
Iron Metabolism in the Disorders of Heme Biosynthesis
 , , , and
, , , and
Abstract
:1. Introduction
2. Role of Iron in the Biosynthesis of Heme
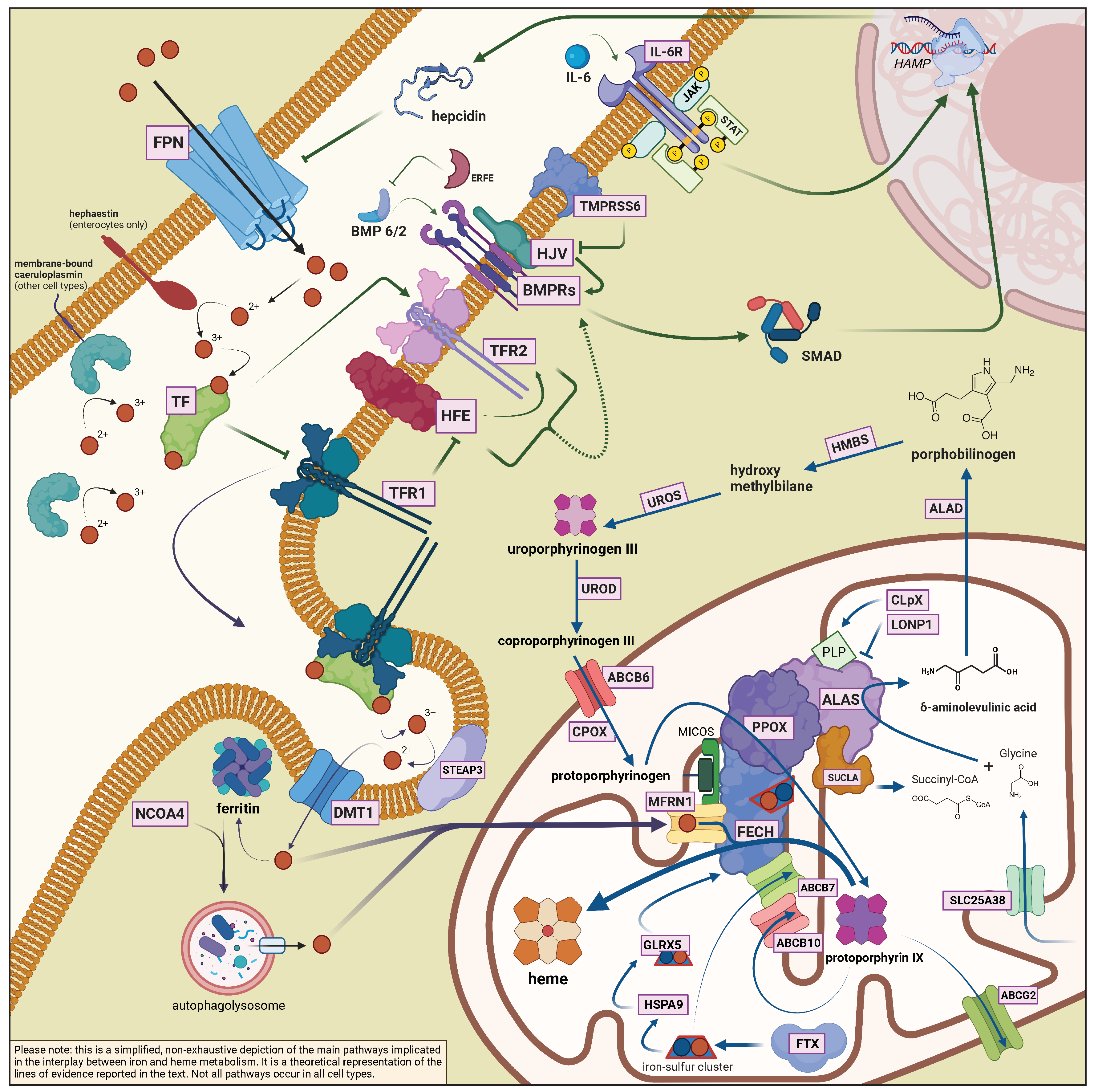
2.1. General Aspects of Iron Metabolism in Mammals
2.2. The Interplay between Heme Biosynthesis and Iron

{kind=link}
| Protein | Gene | Function | Associated Diseases |
|---|---|---|---|
| Bone morphogenetic protein 2 | BMP2 | Ligand of the BMP-SMAD signaling pathway regulating hepcidin expression in response to iron | |
| Bone morphogenetic protein 6 | BMP6 | Ligand of the BMP-SMAD signaling pathway regulating hepcidin expression in response to iron | BMP6-associated iron-overload |
| Caeruloplasmin | CP | Soluble/membrane-bound copper-carrying ferroxidase | Aceruloplasminemia |
| Divalent metal transporter 1 | DMT1 or SLC11A2 | Ferrous iron importer | DMT1 deficiency |
| Duodenal cytochrome B | DCYTB | Reduces dietary ferric iron to ferrous form at the apical border of enterocytes | |
| Erythroferrone | ERFE | Hepcidin inhibitor, produced by the bone marrow in response to erythropoietin | |
| Ferritin heavy chain | FTH1 | Subunit of ferritin, with ferroxidase activity | FTH-related iron-overload |
| Ferritin light chain | FTL | Subunit of ferritin, with iron storage properties | Hyperferritinemia-cataract syndrome Hereditary benign hyperferritinemia Neuroferritinopathy L-ferritin deficiency [66] |
| Ferroportin | SLC40A1 | Ferrous iron exporter | Ferroportin disease (loss-of-function)SLC40A1-related hemochromatosis (gain-of-function) |
| Frataxin | FXN | Iron carrier, participates in iron-sulfur cluster biogenesis | Friedreich’s ataxia |
| Haephastin | HEPH | Membrane-bound ferroxidase | |
| Hemojuvelin | HJV or HFE2 | BMP co-receptor, involved in the iron-sensing pathway which regulates hepcidin | HJV-related hemochromatosis |
| Hepcidin | HAMP | Iron regulating hormone; internalises ferroportin | HAMP-related hemochromatosis |
| Human homeostatic iron regulator protein | HFE | Protein involved in the iron-sensing pathway which regulates hepcidin | HFE-related HH |
| IRE binding protein 1 | IREB1 or IRP1 or ACO1 | Iron-sensing regulator of translation; aconitase activity in the presence of iron | |
| IRE binding protein 2 | IREB2 or IRP2 | Iron-sensing regulator of translation | IRP2-related protoporphyria |
| Matriptase | TMPRSS6 | Cleaves membrane-bound HJV; negative regulator of hepcidin in response to iron deficiency | Iron-deficient iron refractory anaemia (IRIDA) |
| Mitoferrin-1 | MFRN1 | Intramitochondrial iron carrier | |
| Nuclear receptor coactivator 4 | NCOA4 | Delivers ferritin to autophagolysosomes (ferritinophagy) | |
| Six-transmembrane epithelial antigen of prostate 3 | STEAP3 | Membrane-bound metalloreductase | Sideroblastic anaemia with primary hypogonadism |
| Transferrin | TF | Ferric iron carrier | Hypo/Atransferrinemia |
| Transferrin receptor | TFRC | Receptor for endocytosis-mediated iron uptake; one of the plasma iron sensors | TFRC-related combined immunodeficiency [67] |
| Transferrin receptor 2 | TFR2 | Protein involved in the iron-sensing pathway which regulates hepcidin | TFR2-related hemochromatosis |
| Protein | Gene | Function | Associated Diseases |
|---|---|---|---|
| -aminolaevulinate dehydratase | ALAD | Dehydrates ALA to yield PBG | ALAD-deficiency (Doss) porphyria |
| -aminolaevulinate synthase 1 | ALAS1 | Condenses glycine and succinyl-CoA to yield ALA | |
| -aminolaevulinate synthase 2 | ALAS2 | Condenses glycine and succinyl-CoA to yield ALA (erythroid-specific isoform) | X-linked congenital sideroblastic anaemia (loss-of-function) X-linked erythropoietic protoporphyria (gain-of-function) |
| ATP-binding cassette super-family B member 6 | ABCB6 | Imports porphyrins into mithocondria | Phenotype modifier in porphyrias [68] |
| ATP-binding cassette super-family B member 7 | ABCB7 | Mitochondrial [Fe-S] cluster exporter | X-linked sideroblastic anaemia with ataxia |
| ATP-binding cassette super-family B member 10 | ABCB10 | Mitochondrial exporter with putative roles in porphyrin or iron transport; complexes with MFRN1 and FECH to enhance heme biosynthesis | |
| ATP-binding cassette super-family G member 2 | ABCG2 | Cytosolic and mitochondrial exporter of protoporphyrin IX; also involved in the export of heme | |
| caseinolytic mitochondrial matrix peptidase chaperone subunit X | CLpX | Mitochondrial protein with ATP-dependent protease and unfoldase activity; regulates ALAS turnover; activates ALAS catalyzing PLP insertion | Phenotype modifier in protoporphyria (see Section 3.3) |
| coproporphyrinogen III oxidase | CPOX | Eliminates two carboxyl groups from coproporphyrinogen III side chains to yield protoporphyrinogen IX | Hereditary coproporphyria (autosomal dominant) Harderoporphyria (autosomal recessive) |
| ferrochelatase | FECH | Chelates iron into protoporphyrin IX to yield heme | Erythropoietic protoporphyria |
| glutaredoxin 5 | GLRX5 | Mitochondrial protein with thiol reductase activity; involved in [Fe-S] cluster assembly | Autosomal recessive sideroblastic anaemia |
| hydroxymethylbilane synthase | HMBS | Condensates four PBG molecules into HMB | Acute intermittent porphyria (AIP)Autosomal recessive AIP |
| heme oxygenase 1 | HO-1 | Cleaves heme into biliverdin IX, releasing CO and ferrous iron (inducible isoform) | HO-1 deficiency |
| heme oxygenase 2 | HO-2 | Cleaves heme into biliverdin IX, releasing CO and ferrous iron (constitutive isoform); involved in the CO signalling pathway | |
| hemopexin | HPX | Heme scavenger in the plasma | |
| heat shock protein family A member 9 | HSPA9 | Mitochondrial protein with chaperone activity for [Fe-S] clusters | Autosomal recessive sideroblastic anaemia |
| lon peptidase 1, mitochondrial | LONP1 | ATP-dependent protease involved in the turnover of mitochondrial matrix protein | CODAS (Cerebral, Ocular, Dental, Auricular and Skeletal) syndrome [69] |
| protoporphyrinogen oxidase | PPOX | Dehydrogenates protoporphyrinogen IX to yield protoporphyrin IX | Variegate porphyria (VP) Autosomal recessive VP |
| mitochondrial solute carrier family 25 member A38 | SLC25A38 | Mitochondrial glycine transporter | Autosomal recessive sideroblastic anaemia |
| succinyl-CoA synthase | SUCLA | Controls the flux of heme precursors catalyzing a reversible conversion from succinate + coenzime A to succynil-CoA (precursor of ALA) | |
