
An Improved Method to Quantify Short-Chain Fatty Acids in Biological Samples Using Gas Chromatography–Mass Spectrometry



Abstract
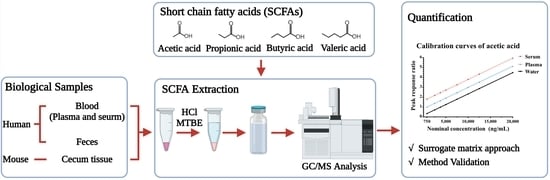
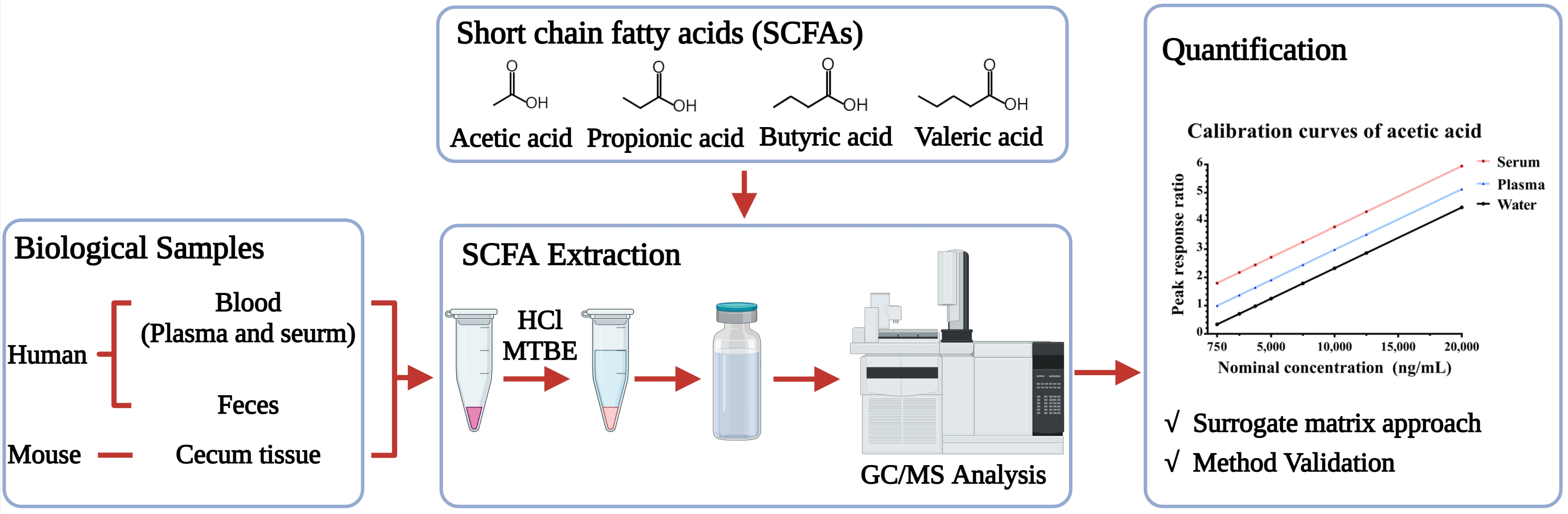
:
1. Introduction
2. Results and Discussion
2.1. Method Optimization
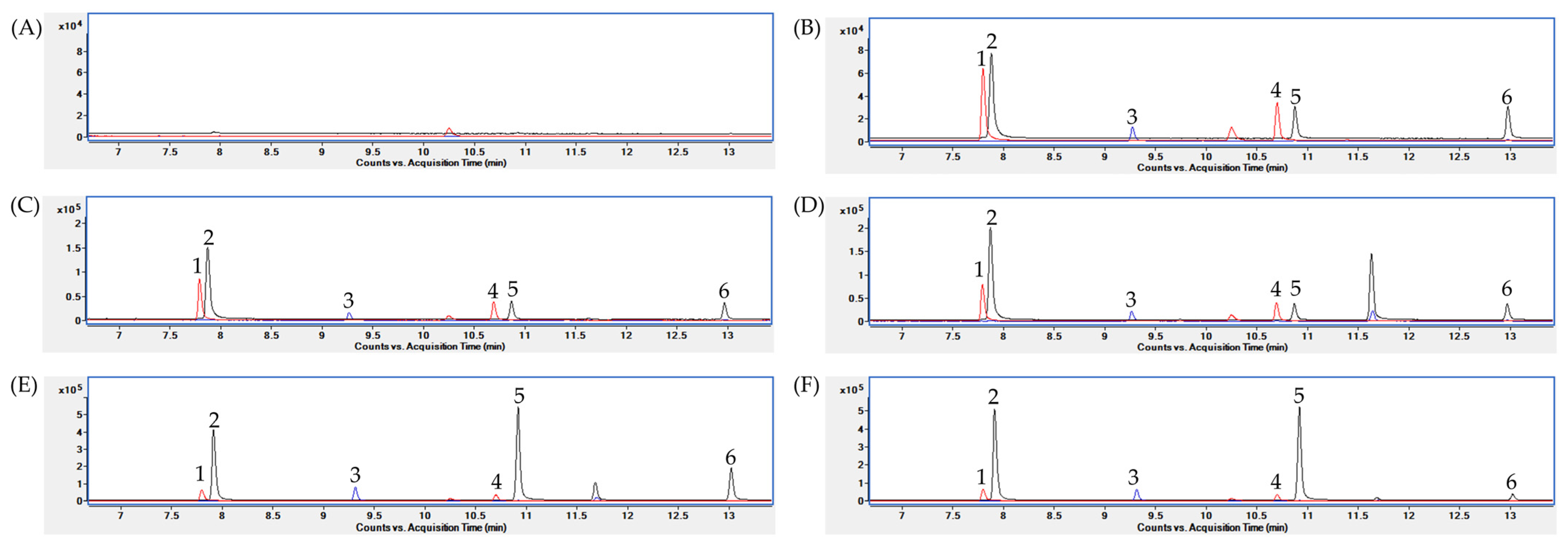
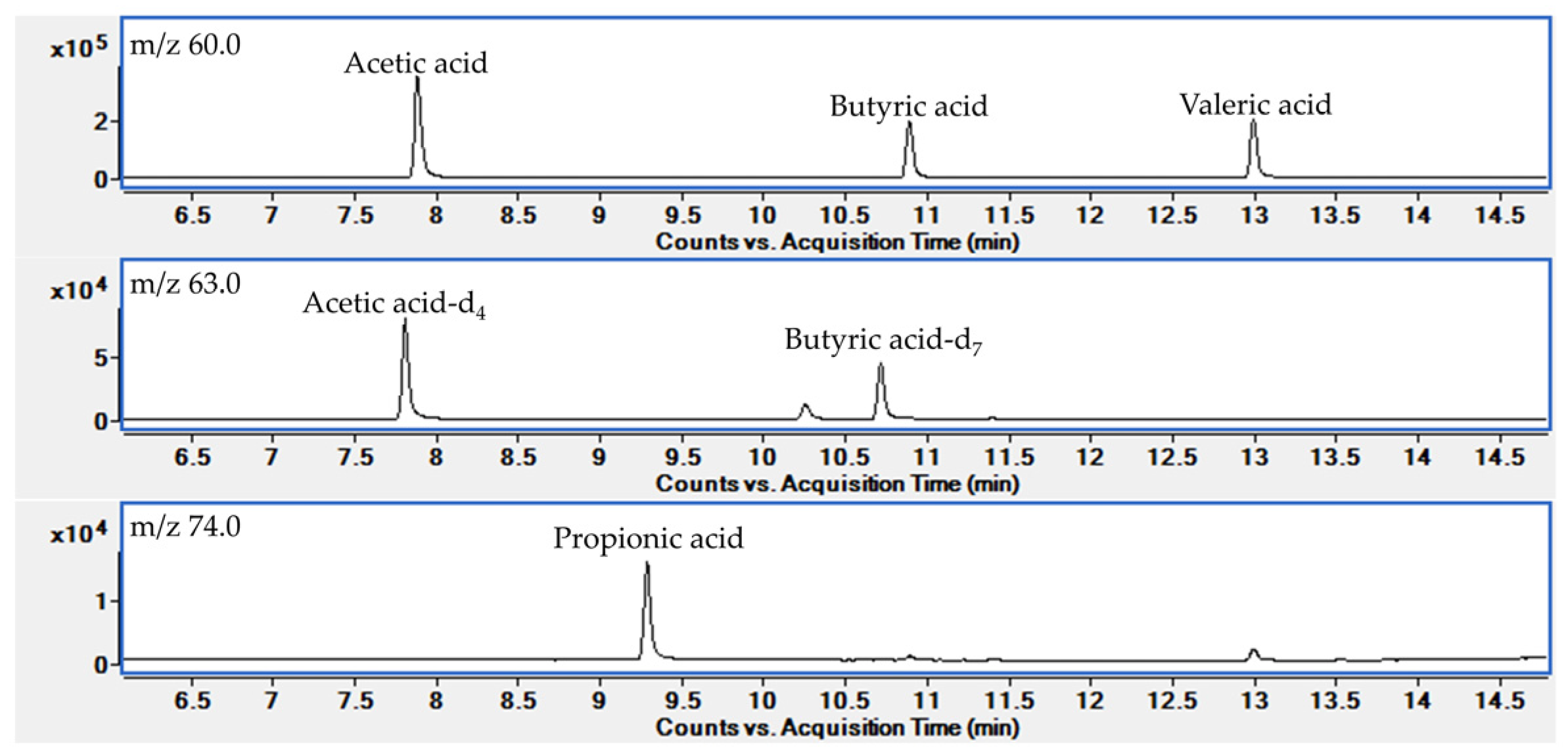
2.1.1. GC/MS Condition
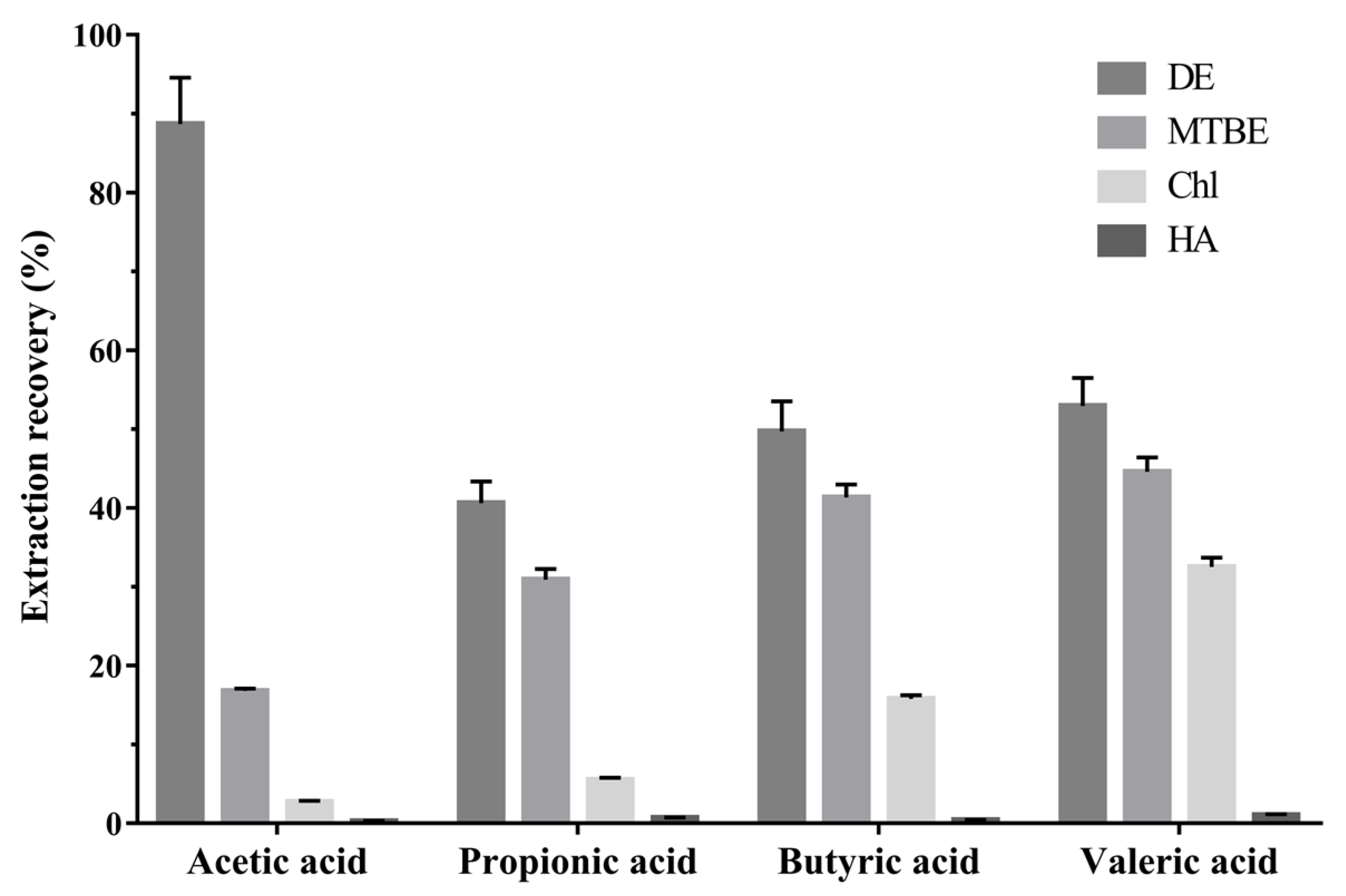
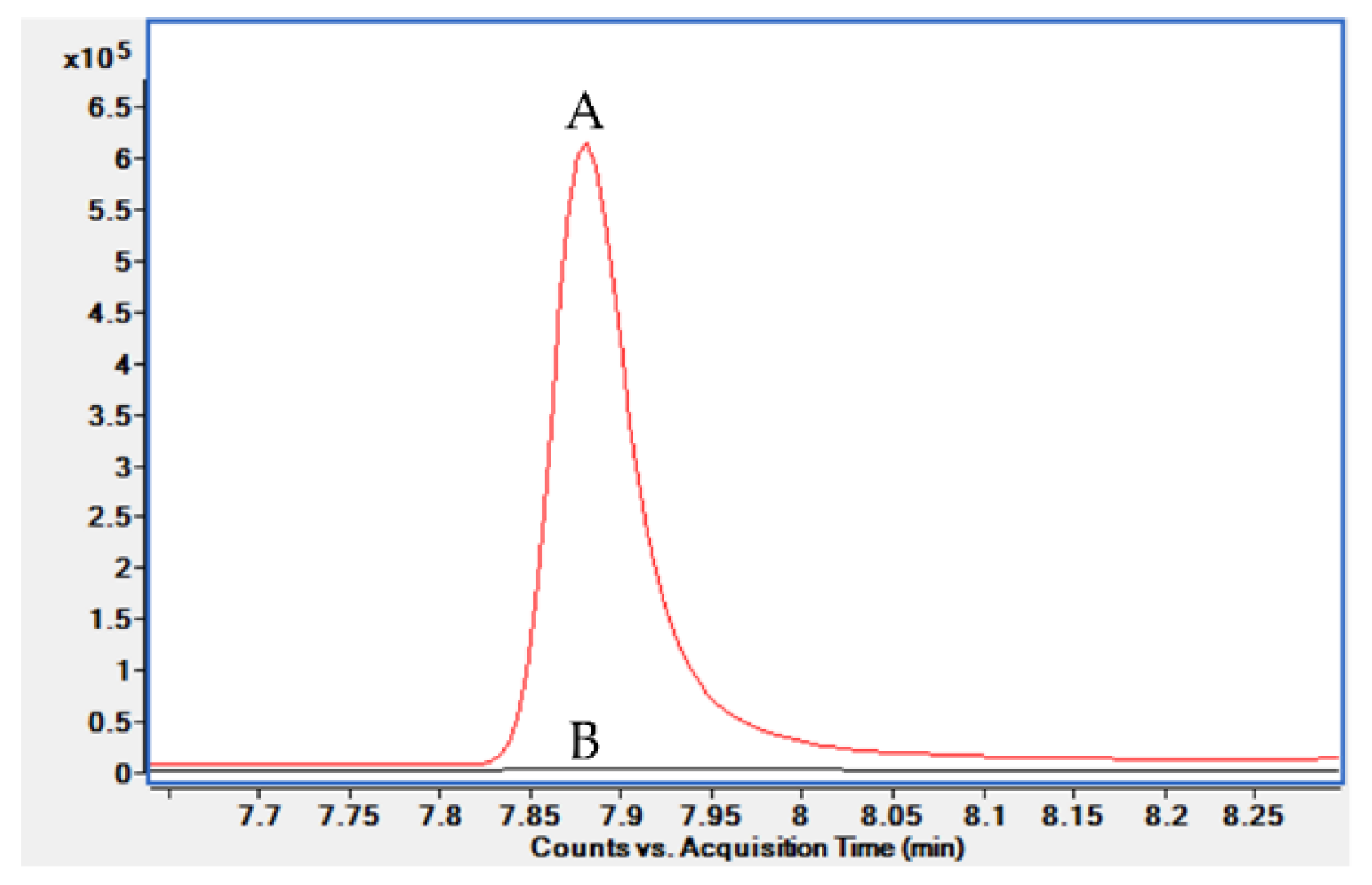
2.1.2. Extraction Condition Optimization
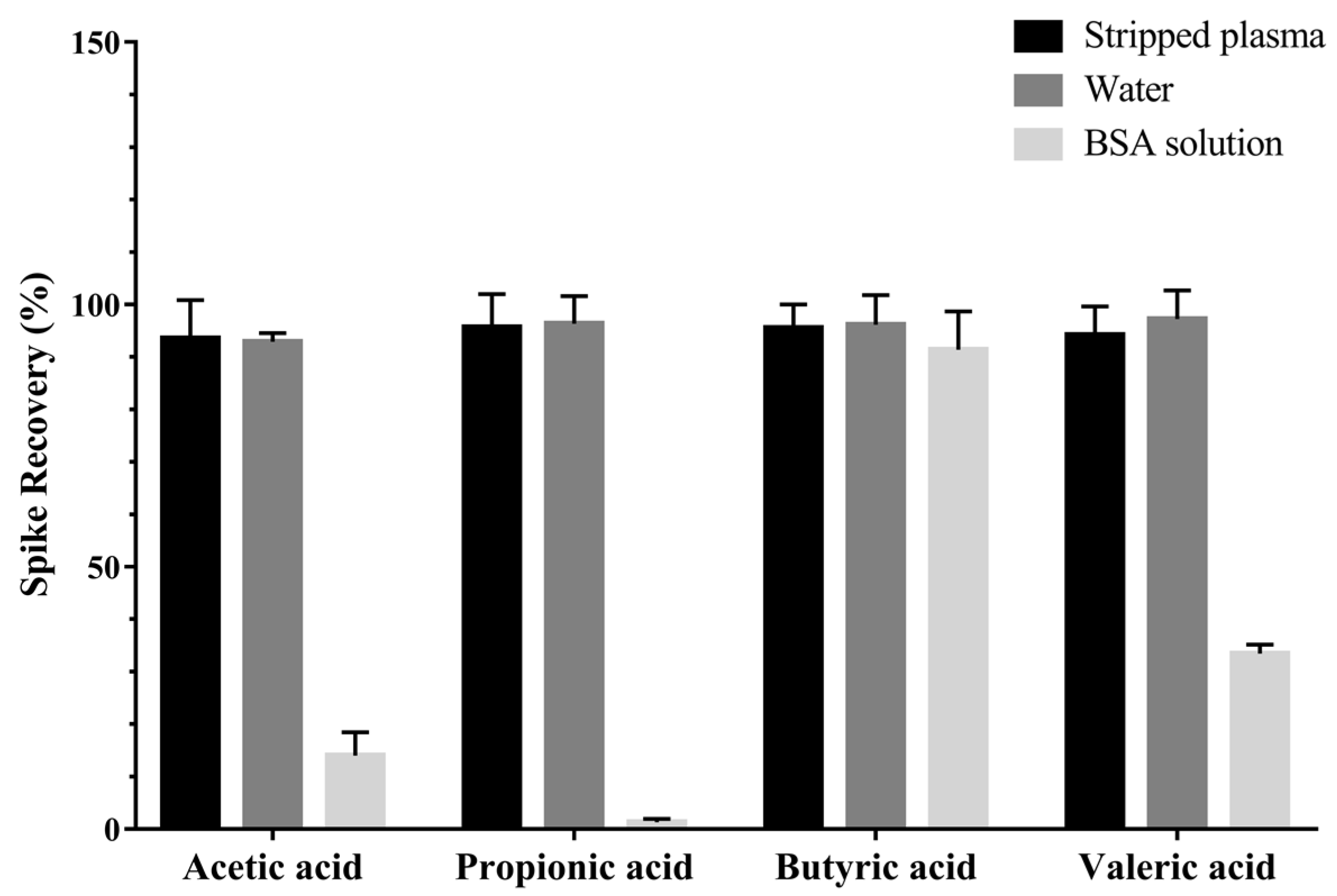
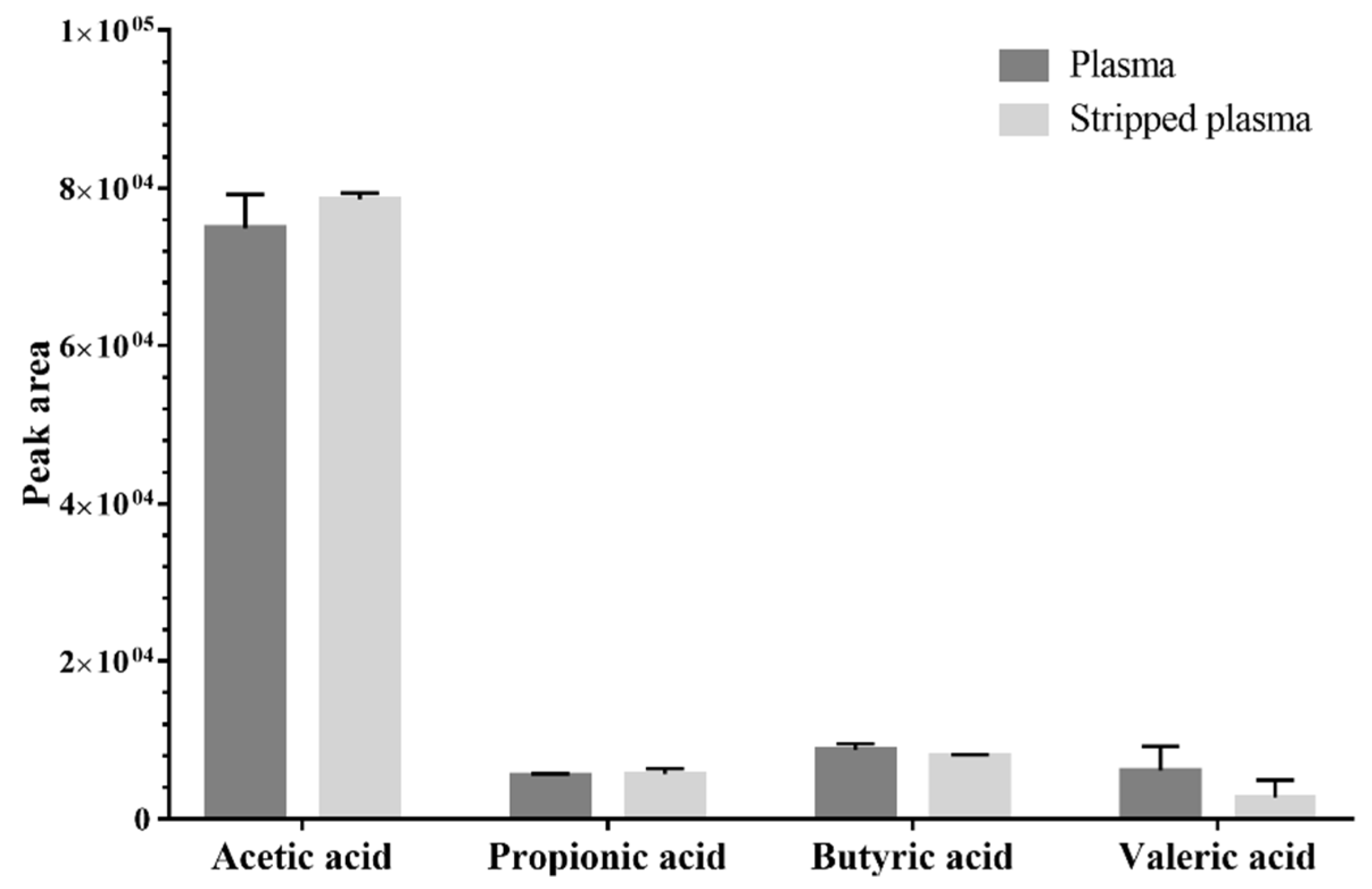
2.1.3. Surrogate Matrix Selection
2.2. Method Validation
2.2.1. Calibration Curve and Precision
2.2.2. Parallelism
2.2.3. Recovery and Matrix Effect
2.3. Quantification of the SCFAs in the Biological Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of Standard Solutions
3.3. Biological Sample Preparation
3.4. Extraction Procedure
3.5. Extraction Recovery
3.6. Surrogate Matrix Selection
3.7. GC/MS Analysis
3.8. Method Validation
3.8.1. Calibration Curve, Linearity, and Precision
3.8.2. Parallelism
