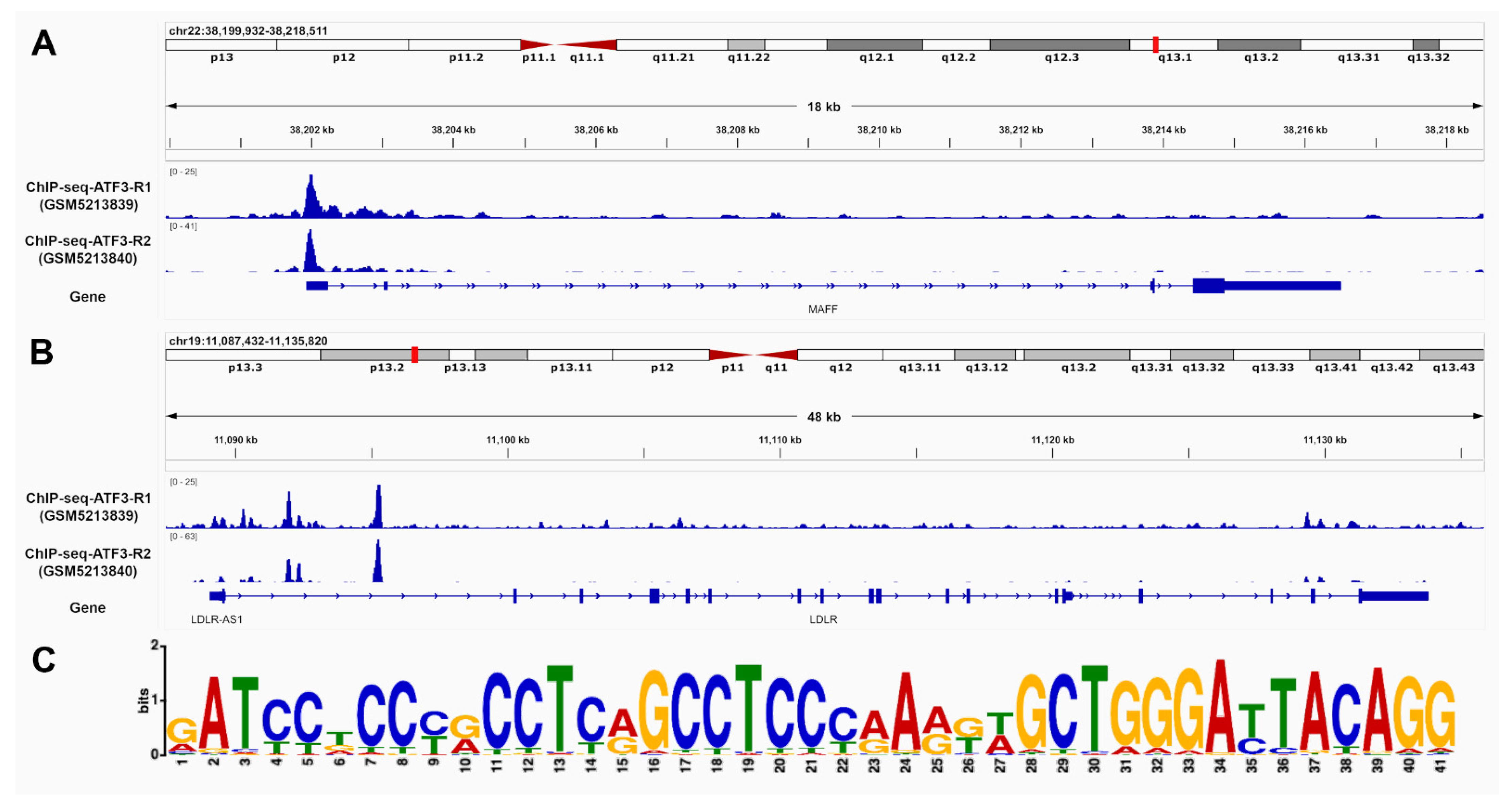
2.3. ATF3 Has the Potential to Bind to MAFF and LDLR Promoter and Intronic Regions
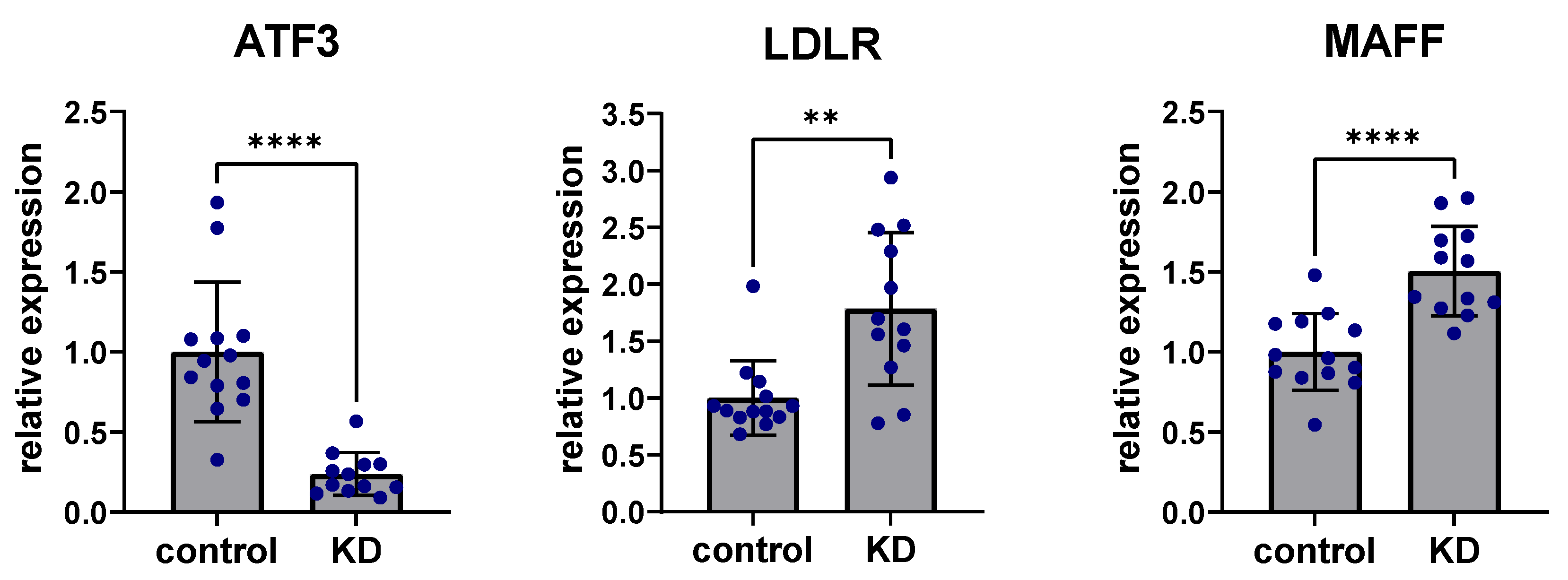
2.4. In Vitro Experiments Clarify the Regulatory Role of ATF3 on MAFF and LDLR Expression
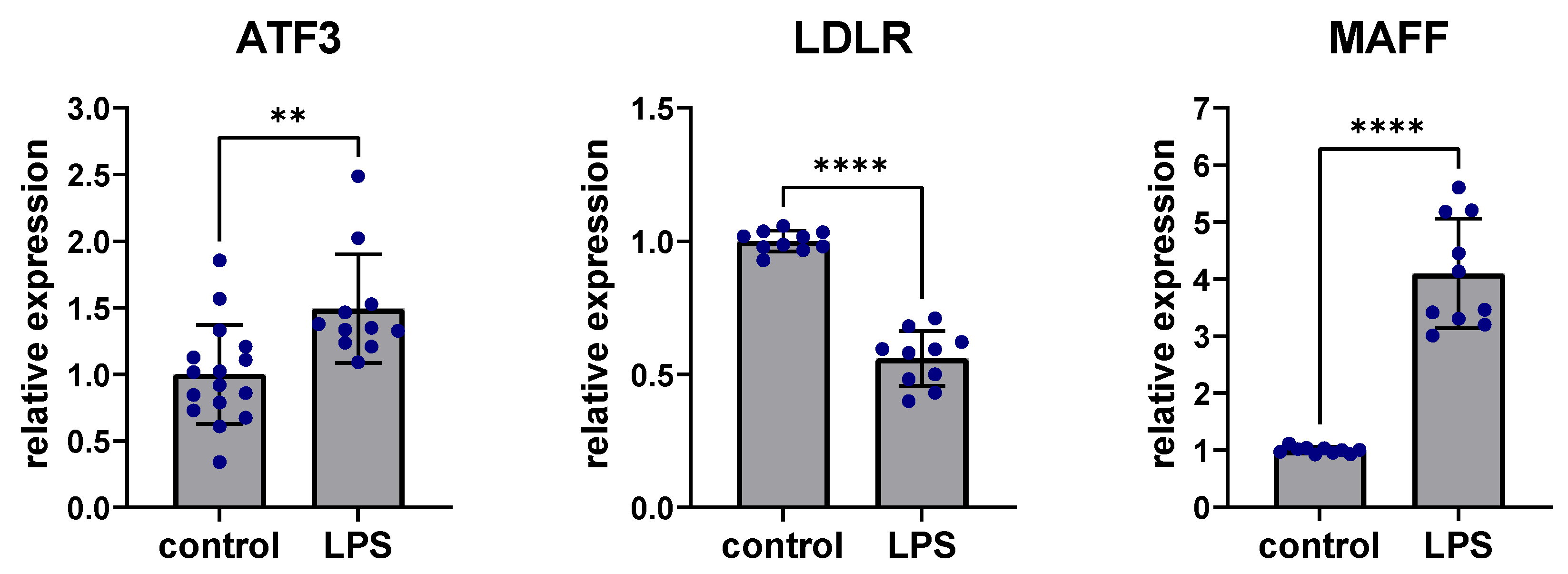
2.5. Inflammatory Effects on ATF3, LDLR and MAFF Expression in Liver Tissue
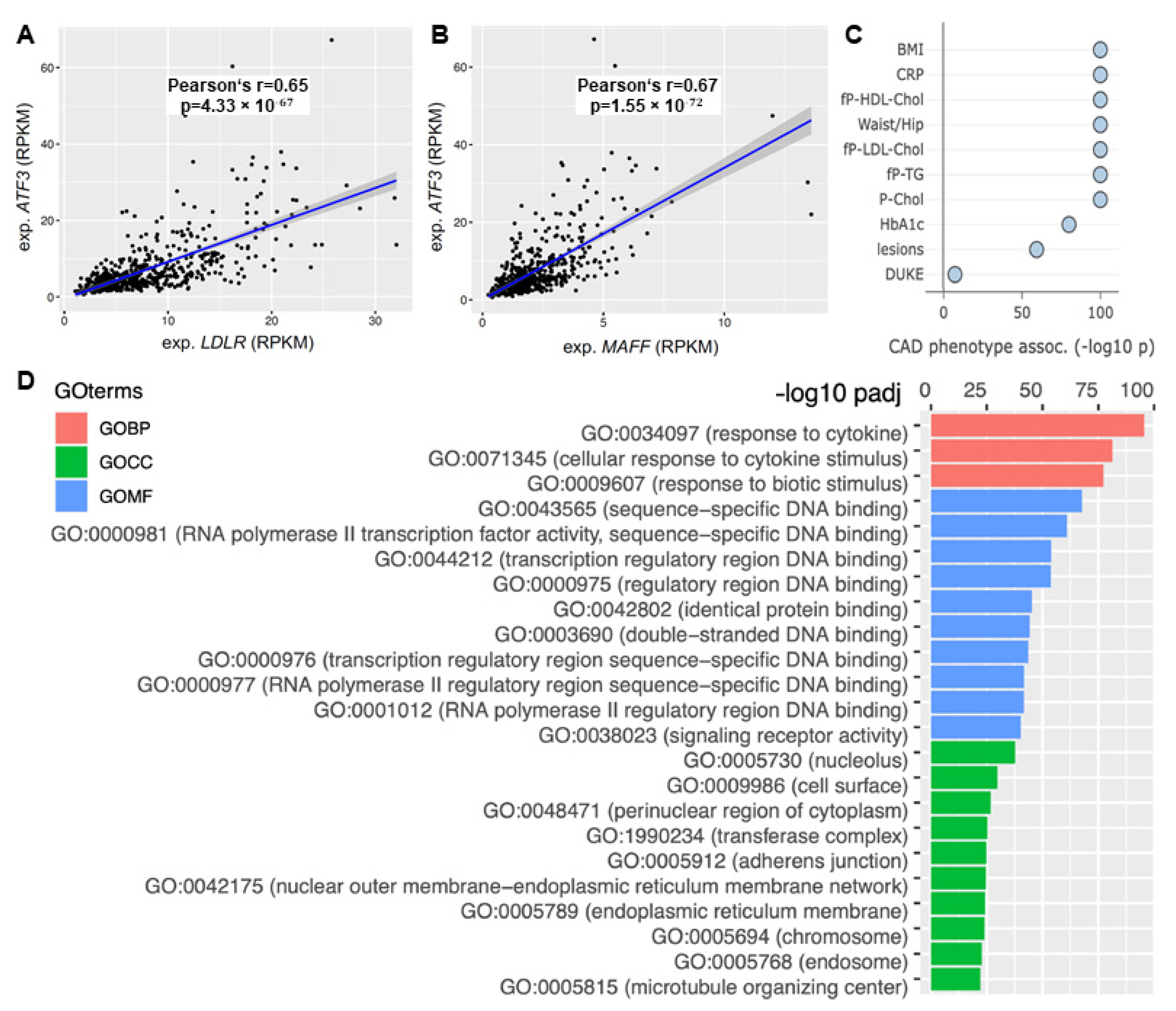
2.6. In Vivo Data of Human and Mouse Confirm the Importance of ATF3 for Cholesterol Metabolism
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [PubMed]
- Townsend, N.; Wilson, L.; Bhatnagar, P.; Wickramasinghe, K.; Rayner, M.; Nichols, M. Cardiovascular disease in Europe: Epidemiological update 2016. Eur. Heart J. 2016, 37, 3232–3245. [Google Scholar] [CrossRef] [PubMed]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Otsuka, F.; Finn, A.V.; Davis, H.R.; Joner, M.; Virmani, R. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat. Rev. Cardiol. 2016, 13, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Kokkinos, P.; Nyelin, E. Physical activity, cardiorespiratory fitness, and the metabolic syndrome. Nutrients 2019, 11, 1652. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Novel invasive and noninvasive cardiac-specific biomarkers in obesity and cardiovascular diseases. Metab. Syndr. Relat. Disord. 2020, 18, 10–30. [Google Scholar] [CrossRef] [PubMed]
- Padro, T.; Muñoz-Garcia, N.; Badimon, L. The role of triglycerides in the origin and progression of atherosclerosis. Clín. Investig. Arterioscler. 2021, 33, 20–28. [Google Scholar] [CrossRef]
- Silveira Rossi, J.L.; Barbalho, S.M.; Reverete de Araujo, R.; Bechara, M.D.; Sloan, K.P.; Sloan, L.A. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. Diabetes/Metab. Res. Rev. 2022, 38, e3502. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Vilne, B.; Schunkert, H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol. Med. 2016, 8, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Chan, K.H.K.; Zhang, G.; Huan, T.; Kurt, Z.; Zhao, Y.; Codoni, V.; Trégouët, D.