| uroporphyrinogen III decarboxylase | UROD | Eliminates four carboxyl groups from uroporphyrinogen III side chains to yield coproporphyrinogen III | Porphyria cutanea tarda (sporadic or familial)Hepatoerythropoietic porphyria |
| uroporphyrinogen III synthase | UROS | Converts linear PBG to cyclic uroporphyrinogen III | Congenital erythropoietic porphyria (Günther disease) |
3. Clinical and Experimental Aspects of the Role of Iron in Porphyrias
3.1. Porphyria Cutanea Tarda
3.2. Congenital Erythropoietic Porphyria
3.3. Erythropoietic and X-Linked Protoporphyria
IRP2-Related Protoporphyria
3.4. Acute Hepatic Porphyrias
4. Clinical and Experimental Aspects of the Role of Iron in Congenital Sideroblastic Anaemias
4.1. ALAS2-Related X-Linked Sideroblastic Anaemia
4.1.1. X-Linked Sideroblastic Anaemia with Ataxia
4.2. Autosomal Recessive Sideroblastic Anaemias
4.2.1. SLC25A38 Mutations
4.2.2. GLRX5, HSPA9, HSCB Mutations
4.2.3. STEAP3-Related Sideroblastic Anaemia with Primary Hypogonadism and DMT1 Deficiency
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-UTR | 5 untranslated region |
| ABCB | adenosine triphosphate-binding cassette subfamily B |
| ACTR | activin receptor |
| AIP | acute intermittent porphyria |
| AHPs | acute hepatic porphyrias |
| ALA | -aminolevulinic acid |
| ALA· | enoyl (ALA) radical |
| ALAD | ALA dehydratase |
| ALAS1 | ALA synthase 1 |
| ALAS2 | ALA synthase 2 |
| ALK | activin receptor-like kinase |
| APA | acute porphyric attack |
| ARCSA | autosomal recessive sideroblastic anaemia |
| BMP | bone morphogenetic protein |
| CEP | congenital erythropoietic porphyria |
| ClpX | caseinolytic mitochondrial matrix peptidase chaperone subunit X |
| CP | ceruloplasmin |
| CPOX | coproporphyrinogen III oxidase |
| CSA | congenital sideroblastic anaemias |
| CYP1A2 | cytochrome 1A2 of the P450 family |
| DCYTB | duodenal cytochrome B |
| DFO | deferoxamine |
| DMT1 | divalent metal transporter 1 |
| DOVA | 4,5-dioxovaleric acid |
| EPP | erythropoietic protoporphyria |
| EPO | erythropoietin |
| ERFE | erythroferrone |
| Fe | iron |
| FECH | ferrochelatase |
| Fe-S cluster | iron-sulfur cluster |
| F-PCT | familial porphyria cutanea tarda |
| FPN1 | ferroportin (protein) |
| FTH1 | ferritin heavy chain |
| FTL | ferritin light chain |
| FXN | frataxin |
| GATA1 | GATA-binding factor 1 |
| GLRX5 | glutaredoxin 5 |
| GDF15 | growth/differentation factor 15 |
| GRX | glutaredoxin |
| HAMP | hepcidin gene |
| HCP | hereditary coproporphyria |
| HC | hemochromatosis |
| HEPH | hephaestin |
| HJV or HFE2 | hemojuvelin gene |
| HFE | human homeostatic iron regulator protein |
| HMB | hydroxymethylbilane |
| HMBS | HMB synthase |
| HO | heme oxygenase |
| HSCB | heat-shock cognate B |
| HSPA9 | heat-shock protein family A member 9 |
| IL-6 | interleukin 6 |
| IRE | iron responsive element |
| IRIDA | iron deficiency-iron refractory anaemia |
| IRP1 | IRE-binding protein 1 |
| IRP2 | IRE-binding protein 2 |
| LIC | liver iron content |
| LONP1 | lon peptidase 1, mitochondrial |
| LSEC | liver sinusoidal endothelial cell |
| Mfrn1 | mitoferrin 1 |
| MtF | mitochondrial ferritin |
| NCOA4 | nuclear receptor coactivator 4 |
| PBG | porphobilinogen |
| PCBP1 | poly rC–binding protein 1 |
| PCT | porphyria cutanea tarda |
| PLP | pyridoxal phosphate |
| PPOX | protoporphyrinogen oxidase |
| RISC | RNA-induced silencing complex |
| ROS | reactive oxygen species |
| S-PCT | sporadic porphyria cutanea tarda |
| SLC40A1 | ferroportin (gene) |
| SMAD | suppressor of mothers against decapentaplegic |
| STAT3 | signal transducer and activator of transcription 3 |
| STEAP3 | six-transmembrane epithelial antigen of prostate 3 |
| TF | transferrin |
| TFR1 | transferrin receptor 1 (protein) |
| TFR2 | transferrin receptor 2 |
| TFRC | transferrin receptor (gene) |
| TMPRSS6 | transmembrane protease serine 6 |
| UROD | uroporphyrinogen decarboxylase |
| UROS | uroporphyrinogen III synthase |
| UTR | untranslated region |
| VP | variegate porphyria |
| XLP | X-linked protoporphyria |
| XLSA | X-linked sideroblastic anaemia |
| XLSA/A | XLSA with ataxia |
References
- Pietrangelo, A. Mechanisms of iron hepatotoxicity. J. Hepatol. 2016, 65, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood J. Am. Soc. Hematol. 2012, 120, 4496–4504. [Google Scholar]
- Camaschella, C. Hereditary sideroblastic anemias: Pathophysiology, diagnosis, and treatment. Semin. Hematol. 2009, 46, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, E.; Granata, F. Nutrients and Porphyria: An Intriguing Crosstalk. Int. J. Mol. Sci. 2020, 21, 3462. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Ventura, P.; Corradini, E. Iron in Porphyrias: Friend or Foe? Diagnostics 2022, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Schmitt, C.; Moulouel, B.; Mirmiran, A.; Puy, H.; Lefèbvre, T.; Gouya, L. Iron, Heme Synthesis and Erythropoietic Porphyrias: A Complex Interplay. Metabolites 2021, 11, 798. [Google Scholar] [CrossRef]
- West, A.R.; Oates, P.S. Mechanisms of heme iron absorption: Current questions and controversies. World J. Gastroenterol. WJG 2008, 14, 4101. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020, 75, 100866. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Alfaro-Magallanes, V.M.; Babitt, J.L. Physiological and pathophysiological mechanisms of hepcidin regulation: Clinical implications for iron disorders. Br. J. Haematol. 2021, 193, 882–893. [Google Scholar] [CrossRef]
- Andriopoulos Jr, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Fisher, A.L.; Babitt, J.L. Coordination of iron homeostasis by bone morphogenetic proteins: Current understanding and unanswered questions. Dev. Dyn. 2022, 251, 26–46. [Google Scholar] [CrossRef]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta BBA Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Ryu, M.S.; Zhang, D.; Protchenko, O.; Shakoury-Elizeh, M.; Philpott, C.C. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Investig. 2017, 127, 1786–1797. [Google Scholar] [CrossRef]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. Intracellular kinetics of iron in reticulocytes: Evidence for endosome involvement in iron targeting to mitochondria. Blood 2005, 105, 368–375. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Zhang, A.S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood J. Am. Soc. Hematol. 2007, 110, 125–132. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP Regulatory System: An Update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schranzhofer, M.; Schifrer, M.; Cabrera, J.A.; Kopp, S.; Chiba, P.; Beug, H.; Müllner, E.W. Remodeling the regulation of iron metabolism during erythroid differentiation to ensure efficient heme biosynthesis. Blood 2006, 107, 4159–4167. [Google Scholar] [CrossRef]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Buzzetti, E.; Pietrangelo, A. Genetic iron overload disorders. Mol. Asp. Med. 2020, 75, 100896. [Google Scholar] [CrossRef] [PubMed]