3.8.3. Recovery and Matrix Effect
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SCFAs | Serum | Feces | Cecum |
|---|---|---|---|
| Acetic acid | 92.78 ± 0.96 | 98.26 ± 1.21 | 100.27 ± 1.17 |
| Propionic acid | 96.04 ± 1.17 | 105.33 ± 1.87 | 105.77 ± 6.35 |
| Butyric acid | 100.08 ± 1.02 | 104.67 ± 3.64 | 105.16 ± 5.74 |
| Valeric acid | 97.09 ± 0.88 | 104.3 ± 5.05 | 108.29 ± 4.24 |
| SCFAs | Spiked Concentration Levels for Calibration Ranges of Plasma and Serum (µg/mL) | Spiked Concentration Levels for Calibration Ranges of Feces and Cecum Tissue (µg/mL) | ||||
|---|---|---|---|---|---|---|
| Low | Medium | High | Low | Medium | High | |
| Acetic acid | 2.25 | 8 | 15 | 60 | 250 | 375 |
| Propionic acid | 0.15 | 0.8 | 1.5 | 6 | 90 | 150 |
| Butyric acid | 0.075 | 0.8 | 1.5 | 6 | 90 | 150 |
| Valeric acid | 0.045 | 0.8 | 1.5 | 3 | 30 | 150 |
References
- McNeil, N.I.; Cummings, J.; James, W. Short chain fatty acid absorption by the human large intestine. Gut 1978, 19, 819–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Li, T.; Li, M.; Huang, S.; Qiu, Y.; Feng, R.; Zhang, S.; Chen, M.; Xiong, L.; Zeng, Z. Systematic