-A.; Consortium, C.; Yang, J. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017, 13, e1007040. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Vivar, J.; Nelson, C.P.; Willenborg, C.; Segrè, A.V.; Mäkinen, V.-P.; Nikpay, M.; Erdmann, J.; Blankenberg, S.; O’Donnell, C. Systems genetics analysis of genome-wide association study reveals novel associations between key biological processes and coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Ghosh, S.; Vivar, J.; Nikpay, M.; Stewart, A. Integrative Genomics Reveals Novel Molecular Pathways and Gene Networks for Coronary Artery Disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar]
- von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab. 2017, 25, 248–261. [Google Scholar] [CrossRef]
- Vos, D.; Kuivenhoven, J.A.; van de Sluis, B. Recycling the LDL receptor to combat atherosclerosis. Aging 2018, 10, 3638–3640. [Google Scholar] [CrossRef]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef]
- Attie, A.D.; Seidah, N.G. Dual regulation of the LDL receptor—Some clarity and new questions. Cell Metab. 2005, 1, 290–292. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Soutar, A.K.; Naoumova, R.P. Mechanisms of Disease: Genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 214–225. [Google Scholar] [CrossRef]
- Von Scheidt, M.; Zhao, Y.; de Aguiar Vallim, T.Q.; Che, N.; Wierer, M.; Seldin, M.M.; Franzén, O.; Kurt, Z.; Pang, S.; Bongiovanni, D. Transcription factor MAFF (MAF basic leucine zipper transcription factor F) regulates an atherosclerosis relevant network connecting inflammation and cholesterol metabolism. Circulation 2021, 143, 1809–1823. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, M.; Thorsson, V.; Li, B.; Rust, A.G.; Korb, M.; Kennedy, K.; Hai, T.; Bolouri, H.; Aderem, A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 2006, 441, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, E.J.; Qi, L.; Zhu, M.X.; Mirmira, R.G.; Montminy, M.R.; Hai, T. The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic β-cell dysfunction. Mol. Endocrinol. 2010, 24, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Jadhav, K.; Pan, X.; Zhu, Y.; Hu, S.; Chen, S.; Chen, L.; Tang, Y.; Wang, H.H. Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism. Nat. Metab. 2021, 3, 59–74. [Google Scholar] [CrossRef]