- Zoller, H.; Schaefer, B.; Vanclooster, A.; Griffiths, B.; Bardou-Jacquet, E.; Corradini, E.; Porto, G.; Ryan, J.; Cornberg, M. EASL Clinical Practice Guidelines on haemochromatosis. J. Hepatol. 2022, 77, 479–502. [Google Scholar] [CrossRef]
- Ramsay, A.J.; Quesada, V.; Sanchez, M.; Garabaya, C.; Sardà, M.P.; Baiget, M.; Remacha, A.; Velasco, G.; López-Otín, C. Matriptase-2 mutations in iron-refractory iron deficiency anemia patients provide new insights into protease activation mechanisms. Hum. Mol. Genet. 2009, 18, 3673–3683. [Google Scholar] [CrossRef]
- Gitlin, J.D. Aceruloplasminemia. Pediatr. Res. 1998, 44, 271–276. [Google Scholar] [CrossRef]
- Heinemann, I.U.; Jahn, M.; Jahn, D. The biochemistry of heme biosynthesis. Arch. Biochem. Biophys. 2008, 474, 238–251. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Ângelo, C.; Ferreira, A.; et al. A Central Role for Free Heme in the Pathogenesis of Severe Sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; McCulloh, R. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Haggard, J.J.; Croatt, A.J.; Grande, J.P.; Poss, K.D.; Alam, J. The Indispensability of Heme Oxygenase-1 in Protecting against Acute Heme Protein-Induced Toxicity in Vivo. Am. J. Pathol. 2000, 156, 1527–1535. [Google Scholar] [CrossRef]
- Deuel, J.W.; Schaer, C.A.; Boretti, F.S.; Opitz, L.; García-Rubio, I.; Baek, J.; Spahn, D.R.; Buehler, P.W.; Schaer, D.J. Hemoglobinuria-related acute kidney injury is driven by intrarenal oxidative reactions triggering a heme toxicity response. Cell Death Dis. 2016, 7, e2064. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.J.; Bossenmaier, I.; Cardinal, R.; Petryka, Z.; Watson, C. Transitory renal failure following rapid administration of a relatively large amount of hematin in a patient with acute intermittent porphyria in clinical remission. Acta Medica Scand. 1978, 203, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, N.; Durante, W.; Kroll, M.H.; Schafer, A.I. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase–stimulatory carbon monoxide. Circulation 1995, 91, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.L.; Johns, D.G.; Kriegsfeld, L.J.; Klein, S.L.; Calvin, D.C.; Demas, G.E.; Schramm, L.P.; Tonegawa, S.; Nelson, R.J.; Snyder, S.H.; et al. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat. Med. 1998, 4, 84–87. [Google Scholar] [CrossRef]
- Szlendak, U.; Bykowska, K.; Lipniacka, A. Clinical, Biochemical and Molecular Characteristics of the Main Types of Porphyria. Adv. Clin. Exp. Med. 2016, 25, 361–368. [Google Scholar] [CrossRef]
- Woods, J. Regulation of porphyrin and heme metabolism in the kidney. Semin. Hematol. 1988, 25, 336–348. [Google Scholar]
- Phillips, J.D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 2019, 128, 164–177. [Google Scholar] [CrossRef]
- Melefors, O.; Goossen, B.; Johansson, H.E.; Stripecke, R.; Gray, N.K.; Hentze, M. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J. Biol. Chem. 1993, 268, 5974–5978. [Google Scholar] [CrossRef]
- Sadlon, T.J.; Dell’Oso, T.; Surinya, K.H.; May, B.K. Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int. J. Biochem. Cell Biol. 1999, 31, 1153–1167. [Google Scholar] [CrossRef]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J. Biol. Chem. 2011, 286, 26424–26430. [Google Scholar] [CrossRef]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of heme biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef] [PubMed]
- Rondelli, C.M.; Perfetto, M.; Danoff, A.; Bergonia, H.; Gillis, S.; O’Neill, L.; Jackson, L.; Nicolas, G.; Puy, H.; West, R.; et al. The ubiquitous mitochondrial protein unfoldase CLPX regulates erythroid heme synthesis by control of iron utilization and heme synthesis enzyme activation and turnover. J. Biol. Chem. 2021, 297, 100972. [Google Scholar] [CrossRef]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell 2015, 161, 858–867. [Google Scholar] [CrossRef]
- Harbin, B.M.; Dailey, H.A. Orientation of ferrochelatase in bovine liver mitochondria. Biochemistry 1985, 24, 366–370. [Google Scholar] [CrossRef]
- Obi, C.D.; Bhuiyan, T.; Dailey, H.A.; Medlock, A.E. Ferrochelatase: Mapping the intersection of iron and porphyrin metabolism in the mitochondria. Front. Cell Dev. Biol. 2022, 10, 894591. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H.; Kafina, M.D.; Coughlin, E.E.; Minogue, C.E.; Hebert, A.S.; et al. Erythropoietin signaling regulates heme biosynthesis. eLife 2017, 6, e24767. [Google Scholar] [CrossRef]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef]
- Shah, D.I.; Takahashi-Makise, N.; Cooney, J.D.; Li, L.; Schultz, I.J.; Pierce, E.L.; Narla, A.; Seguin, A.; Hattangadi, S.M.; Medlock, A.E.; et al. Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature 2012, 491, 608–612. [Google Scholar] [CrossRef]
- Shaw, G.; Cope, J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.; Gwynn, B.; Lambert, A.; Wingert, R.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef]
- Bencze, K.Z.; Yoon, T.; Millán-Pacheco, C.; Bradley, P.B.; Pastor, N.; Cowan, J.; Stemmler, T.L. Human frataxin: Iron and ferrochelatase binding surface. Chem. Commun. 2007, 1798–1800. [Google Scholar] [CrossRef]
- Schmucker, S.; Puccio, H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum. Mol. Genet. 2010, 19, R103–R110. [Google Scholar] [CrossRef]
- Dietz, J.V.; Willoughby, M.M.; Piel, R.B.; Ross, T.A.; Bohovych, I.; Addis, H.G.; Fox, J.L.; Lanzilotta, W.N.; Dailey, H.A.; Wohlschlegel, J.A.; et al. Mitochondrial contact site and cristae organizing system (MICOS) machinery supports heme biosynthesis by enabling optimal performance of ferrochelatase. Redox Biol. 2021, 46, 102125. [Google Scholar] [CrossRef]