Review and Meta-analysis: Short-Chain Fatty Acid Characterization in Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1751–1763. [Google Scholar] [CrossRef]
- Holmes, Z.C.; Silverman, J.D.; Dressman, H.K.; Wei, Z.; Dallow, E.P.; Armstrong, S.C.; Seed, P.C.; Rawls, J.F.; David, L.A. Short-Chain Fatty Acid Production by Gut Microbiota from Children with Obesity Differs According to Prebiotic Choice and Bacterial Community Composition. MBio 2020, 11, e00914-20. [Google Scholar] [CrossRef]
- Rahman, M.N.; Diantini, A.; Fattah, M.; Barliana, M.I.; Wijaya, A. A highly sensitive, simple, and fast gas chromatography–mass spectrometry method for the quantification of serum short-chain fatty acids and their potential features in central obesity. Anal. Bioanal. Chem. 2021, 413, 6837–6844. [Google Scholar] [CrossRef]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef]
- Remely, M.; Aumueller, E.; Merold, C.; Dworzak, S.; Hippe, B.; Zanner, J.; Pointner, A.; Brath, H.; Haslberger, A.G. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene 2014, 537, 85–92. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosser, E.C.; Piper, C.J.M.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851.e10. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Barone, M.; La Rosa, F.; Piancone, F.; Mendozzi, L.; d’Arma, A.; Rossi, V.; Pugnetti, L.; Roda, G.; et al. Alterations in Circulating Fatty Acid Are Associated With Gut Microbiota Dysbiosis and Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, H.; Zhu, M.J. A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta 2019, 196, 249–254. [Google Scholar] [CrossRef]
- Li, M.; Zhu, R.; Song, X.; Wang, Z.; Weng, H.; Liang, J. A sensitive method for the quantification of short-chain fatty acids by benzyl chloroformate derivatization combined with GC-MS. Analyst 2020, 145, 2692–2700. [Google Scholar] [CrossRef]
- He, L.; Prodhan, M.A.I.; Yuan, F.; Yin, X.; Lorkiewicz, P.K.; Wei, X.; Feng, W.; McClain, C.; Zhang, X. Simultaneous quantification of straight-chain and branched-chain short chain fatty acids by gas chromatography mass spectrometry. J. Chromatogr. B 2018, 1092, 359–367. [Google Scholar] [CrossRef]