- Ku, H.-C.; Cheng, C.-F. Master regulator activating transcription factor 3 (ATF3) in metabolic homeostasis and cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Jadhav, K.; Zhang, Y. Activating transcription factor 3 in immune response and metabolic regulation. Liver Res. 2017, 1, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, N.; Yuan, Y.; Jin, Y.-G.; Guo, H.; Deng, W.; Tang, Q.-Z. Activating transcription factor 3 in cardiovascular diseases: A potential therapeutic target. Basic Res. Cardiol. 2018, 113, 37. [Google Scholar] [CrossRef]
- Thompson, M.R.; Xu, D.; Williams, B.R. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 2009, 87, 1053–1060. [Google Scholar] [CrossRef]
- Hai, T.; Wolford, C.C.; Chang, Y.-S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. J. Liver Res. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. J. Liver Res. 1999, 7, 321–335. [Google Scholar]
- Nawa, T.; Nawa, M.T.; Adachi, M.T.; Uchimura, I.; Shimokawa, R.; Fujisawa, K.; Tanaka, A.; Numano, F.; Kitajima, S. Expression of transcriptional repressor ATF3/LRF1 in human atherosclerosis: Colocalization and possible involvement in cell death of vascular endothelial cells. Atherosclerosis 2002, 161, 281–291. [Google Scholar] [CrossRef]
- Gold, E.S.; Ramsey, S.A.; Sartain, M.J.; Selinummi, J.; Podolsky, I.; Rodriguez, D.J.; Moritz, R.L.; Aderem, A. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol–induced lipid body formation. J. Exp. Med. 2012, 209, 807–817. [Google Scholar] [CrossRef]
- Schunkert, H.; König, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.; Barbalic, M.; Gieger, C. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat. Genet. 2011, 43, 339–344. [Google Scholar] [CrossRef]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.-E. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nikpay, M.; Goel, A.; Won, H.-H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of coronary artery disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef]