- Medlock, A.E.; Carter, M.; Dailey, T.A.; Dailey, H.A.; Lanzilotta, W.N. Product release rather than chelation determines metal specificity for ferrochelatase. J. Mol. Biol. 2009, 393, 308–319. [Google Scholar] [CrossRef]
- Troadec, M.B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Chung, J.; Anderson, S.A.; Gwynn, B.; Deck, K.M.; Chen, M.J.; Langer, N.B.; Shaw, G.C.; Huston, N.C.; Boyer, L.F.; Datta, S.; et al. Iron regulatory protein-1 protects against mitoferrin-1-deficient porphyria. J. Biol. Chem. 2014, 289, 7835–7843. [Google Scholar] [CrossRef]
- Phillips, J.; Farrell, C.; Wang, Y.; Singal, A.K.; Anderson, K.; Balwani, M.; Bissell, M.; Bonkovsky, H.; Seay, T.; Paw, B.; et al. Strong correlation of ferrochelatase enzymatic activity with Mitoferrin-1 mRNA in lymphoblasts of patients with protoporphyria. Mol. Genet. Metab. 2019, 128, 391–395. [Google Scholar] [CrossRef]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J. Clin. Investig. 2020, 130, 5245–5256. [Google Scholar] [CrossRef]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the mitochondrial heme metabolism complex. PLoS ONE 2015, 10, e0135896. [Google Scholar]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Bishop, D.F.; Tchaikovskii, V.; Hoffbrand, A.V.; Fraser, M.E.; Margolis, S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 2012, 287, 28943–28955. [Google Scholar] [CrossRef]
- Furuyama, K.; Sassa, S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Investig. 2000, 105, 757–764. [Google Scholar] [CrossRef]
- Cadenas, B.; Fita-Torró, J.; Bermúdez-Cortés, M.; Hernandez-Rodriguez, I.; Fuster, J.L.; Llinares, M.E.; Galera, A.M.; Romero, J.L.; Pérez-Montero, S.; Tornador, C.; et al. L-Ferritin: One Gene, Five Diseases; from Hereditary Hyperferritinemia to Hypoferritinemia—Report of New Cases. Pharmaceuticals 2019, 12, 17. [Google Scholar] [CrossRef]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef]
- Fukuda, Y.; Cheong, P.L.; Lynch, J.; Brighton, C.; Frase, S.; Kargas, V.; Rampersaud, E.; Wang, Y.; Sankaran, V.G.; Yu, B.; et al. The severity of hereditary porphyria is modulated by the porphyrin exporter and Lan antigen ABCB6. Nat. Commun. 2016, 7, 1–9. [Google Scholar]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A 2015, 167, 1501–1509. [Google Scholar] [CrossRef]
- Elder, G.H. Porphyria cutanea tarda. Semin. Liver Dis. 1998, 18, 67–75. [Google Scholar] [CrossRef]
- Badenas, C.; To-Figueras, J.; Phillips, J.; Warby, C.; Munoz, C.; Herrero, C. Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives. Clin. Genet. 2009, 75, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.; Baker, H.; Vernon-Roberts, B.; Magnus, I.A. Iron Metabolism in Porphyria Cutanea Tarda and in Erythropoietic Protoporphyria. QJM Int. J. Med. 1973, 42, 341–355. [Google Scholar] [CrossRef]
- Alla, V.; Bonkovsky, H.L. Iron in nonhemochromatotic liver disorders. Semin. Liver Dis. 2005, 25, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, Z.J.; Phillips, J.D.; Ajioka, R.S.; Franklin, M.R.; Griffen, L.M.; Guinee, D.J.; Edwards, C.Q.; Kushner, J.P. Hemochromatosis genes and other factors contributing to the pathogenesis of porphyria cutanea tarda. Blood 2000, 95, 1565–1571. [Google Scholar] [CrossRef]
- Ryan Caballes, F.; Sendi, H.; Bonkovsky, H.L. Hepatitis C, porphyria cutanea tarda and liver iron: An update. Liver Int. 2012, 32, 880–893. [Google Scholar] [CrossRef]
- Lundvall, O.; Weinfeld, A. Studies of the clinical and metabolic effects of phlebotomy treatment in porphyria cutanea tarda. Acta Med. Scand. 1968, 184, 191–199. [Google Scholar] [CrossRef]
- Felsher, B.F.; Jones, M.L.; Redeker, A.G. Iron and Hepatic Uroporphyrin Synthesis: Relation in Porphyria Cutanea Tarda. JAMA 1973, 226, 663–665. [Google Scholar] [CrossRef]
- Dabrowska, E.; Jabłońska-Kaszewska, I.; Falkiewicz, B. Effect of high fiber vegetable-fruit diet on the activity of liver damage and serum iron level in porphyria cutanea tarda (PCT). Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2001, 7, 282–286. [Google Scholar]
- Roberts, A.G.; Whatley, S.D.; Nicklin, S.; Worwood, M.; Pointon, J.J.; Stone, C.; Elder, G.H. The frequency of hemochromatosis-associated alleles is increased in British patients with sporadic porphyria cutanea tarda. Hepatology 1997, 25, 159–161. [Google Scholar] [CrossRef]
- Stuart, K.A.; Busfield, F.; Jazwinska, E.C.; Gibson, P.; Butterworth, L.A.; Cooksley, W.G.; Powell, L.W.; Crawford, D.H. The C282Y mutation in the haemochromatosis gene (HFE) and hepatitis C virus infection are independent cofactors for porphyria cutanea tarda in Australian patients. J. Hepatol. 1998, 28, 404–409. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Poh-Fitzpatrick, M.; Pimstone, N.; Obando, J.; Di Bisceglie, A.; Tattrie, C.; Tortorelli, K.; LeClair, P.; Mercurio, M.G.; Lambrecht, R.W. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology 1998, 27, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Sampietro, M.; Fiorelli, G.; Fargion, S. Iron overload in porphyria cutanea tarda. Haematologica 1999, 84, 248–253. [Google Scholar] [PubMed]
- Tannapfel, A.; Stölzel, U.; Köstler, E.; Melz, S.; Richter, M.; Keim, V.; Schuppan, D.; Wittekind, C. C282Y and H63D mutation of the hemochromatosis gene in German porphyria cutanea tarda patients. Virchows Arch. 2001, 439, 1–5. [Google Scholar] [CrossRef]
- Egger, N.G.; Goeger, D.E.; Payne, D.A.; Miskovsky, E.P.; Weinman, S.A.; Anderson, K.E. Porphyria cutanea tarda: Multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig. Dis. Sci. 2002, 47, 419–426. [Google Scholar] [CrossRef]
- Phillips, J.D.; Jackson, L.K.; Bunting, M.; Franklin, M.R.; Thomas, K.R.; Levy, J.E.; Andrews, N.C.; Kushner, J.P. A mouse model of familial porphyria cutanea tarda. Proc. Natl. Acad. Sci. USA 2001, 98, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kushner, J.P.; Barbuto, A.J.; Lee, G.R. An inherited enzymatic defect in porphyria cutanea tarda: Decreased uroporphyrinogen decarboxylase activity. J. Clin. Investig. 1976, 58, 1089–1097. [Google Scholar] [CrossRef]
- Elder, G.H.; Lee, G.B.; Tovey, J.A. Decreased Activity of Hepatic Uroporphyrinogen Decarboxylase in Sporadic Porphyria Cutanea Tarda. N. Eng. J. Med. 1978, 299, 274–278. [Google Scholar] [CrossRef]