- Zheng, X.; Qiu, Y.; Zhong, W.; Baxter, S.; Su, M.; Li, Q.; Xie, G.; Ore, B.M.; Qiao, S.; Spencer, M.D.; et al. A targeted metabolomic protocol for short-chain fatty acids and branched-chain amino acids. Metabolomics 2013, 9, 818–827. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Nyman, M.; Jonsson, J.A. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed. Chromatogr. 2006, 20, 674–682. [Google Scholar] [CrossRef]
- Wang, C.Y.; Chen, Y.W.; Tain, Y.L.; Chang, S.K.C.; Huang, L.T.; Hsieh, C.W.; Hou, C.Y. Fast quantification of short-chain fatty acids in rat plasma by gas chromatography. J. Food Sci. 2020, 85, 1932–1938. [Google Scholar] [CrossRef]
- Lotti, C.; Rubert, J.; Fava, F.; Tuohy, K.; Mattivi, F.; Vrhovsek, U. Development of a fast and cost-effective gas chromatography-mass spectrometry method for the quantification of short-chain and medium-chain fatty acids in human biofluids. Anal. Bioanal. Chem. 2017, 409, 5555–5567. [Google Scholar] [CrossRef]
- Yamabe, S.; Tsuchida, N.; Hayashida, Y. Reaction paths of the water-assisted neutral hydrolysis of ethyl acetate. J. Phys. Chem. A 2005, 109, 7216–7224. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Nayak, J. Acetic acid production and purification: Critical review towards process intensification. Sep. Purif. Rev. 2017, 46, 44–61. [Google Scholar] [CrossRef]
- Jones, B.R.; Schultz, G.A.; Eckstein, J.A.; Ackermann, B.L. Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis 2012, 4, 2343–2356. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Ramanathan, R.; Horlbogen, L.; Mathialagan, S.; Costales, C.; Vourvahis, M.; Holliman, C.L.; Rodrigues, A.D. A multiplexed HILIC-MS/HRMS assay for the assessment of transporter inhibition biomarkers in phase i clinical trials: Isobutyryl-carnitine as an organic cation transporter (OCT1) biomarker. Anal. Chem. 2020, 92, 9745–9754. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation—Guidance for Industry. 2018. Available online: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (accessed on 3 April 2022).
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation Step 2b. 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf (accessed on 3 April 2022).