- Howson, J.M.; Zhao, W.; Barnes, D.R.; Ho, W.-K.; Young, R.; Paul, D.S.; Waite, L.L.; Freitag, D.F.; Fauman, E.B.; Salfati, E.L. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat. Genet. 2017, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Brænne, I.; Civelek, M.; Vilne, B.; Di Narzo, A.; Johnson, A.D.; Zhao, Y.; Reiz, B.; Codoni, V.; Webb, T.R.; Foroughi Asl, H. Prediction of causal candidate genes in coronary artery disease loci. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Schunkert, H.; Götz, A.; Braund, P.; McGinnis, R.; Tregouet, D.-A.; Mangino, M.; Linsel-Nitschke, P.; Cambien, F.; Hengstenberg, C.; Stark, K. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21. 3 and coronary artery disease. Circulation 2008, 117, 1675–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaterra, G.A.; Zügel, S.; Thogersen, J.; Walter, S.A.; Haberkorn, U.; Strelau, J.; Kinscherf, R. Growth differentiation factor-15 deficiency inhibits atherosclerosis progression by regulating interleukin-6–dependent inflammatory response to vascular injury. J. Am. Heart Assoc. 2012, 1, e002550. [Google Scholar] [CrossRef]
- Qing, H.; Liu, Y.; Zhao, Y.; Aono, J.; Jones, K.L.; Heywood, E.B.; Howatt, D.; Binkley, C.M.; Daugherty, A.; Liang, Y. Deficiency of the NR4A orphan nuclear receptor NOR1 in hematopoietic stem cells accelerates atherosclerosis. Stem Cells 2014, 32, 2419–2429. [Google Scholar] [CrossRef]
- Arndt, L.; Dokas, J.; Jeromin, F.; Thiery, J.; Burkhardt, R. Heterozygous deficiency of Tribbles homolog-1 gene (Trib1) increases atherosclerotic lesions in ApoE-knockout mice. Clin. Chem. Lab. Med. 2015, 53, P068. [Google Scholar]
- Kubota, M.; Yoshida, Y.; Kobayashi, E.; Matsutani, T.; Li, S.-Y.; Zhang, B.-S.; Mine, S.; Machida, T.; Takizawa, H.; Hiwasa, T.; et al. Serum anti-SERPINE1 antibody as a potential biomarker of acute cerebral infarction. Sci. Rep. 2021, 11, 21772. [Google Scholar] [CrossRef]