- Phillips, J.D.; Bergonia, H.A.; Reilly, C.A.; Franklin, M.R.; Kushner, J.P. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc. Natl. Acad. Sci. USA 2007, 104, 5079–5084. [Google Scholar] [CrossRef]
- Sinclair, R.P.; Gorman, N.; Dalton, T.; Walton, S.H.; Bement, J.W.; Sinclair, F.J.; Smith, G.A.; Nebert, W.D. Uroporphyria produced in mice by iron and 5-aminolaevulinic acid does not occur in Cyp1a2 (-/-) null mutant mice. Biochem. J. 1998, 330, 149–153. [Google Scholar] [CrossRef]
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital erythropoietic porphyria due to a mutation in GATA1: The first trans-acting mutation causative for a human porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef]
- Di Pierro, E.; Brancaleoni, V.; Granata, F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br. J. Haematol. 2016, 173, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L.; Desnick, R.J. Congenital erythropoietic porphyria: Recent advances. Mol. Genet. Metab. 2019, 128, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Podlipnik, S.; Guijarro, F.; Combalia, A.; To-Figueras, J.; Badenas, C.; Costa, D.; Rozman, M.; Jorge, S.; Aguilera, P.; Gaya, A. Acquired erythropoietic uroporphyria secondary to myelodysplastic syndrome with chromosome 3 alterations: A case report. Br. J. Dermatol. 2018, 179, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Serra-García, L.; Morgado-Carrasco, D.; Pérez-Valencia, A.I.; Castaño-Díez, S.; Alamon-Reig, F.; Badenas, C.; To-Figueras, J.; Aguilera, P. Acquired Erythropoietic Uroporphyria secondary to Myeloid Malignancy: Case report and literature review. Photodermatol. Photoimmunol. Photomed. 2021, 38, 86–91. [Google Scholar] [CrossRef]
- Egan, D.N.; Yang, Z.; Phillips, J.; Abkowitz, J.L. Inducing iron deficiency improves erythropoiesis and photosensitivity in congenital erythropoietic porphyria. Blood J. Am. Soc. Hematol. 2015, 126, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Katugampola, R.; Anstey, A.; Finlay, A.; Whatley, S.; Woolf, J.; Mason, N.; Deybach, J.; Puy, H.; Ged, C.; de Verneuil, H.; et al. A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases. Br. J. Dermatol. 2012, 167, 888–900. [Google Scholar] [CrossRef]
- Piomelli, S.; Poh-Fitzpatrick, M.B.; Seaman, C.; Skolnick, L.M.; Berdon, W.E. Complete Suppression of the Symptoms of Congenital Erythropoietic Porphyria by Long-Term Treatment with High-Level Transfusions. N. Eng. J. Med. 1986, 314, 1029–1031. [Google Scholar] [CrossRef]
- Lange, B.; Hofweber, K.; Waldherr, R.; Schärer, K. Congenital erythropoietic porphyria associated with nephrotic syndrome and renal siderosis. Acta Pædiatr. 1995, 84, 1325–1328. [Google Scholar] [CrossRef]
- Mirmiran, A.; Poli, A.; Ged, C.; Schmitt, C.; Lefebvre, T.; Manceau, H.; Daher, R.; Moulouel, B.; Peoc’h, K.; Simonin, S.; et al. Phlebotomy as an efficient long-term treatment of congenital erythropoietic porphyria. Haematologica 2021, 106, 913. [Google Scholar] [CrossRef]
- Blouin, J.M.; Ged, C.; Bernardo-Seisdedos, G.; Cabantous, T.; Pinson, B.; Poli, A.; Puy, H.; Millet, O.; Gouya, L.; Morice-Picard, F.; et al. Identification of novel UROS mutations in a patient with congenital erythropoietic porphyria and efficient treatment by phlebotomy. Mol. Genet. Metab. Rep. 2021, 27, 100722. [Google Scholar] [CrossRef]
- Ged, C.; Mendez, M.; Robert, E.; Lalanne, M.; Lamrissi-Garcia, I.; Costet, P.; Daniel, J.; Dubus, P.; Mazurier, F.; Moreau-Gaudry, F.; et al. A knock-in mouse model of congenital erythropoietic porphyria. Genomics 2006, 87, 84–92. [Google Scholar] [CrossRef]
- Millot, S.; Delaby, C.; Moulouel, B.; Lefebvre, T.; Pilard, N.; Ducrot, N.; Ged, C.; Lettéron, P.; De Franceschi, L.; Deybach, J.C.; et al. Hemolytic anemia repressed hepcidin level without hepatocyte iron overload: Lesson from Günther disease model. Haematologica 2017, 102, 260. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Millot, S.; Richard, E.; Blouin, J.M.; Lalanne, M.; Lamrissi-Garcia, I.; Costet, P.; Lyoumi, S.; Gouya, L.; Puy, H.; et al. Genetic background influences hepcidin response to iron imbalance in a mouse model of hemolytic anemia (Congenital erythropoietic porphyria). Biochem. Biophys. Res. Commun. 2019, 520, 297–303. [Google Scholar] [CrossRef] [PubMed]
- To-Figueras, J.; Ducamp, S.; Clayton, J.; Badenas, C.; Delaby, C.; Ged, C.; Lyoumi, S.; Gouya, L.; de Verneuil, H.; Beaumont, C.; et al. ALAS2 acts as a modifier gene in patients with congenital erythropoietic porphyria. Blood 2011, 118, 1443–1451. [Google Scholar] [CrossRef]
- Peoc’h, K.; Nicolas, G.; Schmitt, C.; Mirmiran, A.; Daher, R.; Lefebvre, T.; Gouya, L.; Karim, Z.; Puy, H. Regulation and tissue-specific expression of δ-aminolevulinic acid synthases in non-syndromic sideroblastic anemias and porphyrias. Mol. Genet. Metab. 2019, 128, 190–197. [Google Scholar] [CrossRef]
- Blouin, J.M.; Ged, C.; Lalanne, M.; Lamrissi-Garcia, I.; Morice-Picard, F.; Costet, P.; Daher, R.; Moreau-Gaudry, F.; Bedel, A.; Puy, H.; et al. Iron chelation rescues hemolytic anemia and skin photosensitivity in congenital erythropoietic porphyria. Blood 2020, 136, 2457–2468. [Google Scholar] [CrossRef]
- Di Pierro, E.; Granata, F.; De Canio, M.; Rossi, M.; Ricci, A.; Marcacci, M.; De Luca, G.; Sarno, L.; Barbieri, L.; Ventura, P.; et al. Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria. Diagnostics 2022, 12, 151. [Google Scholar] [CrossRef]
- Gouya, L.; Puy, H.; Robreau, A.M.; Bourgeois, M.; Lamoril, J.; Da Silva, V.; Grandchamp, B.; Deybach, J.C. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat. Genet. 2002, 30, 27–28. [Google Scholar] [CrossRef]
- Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 2019, 128, 298–303. [Google Scholar] [CrossRef]
- Delaby, C.; Lyoumi, S.; Ducamp, S.; Martin-Schmitt, C.; Gouya, L.; Deybach, J.; Beaumont, C.; Puy, H. Excessive erythrocyte PPIX influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Cell. Mol. Biol. 2009, 55, 45–52. [Google Scholar]
- Schneider-Yin, X.; Minder, E.I. Erythropoietic protoporphyria and X-linked dominant protoporphyria. In Handbook of Porphyrin Science (Volume 29) with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine—Volume 29: Porphyrias and Sideroblastic Anemias; World Scientific: Singapore, 2014; pp. 299–328. [Google Scholar]