- Hurst, N.R.; Kendig, D.M.; Murthy, K.S.; Grider, J.R. The short chain fatty acids, butyrate and propionate, have differential effects on the motility of the guinea pig colon. Neurogastroenterol. Motil. 2014, 26, 1586–1596. [Google Scholar] [CrossRef]
- Wieling, J.; Dijkstra, H.; Mensink, C.; Jonkman, J.; Coenegracht, P.; Duineveld, C.; Doornbos, D. Chemometrics in bioanalytical sample preparation: A fractionated combined mixture and factorial design for the modelling of the recovery of five tricyclic amines from plasma after liquid—liquid extraction prior to high-performance liquid chromatography. J. Chromatogr. A 1993, 629, 181–199. [Google Scholar] [CrossRef]
- Di Tommaso, S.; Rotureau, P.; Crescenzi, O.; Adamo, C. Oxidation mechanism of diethyl ether: A complex process for a simple molecule. Phys. Chem. Chem. Phys. 2011, 13, 14636–14645. [Google Scholar] [CrossRef]
- Garcia-Villalba, R.; Gimenez-Bastida, J.A.; Garcia-Conesa, M.T.; Tomas-Barberan, F.A.; Carlos Espin, J.; Larrosa, M. Alternative method for gas chromatography-mass spectrometry analysis of short-chain fatty acids in faecal samples. J. Sep. Sci. 2012, 35, 1906–1913. [Google Scholar] [CrossRef]
- Jaochico, A.; Sangaraju, D.; Shahidi-Latham, S.K. A rapid derivatization based LC–MS/MS method for quantitation of short chain fatty acids in human plasma and urine. Bioanalysis 2019, 11, 741–753. [Google Scholar] [CrossRef]
- Han, J.; Lin, K.; Sequeira, C.; Borchers, C.H. An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2015, 854, 86–94. [Google Scholar] [CrossRef]
- Laffin, M.; Fedorak, R.; Zalasky, A.; Park, H.; Gill, A.; Agrawal, A.; Keshteli, A.; Hotte, N.; Madsen, K.L. A high-sugar diet rapidly enhances susceptibility to colitis via depletion of luminal short-chain fatty acids in mice. Sci. Rep. 2019, 9, 12294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Pomare, E.; Branch, W.; Naylor, C.; Macfarlane, G. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.E.; Lee, H.Y.; Kim, S.J.; Back, S.H.; Yoo, H.J. A Facile Profiling Method of Short Chain Fatty Acids Using Liquid Chromatography-Mass Spectrometry. Metabolites 2019, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Validation of Analytical Procedures: Text and Methodology Q2 (R1). 1996. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf (accessed on 3 April 2022).


| Analytes | Retention Time (min) | Target Ion (m/z) | Confirmative Ion (m/z) |
|---|---|---|---|
| Acetic acid | 7.92 | 60.0 | 43.0 |
| Propionic acid | 9.32 | 74.0 | 45.0 |
| Butyric acid | 10.92 | 60.0 | 73.0 |
| Valeric acid | 13.02 | 60.0 | 73.0 |
| Acetic acid-d4 a | 7.84 | 63.0 | 46.0 |
| Butyric acid-d7 b | 10.74 | 63.0 | 77.0 |
| SCFAs | Calibration Range (µg/mL) | LOD (ng/mL) | LOQ (ng/mL) | Inter-Day Precision (n = 15, RSD%) | ||
|---|---|---|---|---|---|---|
| Low | Medium | High | ||||
| Acetic acid a | 0.75–20 | 176.15 | 528.45 | 6.22 | 5.13 | 5.02 |
| Propionic acid a | 0.05–2 | 9.35 | 28.06 | 6.18 | 6.96 | 8.18 |
| Butyric acid a | 0.025–2 | 6.66 | 19.99 | 4.44 | 2 | 3.26 |
| Valeric acid a | 0.015–2 | 4.22 | 12.67 | 9.76 | 6.10 | 8.2 |
| Acetic acid b | 20–500 | 159.69 | 526.98 | 1.92 | 1.35 | 1.51 |
| Propionic acid b | 2–200 | 225.99 | 745.77 | 3.82 | 4.03 | 2.72 |
| Butyric acid b | 2–200 | 31.27 | 103.19 | 4.5 | 4.91 | 3.29 |
| Valeric acid b | 1–200 | 42.69 | 140.88 | 4.46 | 5.63 | 6.11 |
| Matrix | SCFAs | Parallelism (n = 6) | |
|---|---|---|---|
| Slope (Water/Biological Matrix) | RE% of SCFA Concentration | ||
| Plasma | Acetic acid | 0.98 ± 0.02 | 6.59 ± 0.04 |
| Propionic acid | 1.02 ± 0.01 | 9.98 ± 0.12 | |
| Butyric acid | 0.99 ± 0.01 | 6.36 ± 0.04 | |
| Valeric acid | 0.97 ± 0.02 | 6.17 ± 0.03 | |
| Serum | Acetic acid | 0.98 ± 0.01 | 4.01 ± 3.36 |
| Propionic acid | 0.97 ± 0.02 | 6.89 ± 4.58 | |
| Butyric acid | 0.98 ± 0.01 | 3.73 ± 1.51 | |
| Valeric acid | 0.98 ± 0.02 | 6.43 ±3.33 | |