- Varbo, A.; Benn, M.; Tybjærg-Hansen, A.; Grande, P.; Nordestgaard, B.G. TRIB1 and GCKR polymorphisms, lipid levels, and risk of ischemic heart disease in the general population. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 451–457. [Google Scholar] [CrossRef]
- Johnston, J.M.; Angyal, A.; Bauer, R.C.; Hamby, S.; Suvarna, S.K.; Baidžajevas, K.; Hegedus, Z.; Dear, T.N.; Turner, M.; Consortium, C. Myeloid Tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci. Adv. 2019, 5, eaax9183. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Zhang, N.; Yang, D.; Jiao, Q.; Qu, P.; Zhang, Y. High expression of SGK1 in thrombosis of acute ST-segment elevation myocardial infarction: Based on proteomics analysis of intracoronary thrombosis. Rev. Port. Cardiol. 2022, 41, 271–279. [Google Scholar] [CrossRef]
- Yasuda, H.; Kamide, K.; Takiuchi, S.; Matayoshi, T.; Hanada, H.; Kada, A.; Yang, J.; Miwa, Y.; Yoshii, M.; Horio, T. Association of single nucleotide polymorphisms in endothelin family genes with the progression of atherosclerosis in patients with essential hypertension. J. Hum. Hypertens. 2007, 21, 883–892. [Google Scholar] [CrossRef]
- Marcos-Jubilar, M.; Orbe, J.; Roncal, C.; Machado, F.J.; Rodriguez, J.A.; Fernández-Montero, A.; Colina, I.; Rodil, R.; Pastrana, J.C.; Páramo, J.A. Association of SDF1 and MMP12 with Atherosclerosis and Inflammation: Clinical and Experimental Study. Life 2021, 11, 414. [Google Scholar] [CrossRef]
- Blaschke, F.; Bruemmer, D.; Law, R.E. Egr-1 is a major vascular pathogenic transcription factor in atherosclerosis and restenosis. Rev. Endocr. Metab. Disord. 2004, 5, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Huo, Y. VCAM-1 is critical in atherosclerosis. J. Clin. Investig. 2001, 107, 1209–1210. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.-D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid–induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Hettwer, J.; Hinterdobler, J.; Miritsch, B.; Deutsch, M.-A.; Li, X.; Mauersberger, C.; Moggio, A.; Braster, Q.; Gram, H.; Robertson, A.A. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc. Res. 2021; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Koplev, S.; Seldin, M.; Sukhavasi, K.; Ermel, R.; Pang, S.; Zeng, L.; Bankier, S.; Di Narzo, A.; Cheng, H.; Meda, V. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 2022, 1, 85–100. [Google Scholar] [CrossRef]
- Lusis, A.J.; Seldin, M.M.; Allayee, H.; Bennett, B.J.; Civelek, M.; Davis, R.C.; Eskin, E.; Farber, C.R.; Hui, S.; Mehrabian, M.; et al. The Hybrid Mouse Diversity Panel: A resource for systems genetics analyses of metabolic and cardiovascular traits. J. Lipid Res. 2016, 57, 925–942. [Google Scholar] [CrossRef] [PubMed]
- Ghazalpour, A.; Rau, C.D.; Farber, C.R.; Bennett, B.J.; Orozco, L.D.; van Nas, A.; Pan, C.; Allayee, H.; Beaven, S.W.; Civelek, M. Hybrid mouse diversity panel: A panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm. Genome 2012, 23, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Davis, R.C.; Civelek, M.; Orozco, L.; Wu, J.; Qi, H.; Pan, C.; Packard, R.R.S.; Eskin, E.; Yan, M. Genetic architecture of atherosclerosis in mice: A systems genetics analysis of common inbred strains. PLoS Genet. 2015, 11, e1005711. [Google Scholar] [CrossRef]
- Willemsen, N.; Arigoni, I.; Studencka-Turski, M.; Krüger, E.; Bartelt, A. Proteasome dysfunction disrupts adipogenesis and induces inflammation via ATF3. Mol. Metab. 2022, 62, 101518. [Google Scholar] [CrossRef]
- Peng, J.; Le, C.; Xia, B.; Wang, J.; Liu, J.; Li, Z.; Zhang, Q.; Zhang, Q.; Wang, J.; Wan, C. Research on the correlation between activating transcription factor 3 expression in the human coronary artery and atherosclerotic plaque stability. BMC Cardiovasc. Disord. 2021, 21, 356. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.-F.; Cheng, C.-F.; Lin, H.; Tseng, T.-L.; Chen, H.-H.; Chen, S.-H. ATF3 protects against LPS-induced inflammation in mice via inhibiting HMGB1 expression. Evid.-Based Complementary Altern. Med. 2013, 2013, 716481. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.-W.; Kwon, H.-K.; Shin, H.-J.; Choi, Y.