- Wahlin, S.; Floderus, Y.; Stål, P.; Harper, P. Erythropoietic protoporphyria in Sweden: Demographic, clinical, biochemical and genetic characteristics. J. Intern. Med. 2011, 269, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Graziadei, G.; Duca, L.; Granata, F.; De Luca, G.; De Giovanni, A.; Brancaleoni, V.; Nava, I.; Di Pierro, E. Microcytosis in Erythropoietic Protoporphyria. Front. Physiol. 2022, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Langer, N.B.; Shaw, G.C.; Yang, G.; Li, L.; Kaplan, J.; Paw, B.H.; Bloomer, J.R. Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp. Hematol. 2011, 39, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Bossi, K.; Lee, J.; Schmeltzer, P.; Holburton, E.; Groseclose, G.; Besur, S.; Hwang, S.; Bonkovsky, H.L. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur. J. Clin. Investig. 2015, 45, 1032–1041. [Google Scholar] [CrossRef]
- Barman-Aksoezen, J.; Girelli, D.; Aurizi, C.; Schneider-Yin, X.; Campostrini, N.; Barbieri, L.; Minder, E.I.; Biolcati, G. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J. Inherit. Metab. Dis. 2017, 40, 433–441. [Google Scholar] [CrossRef]
- Landefeld, C.; Kentouche, K.; Gruhn, B.; Stauch, T.; Rößler, S.; Schuppan, D.; Whatley, S.D.; Beck, J.F.; Stölzel, U. X-linked protoporphyria: Iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br. J. Haematol. 2016, 173, 482–484. [Google Scholar] [CrossRef]
- Holme, S.A.; Worwood, M.; Anstey, A.V.; Elder, G.H.; Badminton, M.N. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood 2007, 110, 4108–4110. [Google Scholar] [CrossRef]
- Rademakers, L.H.P.M.; Koningsberger, J.C.; Sorber, C.W.J.; Faille, H.B.D.L.; Hattum, J.V.; Marx, J.J.M. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur. J. Clin. Investig. 1993, 23, 130–138. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Hollowell, M.L.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Mild iron deficiency does not ameliorate the phenotype of a murine erythropoietic protoporphyria model. Am. J. Hematol. 2020, 95, 492–496. [Google Scholar] [CrossRef]
- Lyoumi, S.; Abitbol, M.; Andrieu, V.; Henin, D.; Robert, E.; Schmitt, C.; Gouya, L.; de Verneuil, H.; Deybach, J.C.; Montagutelli, X.; et al. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood 2007, 109, 811–818. [Google Scholar] [CrossRef]
- Hagiwara, S.; Nishida, N.; Ida, H.; Ueshima, K.; Minami, Y.; Takita, M.; Aoki, T.; Morita, M.; Chishina, H.; Komeda, Y.; et al. Role of phlebotomy in the treatment of liver damage related to erythropoietic porphyria. Sci. Rep. 2022, 12, 6100. [Google Scholar] [CrossRef] [PubMed]
- Kniffen, J. Protoporphyrin removal in intrahepatic porphyrastasis. Gastroenterology 1970, 58, 1027. [Google Scholar]
- Gordeuk, V.R.; Brittenham, G.M.; Hawkins, C.W.; Mukhtar, H.; Bickers, D.R. Iron therapy for hepatic dysfunction in erythropoietic protoporphyria. Ann. Intern. Med. 1986, 105, 27–31. [Google Scholar] [CrossRef]
- Holme, S.A.; Thomas, C.L.; Whatley, S.D.; Bentley, D.P.; Anstey, A.V.; Badminton, M.N. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J. Am. Acad. Dermatol. 2007, 56, 1070–1072. [Google Scholar] [CrossRef]
- Milligan, A.; Graham-Brown, R.; Sarkany, I.; Baker, H. Erythropoietic protoporphyria exacerbated by oral iron therapy. Br. J. Dermatol. 1988, 119, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Baker, H. Erythropoietic protoporphyria provoked by iron therapy. Proc. Roy. Soc. Med. 1971, 64, 610–611. [Google Scholar] [CrossRef]
- McClements, B.; Bingham, A.; Callender, M.; Trimble, E.R. Erythropoietic protoporphyria and iron therapy. Br. J. Dermatol. 1990, 122, 423–424. [Google Scholar] [CrossRef]
- Barman-Aksözen, J.; Minder, E.I.; Schubiger, C.; Biolcati, G.; Schneider-Yin, X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol. Dis. 2015, 54, 71–77. [Google Scholar] [CrossRef]
- Barman-Aksözen, J.; Halloy, F.; Iyer, P.S.; Schümperli, D.; Minder, A.E.; Hall, J.; Minder, E.I.; Schneider-Yin, X. Delta-aminolevulinic acid synthase 2 expression in combination with iron as modifiers of disease severity in erythropoietic protoporphyria. Mol. Genet. Metab. 2019, 128, 304–308. [Google Scholar] [CrossRef]
- Inafuku, K.; Takamiyagi, A.; Oshiro, M.; Kinjo, T.; Nakashima, Y.; Nonaka, S. Alteration of mRNA levels of δ-aminolevulinic acid synthase, ferrochelatase and heme oxygenase-1 in griseofulvin induced protoporphyria mice. J. Dermatol. Sci. 1999, 19, 189–198. [Google Scholar] [CrossRef]
- Whitman, J.C.; Paw, B.H.; Chung, J. The role of ClpX in erythropoietic protoporphyria. Hematol. Transfus. Cell Ther. 2018, 40, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of δ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Natl. Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef] [PubMed]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortès, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030. [Google Scholar] [CrossRef]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.R.; Cooperman, S.; Lavaute, T.; Tresser, N.; Ghosh, M.; Meyron-Holtz, E.; Land, W.; Ollivierre, H.; Jortner, B.; Switzer III, R.; et al. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann. N. Y. Acad. Sci. 2004, 1012, 65–83. [Google Scholar] [CrossRef]
- Costain, G.; Ghosh, M.C.; Maio, N.; Carnevale, A.; Si, Y.C.; Rouault, T.A.; Yoon, G. Absence of iron-responsive element-binding protein 2 causes a novel neurodegenerative syndrome. Brain 2019, 142, 1195–1202. [Google Scholar] [CrossRef]
- Marcacci, M.; Ricci, A.; Cuoghi, C.; Marchini, S.; Pietrangelo, A.; Ventura, P. Challenges in diagnosis and management of acute hepatic porphyrias: From an uncommon pediatric onset to innovative treatments and perspectives. Orphanet J. Rare Dis. 2022, 17, 1–10. [Google Scholar] [CrossRef]
- Ricci, A.; Di Pierro, E.; Marcacci, M.; Ventura, P. Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias. Diagnostics 2021, 11, 2205. [Google Scholar] [CrossRef]
- Ricci, A.; Guida, C.C.; Manzini, P.; Cuoghi, C.; Ventura, P. Kidney Involvement in Acute Hepatic Porphyrias: Pathophysiology and Diagnostic Implications. Diagnostics 2021, 11, 2324. [Google Scholar] [CrossRef]
- Ricci, A.; Sandri, G.; Marcacci, M.; Di Pierro, E.; Granata, F.; Cuoghi, C.; Marchini, S.; Pietrangelo, A.; Ventura, P. Endothelial Dysfunction in Acute Hepatic Porphyrias. Diagnostics 2022, 12, 1303. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.; Bechara, E. 5-Aminolevulinic Acid Induces Lipid Peroxidation in Cardiolipin-Rich Liposomes. Arch. Biochem. Biophys. 1993, 305, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.E.; Ferreira, A.M.; Bechara, E.J. Roles of phosphate and an enoyl radical in ferritin iron mobilization by 5-aminolevulinic acid. Free. Radic. Biol. Med. 2000, 29, 1272–1279. [Google Scholar] [CrossRef]