| Feces | Acetic acid | 0.99 ± 0.01 | 3.77 ± 4.12 |
| Propionic acid | 0.98 ± 0.02 | 2.84 ± 2.88 | |
| Butyric acid | 1 ± 0.02 | 3.57 ± 2.85 | |
| Valeric acid | 0.99 ± 0.01 | 3.43 ± 3.02 | |
| Cecum tissue | Acetic acid | 1.01 ± 0.02 | 3.87 ± 3.26 |
| Propionic acid | 1 ± 0.02 | 6.87 ± 4.62 | |
| Butyric acid | 1 ± 0.02 | 4.5 ± 3.14 | |
| Valeric acid | 1 ± 0.02 | 5.81 ± 4.76 | |
| Matrix | SCFAs | Recovery (%, n = 3) | Matrix Effect (%, n = 6) | ||||
|---|---|---|---|---|---|---|---|
| Low | Medium | High | Low | Medium | High | ||
| Plasma | Acetic acid | 102.12 ± 4.41 | 108.95 ± 2.06 | 103.62 ± 3 | 107.19 ± 6.42 | 99.73 ± 3.52 | 99.9 ± 1.81 |
| Propionic acid | 101.62 ± 4.23 | 109.32 ± 2.69 | 103.45 ± 2.82 | 98.26 ± 6.52 | 97.31 ± 6.06 | 102.51 ± 1.28 | |
| Butyric acid | 100.26 ± 3.68 | 106.69 ± 2.61 | 101.8 ± 2.29 | 107.14 ± 5.33 | 99.03 ± 3.29 | 100.68 ± 0.77 | |
| Valeric acid | 97.27 ± 4.21 | 105.84 ± 3.33 | 103.96 ± 2.67 | 105.19 ± 2.88 | 98.93 ± 1.96 | 102 ± 1.15 | |
| Serum | Acetic acid | 95.9 ± 3.45 | 98.98 ± 4.92 | 98.75 ± 2.48 | 101.64 ± 5.97 | 99.76 ± 2.48 | 100.18 ± 1.48 |
| Propionic acid | 97.66 ± 5.41 | 100.36 ± 4.28 | 98.61 ± 3.96 | 108.37 ± 2.97 | 100.29 ± 3.7 | 103.08 ± 1.26 | |
| Butyric acid | 95.65 ± 6.04 | 100.3 ± 3.92 | 98.88 ± 2.55 | 102.25 ± 1.59 | 101.88 ± 2.3 | 100.85 ± 0.36 | |
| Valeric acid | 96.14 ± 5.94 | 102.95 ± 3.58 | 100.07 ± 4.04 | 99.54 ± 1.56 | 100.46 ± 0.94 | 101.69 ± 1.7 | |
| Feces | Acetic acid | 97.7 ± 3.16 | 102.78 ± 4.15 | 107.58 ± 3.7 | 103.91 ± 2.36 | 99.54 ± 2.2 | 101.22 ± 1.11 |
| Propionic acid | 96.59 ± 4.38 | 102.26 ± 4.14 | 107.12 ± 3.91 | 107.78 ± 4.65 | 103.51 ± 5.89 | 97.65 ± 1.57 | |
| Butyric acid | 98.98 ± 2.36 | 102.09 ± 5.13 | 106.43 ± 3.97 | 105.29 ±5.1 | 98.92 ± 4.03 | 97.83 ± 1.54 | |
| Valeric acid | 98.77 ± 5.03 | 102.76 ± 4.46 | 106.6 ± 4.06 | 103.34 ± 2.42 | 99.94 ± 2.51 | 98.59 ± 1.21 | |
| Cecum tissue | Acetic acid | 100.7 ± 2.34 | 96.37 ± 4.72 | 99.47 ± 2.46 | 99.02 ± 2.89 | 97.18 ± 2.32 | 98.7 ± 2.53 |
| Propionic acid | 94.89 ± 5.4 | 95.35 ± 6.05 | 99.74 ± 2.74 | 102.69 ± 2.22 | 99.45 ± 1.12 | 98.5 ± 1.15 | |
| Butyric acid | 98.94 ± 4.26 | 95.98 ± 6.26 | 100.3 ± 2.95 | 103.29 ± 5.82 | 99.96 ± 2.56 | 98.34 ± 1.16 | |
| Valeric acid | 98.21 ± 6.26 | 96.14 ± 6.71 | 100.49 ± 2.88 | 99.67 ± 2.66 | 99.76 ± 1.53 | 99.06 ± 1.53 | |
| SCFAs | Plasma a (ng/mL) n = 10 | Serum (ng/mL) n = 20 | Feces (µg/g) n = 10 | Cecum Tissue (µg/g) n = 6 |
|---|---|---|---|---|
| Acetic acid | 1504.21–2906.72 (2077.55 ± 456.35) | 4788.13–8823.54 (6561.86 ± 1068.53) | 1251.97–4193.4 (2849.87 ± 1040.81) | 2262–4363.38 (2907.17 ± 691.22) |
| Propionic acid | 63.94–184.89 (97.57 ± 37.78) | 247.69–757.96 (516.93 ± 108.27) | 668.82–2398.44 (1406.92 ± 487.19) | 308.13–566.11 (399.31 ± 86.59) |
| Butyric acid | 25.5–63.61 (38.77 ± 13) | 45.01–105.76 (72.78 ± 17.65) | 342.08–1966.7 (1086.97 ± 514.29) | 865.21–1353.25 (1008 ± 168.88) |
| Valeric acid | 18.04–28.34 (22.52 ± 3.35) | 20.44–77.65 (38.26 ± 16.42) | 86.17–412.25 (199.91 ± 107.49) | 43.14–85.53 (65.96 ± 12.73) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-S.; Lee, Y.; Chae, W.; Cho, J.-Y. An Improved Method to Quantify Short-Chain Fatty Acids in Biological Samples Using Gas Chromatography–Mass Spectrometry. Metabolites 2022, 12, 525. https://doi.org/10.3390/metabo12060525
Kim K-S, Lee Y, Chae W, Cho J-Y. An Improved Method to Quantify Short-Chain Fatty Acids in Biological Samples Using Gas Chromatography–Mass Spectrometry. Metabolites. 2022; 12(6):525. https://doi.org/10.3390/metabo12060525
Chicago/Turabian StyleKim, Kyeong-Seog, Yujin Lee, Woori Chae, and Joo-Youn Cho. 2022. "An Improved Method to Quantify Short-Chain Fatty Acids in Biological Samples Using Gas Chromatography–Mass Spectrometry" Metabolites 12, no. 6: 525. https://doi.org/10.3390/metabo12060525