-M.; Anwar, M.A.; Choi, S. Activating transcription factor 3 represses inflammatory responses by binding to the p65 subunit of NF-κB. Sci. Rep. 2015, 5, 14470. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Zou, J.; Zhang, Z.; Pan, R.; Zhang, Z.Y.; Han, S.; Xu, Y.; Gao, Y.; Meng, Z.-X. BAF60a Deficiency in Macrophage Promotes Diet-Induced Obesity and Metabolic Inflammation. Diabetes 2022, db220114. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, K.J.; Hwang, J.-Y.; Kim, G.H.; Lee, D.; Lee, Y.J.; Song, E.H.; Yoo, M.-G.; Kim, B.-J.; Suh, Y.H. Activating transcription factor 3 is a target molecule linking hepatic steatosis to impaired glucose homeostasis. J. Hepatol. 2017, 67, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Belalcazar, M.; Brasier, A.R. Liver gene expression associated with diet and lesion. Physiol Genom. 2004, 19, 131–142. [Google Scholar]
- Zhu, J.; Zhang, B.; Smith, E.N.; Drees, B.; Brem, R.B.; Kruglyak, L.; Bumgarner, R.E.; Schadt, E.E. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008, 40, 854–861. [Google Scholar] [CrossRef]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef]
- Webb, T.; Erdmann, J.; Stirrups, K.; Stitziel, N.; Masca, N. Wellcome Trust Case Control Consortium; MORGAM Investigators; Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Systematic Evaluation of Pleiotropy Identifies 6 Further Loci Associated With Coronary Artery Disease. JACC 2017, 69, 823. [Google Scholar] [CrossRef] [PubMed]
- Derry, J.M.; Zhong, H.; Molony, C.; MacNeil, D.; Guhathakurta, D.; Zhang, B.; Mudgett, J.; Small, K.; El Fertak, L.; Guimond, A. Identification of genes and networks driving cardiovascular and metabolic phenotypes in a mouse F2 intercross. PLoS ONE 2010, 5, e14319. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Schadt, E.E.; Wang, H.; Wang, X.; Ingram-Drake, L.; Shi, W.; Drake, T.A.; Lusis, A.J. Identification of pathways for atherosclerosis in mice: Integration of quantitative trait locus analysis and global gene expression data. Circ. Res. 2007, 101, e11–e30. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Schadt, E.E.; Wang, S.; Wang, H.; Arnold, A.P.; Ingram-Drake, L.; Drake, T.A.; Lusis, A.J. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadt, E.E.; Molony, C.; Chudin, E.; Hao, K.; Yang, X.; Lum, P.Y.; Kasarskis, A.; Zhang, B.; Wang, S.; Suver, C. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008, 6, e107. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Keller, M.P.; Zhang, C.; Rabaglia, M.E.; Greenawalt, D.M.; Yang, X.; Wang, I.-M.; Dai, H.; Bruss, M.D.; Lum, P.Y. Integrative analysis of a cross-loci regulation network identifies App as a gene regulating insulin secretion from pancreatic islets. PLoS Genet. 2012, 8, e1003107. [Google Scholar] [CrossRef]
- Wang, I.M.; Zhang, B.; Yang, X.; Zhu, J.; Stepaniants, S.; Zhang, C.; Meng, Q.; Peters, M.; He, Y.; Ni, C. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 2012, 8, 594. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef]
- Shu, L.; Zhao, Y.; Kurt, Z.; Byars, S.G.; Tukiainen, T.; Kettunen, J.; Orozco, L.D.; Pellegrini, M.; Lusis, A.J.; Ripatti, S. Mergeomics: Multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genom. 2016, 17, 874. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, S.; Eigenmann, J.; Zhao, Y.; Fleig, J.; Hawe, J.S.; Pan, C.; Bongiovanni, D.; Wengert, S.; Ma, A.; Lusis, A.J.; et al. Identification of the Transcription Factor ATF3 as a Direct and Indirect Regulator of the LDLR. Metabolites 2022, 12, 840. https://doi.org/10.3390/metabo12090840
Bauer S, Eigenmann J, Zhao Y, Fleig J, Hawe JS, Pan C, Bongiovanni D, Wengert S, Ma A, Lusis AJ, et al. Identification of the Transcription Factor ATF3 as a Direct and Indirect Regulator of the LDLR. Metabolites. 2022; 12(9):840. https://doi.org/10.3390/metabo12090840
Chicago/Turabian StyleBauer, Sabine, Jana Eigenmann, Yuqi Zhao, Julia Fleig, Johann S. Hawe, Calvin Pan, Dario Bongiovanni, Simon Wengert, Angela Ma, Aldons J. Lusis, and et al. 2022. "Identification of the Transcription Factor ATF3 as a Direct and Indirect Regulator of the LDLR" Metabolites 12, no. 9: 840. https://doi.org/10.3390/metabo12090840