- Douki, T.; Onuki, J.; Medeiros, M.H.; Bechara, E.J.; Cadet, J.; Di Mascio, P. DNA alkylation by 4, 5-dioxovaleric acid, the final oxidation product of 5-aminolevulinic acid. Chem. Res. Toxicol. 1998, 11, 150–157. [Google Scholar] [CrossRef]
- Di Mascio, P.; Teixeira, P.C.; Onuki, J.; Medeiros, M.H.; Dörnemann, D.; Douki, T.; Cadet, J. DNA damage by 5-aminolevulinic and 4, 5-dioxovaleric acids in the presence of ferritin. Arch. Biochem. Biophys. 2000, 373, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.; Kleinman, C.; Demasi, M.; Bechara, E. 5-Aminolevulinic Acid Induces Iron Release from Ferritin. Arch. Biochem. Biophys. 1995, 316, 607–611. [Google Scholar] [CrossRef]
- Rocha, M.E.M.; Dutra, F.; Bandy, B.; Baldini, R.L.; Gomes, S.L.; Faljoni-Alário, A.; Liria, C.W.; Miranda, M.T.M.; Bechara, E.J.H. Oxidative damage to ferritin by 5-aminolevulinic acid. Arch. Biochem. Biophys. 2003, 409, 349–356. [Google Scholar] [CrossRef]
- Rocha, M.E.M.; Bandy, B.; Costa, C.A.; de Barros, M.P.; Pinto, A.M.; Bechara, E.J. Iron mobilization by succinylacetone methyl ester in rats. A model study for hereditary tyrosinemia and porphyrias characterized by 5-Aminolevulinic acid overload. Free Radic. Res. 2000, 32, 343–353. [Google Scholar] [CrossRef]
- Demasi, M.; Penatti, C.A.; Delucia, R.; Bechara, E.J. The prooxidant effect of 5-aminolevulinic acid in the brain tissue of rats: Implications in neuropsychiatric manifestations in porphyrias. Free Radic. Biol. Med. 1996, 20, 291–299. [Google Scholar] [CrossRef]
- Carvalho, H.; Bechara, E.J.H.; Meneghini, R.; Demasi, M. Haem precursor δ-aminolaevulinic acid induces activation of the cytosolic iron regulatory protein 1. Biochem. J. 1997, 328, 827–832. [Google Scholar] [CrossRef]
- Handschin, C.; Lin, J.; Rhee, J.; Peyer, A.K.; Chin, S.; Wu, P.H.; Meyer, U.A.; Spiegelman, B.M. Nutritional Regulation of Hepatic Heme Biosynthesis and Porphyria through PGC-1α. Cell 2005, 122, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, C.; Montosi, G.; Garuti, C.; Corradini, E.; Sabelli, M.; Canali, S.; Pietrangelo, A. Gluconeogenic signals regulate iron homeostasis via hepcidin in mice. Gastroenterology 2014, 146, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Ventura, P.; Bonkovsky, H.L.; Gouya, L.; Aguilera-Peiró, P.; Montgomery Bissell, D.; Stein, P.E.; Balwani, M.; Anderson, D.K.E.; Parker, C.; Kuter, D.J.; et al. Efficacy and safety of givosiran for acute hepatic porphyria: 24-month interim analysis of the randomized phase 3 ENVISION study. Liver Int. 2022, 42, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Ventura, P. Givosiran for the treatment of acute hepatic porphyria. Expert Rev. Clin. Pharmacol. 2022, 15, 383–393. [Google Scholar] [CrossRef]
- To-Figueras, J.; Lopez, R.M.; Deulofeu, R.; Herrero, C. Preliminary report: Hyperhomocysteinemia in patients with acute intermittent porphyria. Metabolism 2010, 59, 1809–1810. [Google Scholar] [CrossRef]
- Ventura, P.; Corradini, E.; Di Pierro, E.; Marchini, S.; Marcacci, M.; Cuoghi, C.; Buzzetti, E.; Pietrangelo, A. Hyperhomocysteinemia in patients with acute porphyrias: A potentially dangerous metabolic crossroad? Eur. J. Intern. Med. 2020, 79, 101–107. [Google Scholar] [CrossRef]
- Fontanellas, A.; Ávila, M.A.; Arranz, E.; de Salamanca, R.E.; Morales-Conejo, M. Acute intermittent porphyria, givosiran, and homocysteine. J. Inherit. Metab. Dis. 2021, 44, 790. [Google Scholar] [CrossRef]
- To-Figueras, J.; Wijngaard, R.; García-Villoria, J.; Aarsand, A.K.; Aguilera, P.; Deulofeu, R.; Brunet, M.; Gómez-Gómez, À.; Pozo, O.J.; Sandberg, S. Dysregulation of homocysteine homeostasis in acute intermittent porphyria patients receiving heme arginate or givosiran. J. Inherit. Metab. Dis. 2021, 44, 961–971. [Google Scholar] [CrossRef]
- Petrides, P.E.; Klein, M.; Schuhmann, E.; Torkler, H.; Molitor, B.; Loehr, C.; Obermeier, Z.; Beykirch, M.K. Severe homocysteinemia in two givosiran-treated porphyria patients: Is free heme deficiency the culprit? Ann. Hematol. 2021, 100, 1685–1693. [Google Scholar] [CrossRef]
- Vassiliou, D.; Sardh, E. Homocysteine elevation in givosiran treatment: Suggested ALAS1 siRNA effect on cystathionine beta-synthase. J. Intern. Med. 2021, 290, 928–930. [Google Scholar] [CrossRef]
- Ricci, A.; Marcacci, M.; Cuoghi, C.; Pietrangelo, A.; Ventura, P. Hyperhomocysteinemia in patients with acute porphyrias: A possible effect of ALAS1 modulation by siRNAm therapy and its control by vitamin supplementation. Eur. J. Intern. Med. 2021, 92, 121–123. [Google Scholar] [CrossRef]
- Bins, S.; Sardh, E.; Langendonk, J.G. Givosiran Likely Inhibits Cytochrome P450 More Substantially Than Reported. Clin. Pharmacol. Ther. 2022, 112, 24. [Google Scholar] [CrossRef]
- Lazareth, H.; Poli, A.; Bignon, Y.; Mirmiran, A.; Rabant, M.; Schmitt, C.; Puy, H.; Karras, A.; Gouya, L.; Pallet, N.; et al. Renal function decline with small interfering RNA silencing ALAS1 for Acute Intermittent Porphyria. Kidney Int. Rep. 2021, 6, 1904–1911. [Google Scholar] [CrossRef]
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood J. Am. Soc. Hematol. 2019, 133, 59–69. [Google Scholar] [CrossRef]
- Abu-Zeinah, G.; DeSancho, M.T. Understanding sideroblastic anemia: An overview of genetics, epidemiology, pathophysiology and current therapeutic options. J. Blood Med. 2020, 11, 305. [Google Scholar] [CrossRef]
- Chiabrando, D.; Bertino, F.; Tolosano, E. Hereditary ataxia: A focus on heme metabolism and fe-s cluster biogenesis. Int. J. Mol. Sci. 2020, 21, 3760. [Google Scholar] [CrossRef]
- Bergmann, A.K.; Campagna, D.R.; McLoughlin, E.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer 2010, 54, 273–278. [Google Scholar] [CrossRef]
- Camaschella, C. Recent advances in the understanding of inherited sideroblastic anaemia. Br. J. Haematol. 2008, 143, 27–38. [Google Scholar] [CrossRef]
- Furuyama, K.; Kaneko, K. Iron metabolism in erythroid cells and patients with congenital sideroblastic anemia. Int. J. Hematol. 2018, 107, 44–54. [Google Scholar] [CrossRef]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood J. Am. Soc. Hematol. 2003, 101, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Cooley, T.B. A severe type of Hereditary Anemia with elliptocytosis-interisting sequence of splecectomy. Am. J. Med. Sci. 1945, 209, 561–568. [Google Scholar] [CrossRef]
- Ducamp, S.; Kannengiesser, C.; Touati, M.; Garçon, L.; Guerci-Bresler, A.; Guichard, J.F.; Vermylen, C.; Dochir, J.; Poirel, H.A.; Fouyssac, F.; et al. Sideroblastic anemia: Molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum. Mutat. 2011, 32, 590–597. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Clin. 2014, 28, 653–670. [Google Scholar]
- Campagna, D.R.; de Bie, C.I.; Schmitz-Abe, K.; Sweeney, M.; Sendamarai, A.K.; Schmidt, P.J.; Heeney, M.M.; Yntema, H.G.; Kannengiesser, C.; Grandchamp, B.; et al. X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am. J. Hematol. 2014, 89, 315–319. [Google Scholar] [CrossRef]
- Kaneko, K.; Furuyama, K.; Fujiwara, T.; Kobayashi, R.; Ishida, H.; Harigae, H.; Shibahara, S. Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica 2014, 99, 252. [Google Scholar] [CrossRef]
- Cazzola, M.; May, A.; Bergamaschi, G.; Cerani, P.; Rosti, V.; Bishop, D.F. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood J. Am. Soc. Hematol. 2000, 96, 4363–4365. [Google Scholar]
- Aivado, M.; Gattermann, N.; Rong, A.; Giagounidis, A.A.; Prall, W.C.; Czibere, A.; Hildebrandt, B.; Haas, R.; Bottomley, S.S. X-linked sideroblastic anemia associated with a novel ALAS2 mutation and unfortunate skewed X-chromosome inactivation patterns. Blood Cells Mol. Dis. 2006, 37, 40–45. [Google Scholar] [CrossRef]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood J. Am. Soc. Hematol. 2000, 96, 3256–3264. [Google Scholar]
- Heeney, M.M.; Berhe, S.; Campagna, D.R.; Oved, J.H.; Kurre, P.; Shaw, P.J.; Teo, J.; Shanap, M.A.; Hassab, H.M.; Glader, B.E.; et al. SLC25A38 congenital sideroblastic anemia: Phenotypes and genotypes of 31 individuals from 24 families, including 11 novel mutations, and a review of the literature. Hum. Mutat. 2021, 42, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Guo, S.; Kang, H.; Zhang, F.; Hu, Y.; Wang, L.; Li, M.; Ru, Y.; Camaschella, C.; Han, B.; et al. Mutation spectrum in Chinese patients affected by congenital sideroblastic anemia and a search for a genotype-phenotype relationship. Haematologica 2013, 98, e158. [Google Scholar] [CrossRef] [PubMed]
- Casas, K.A.; Fischel-Ghodsian, N. Mitochondrial myopathy and sideroblastic anemia. Am. J. Med. Genet. Part A 2004, 125, 201–204. [Google Scholar] [CrossRef]
- Camaschella, C.; Campanella, A.; De Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood J. Am. Soc. Hematol. 2007, 110, 1353–1358. [Google Scholar] [CrossRef]
- Liu, G.; Guo, S.; Anderson, G.J.; Camaschella, C.; Han, B.; Nie, G. Heterozygous missense mutations in the GLRX5 gene cause sideroblastic anemia in a Chinese patient. Blood J. Am. Soc. Hematol. 2014, 124, 2750–2751. [Google Scholar] [CrossRef]
- Daher, R.; Mansouri, A.; Martelli, A.; Bayart, S.; Manceau, H.; Callebaut, I.; Moulouel, B.; Gouya, L.; Puy, H.; Kannengiesser, C.; et al. GLRX5 mutations impair heme biosynthetic enzymes ALA synthase 2 and ferrochelatase in Human congenital sideroblastic anemia. Mol. Genet. Metab. 2019, 128, 342–351. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A.; et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761. [Google Scholar] [CrossRef]
- Schmitz-Abe, K.; Ciesielski, S.J.; Schmidt, P.J.; Campagna, D.R.; Rahimov, F.; Schilke, B.A.; Cuijpers, M.; Rieneck, K.; Lausen, B.; Linenberger, M.L.; et al. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood J. Am. Soc. Hematol. 2015, 126, 2734–2738. [Google Scholar] [CrossRef]
- Crispin, A.; Schmidt, P.; Campagna, D.; Cao, C.; Lichtenstein, D.; Sendamarai, A.; Guo, C.; Chen, C.; Hildick-Smith, G.J.; Huston, N.C.; et al. Hscb, a mitochondrial iron-sulfur cluster assembly co-chaperone, is a novel candidate gene for congenital sideroblastic anemia. Blood 2017, 130, 79. [Google Scholar] [CrossRef]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmidt, B.; et al. Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Donker, A.E.; Raymakers, R.A.; Vlasveld, L.T.; van Barneveld, T.; Terink, R.; Dors, N.; Brons, P.P.; Knoers, N.V.; Swinkels, D.W. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood J. Am. Soc. Hematol. 2014, 123, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Grandchamp, B.; Hetet, G.; Kannengiesser, C.; Oudin, C.; Beaumont, C.; Rodrigues-Ferreira, S.; Amson, R.; Telerman, A.; Nielsen, P.; Kohne, E.; et al. A novel type of congenital hypochromic anemia associated with a nonsense mutation in the STEAP3/TSAP6 gene. Blood J. Am. Soc. Hematol. 2011, 118, 6660–6666. [Google Scholar] [CrossRef]
- Romero-Cortadellas, L.; Hernández, G.; Ferrer-Cortès, X.; Zalba-Jadraque, L.; Fuster, J.L.; Bermúdez-Cortés, M.; Galera-Miñarro, A.M.; Pérez-Montero, S.; Tornador, C.; Sánchez, M. New Cases of Hypochromic Microcytic Anemia Due to Mutations in the SLC11A2 Gene and Functional Characterization of the G75R Mutation. Int. J. Mol. Sci. 2022, 23, 4406. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Trenor, C.C.; Su, M.A.; Foernzler, D.; Beier, D.R.; Dietrich, W.F.; Andrews, N.C. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat. Genet. 1997, 16, 383–386. [Google Scholar] [CrossRef]
- Iolascon, A.; d’Apolito, M.; Servedio, V.; Cimmino, F.; Piga, A.; Camaschella, C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2). Blood 2006, 107, 349–354. [Google Scholar] [CrossRef]
- Iolascon, A.; De Falco, L.; Beaumont, C. Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme synthesis. Haematologica 2009, 94, 395. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, A.; Di Betto, G.; Bergamini, E.; Buzzetti, E.; Corradini, E.; Ventura, P. Iron Metabolism in the Disorders of Heme Biosynthesis. Metabolites 2022, 12, 819. https://doi.org/10.3390/metabo12090819
Ricci A, Di Betto G, Bergamini E, Buzzetti E, Corradini E, Ventura P. Iron Metabolism in the Disorders of Heme Biosynthesis. Metabolites. 2022; 12(9):819. https://doi.org/10.3390/metabo12090819
Chicago/Turabian StyleRicci, Andrea, Giada Di Betto, Elisa Bergamini, Elena Buzzetti, Elena Corradini, and Paolo Ventura. 2022. "Iron Metabolism in the Disorders of Heme Biosynthesis" Metabolites 12, no. 9: 819. https://doi.org/10.3390/metabo12090819