
Network Approaches to Integrate Analyses of Genetics and Metabolomics Data with Applications to Fetal Programming Studies

 , ,
, ,
Abstract
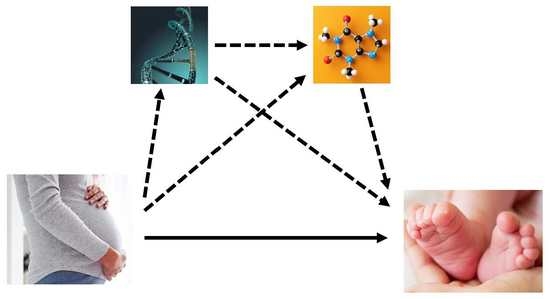
:
1. Introduction
2. Results
2.1. Significant Variant-to-Metabolite Associations
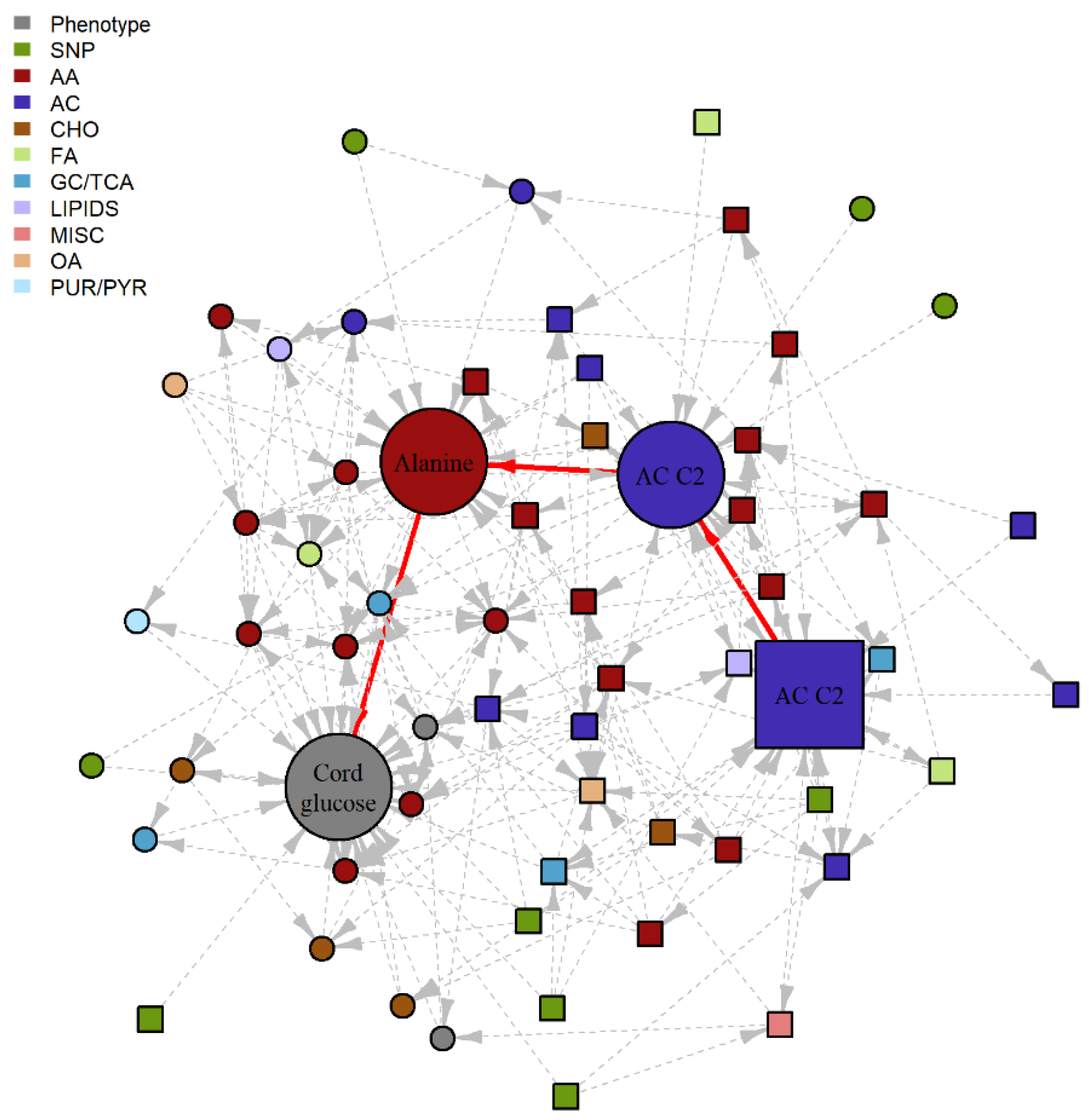
2.2. Bayesian Network Model (BNM)
2.3. Serial Mediation Model (SMM)
3. Discussion
4. Materials and Methods
4.1. Data Sets
4.1.1. Hyperglycemia and Adverse Pregnancy Outcome Study
4.1.2. Metabolomics Data
4.1.3. Genotype Data
4.1.4. Combined Genotype, Metabolomics and Phenotype Training and Validation Data Sets
4.2. Statistical Analyses
4.2.1. Variant-to-Metabolite Associations
4.2.2. Bayesian Network Modeling (BNM)
4.2.3. Serial Mediation Model (SMM)
4.2.4. Network Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, E.J.; Kim, Y.J. What is fetal programming? A lifetime health is under the control of in utero health. Obstet. Gynecol. Sci. 2017, 60, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. In utero programming of chronic disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- HAPO Study Cooperative Research Group. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HAPO Study Cooperative Research Group. Hyperglycaemia and Adverse Pregnancy Outcome (HAPO) Study: Associations with maternal body mass index. BJOG 2010, 117, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.G.; Urbanek, M.; Hivert, M.F.; Armstrong, L.L.; Morrison, J.; Guo, C.; Lowe, L.P.; Scheftner, D.A.; Pluzhnikov, A.; Levine, D.M.; et al. Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies. Diabetes 2013, 62, 3282–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Kuang, A.; Bain, J.R.; Muehlbauer, M.J.; Ilkayeva, O.R.; Lowe, L.P.; Metzger, B.E.; Newgard, C.B.; Scholtens, D.M.; Lowe, W.L. Maternal Metabolites Associated With Gestational Diabetes Mellitus and a Postpartum Disorder of Glucose Metabolism. J. Clin. Endocrinol. Metab. 2021, 106, 3283–3294. [Google Scholar] [CrossRef]
- Kadakia, R.; Nodzenski, M.; Talbot, O.; Kuang, A.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Ilkayeva, O.R.; O’Neal, S.K.; Lowe, L.P.; et al. Maternal metabolites during pregnancy are associated with newborn outcomes and hyperinsulinaemia across ancestries. Diabetologia 2019, 62, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.E.; Nodzenski, M.; Beaumont, R.N.; Talbot, O.; Shields, B.M.; Scholtens, D.M.; Knight, B.A.; Lowe, W.L., Jr.; Hattersley, A.T.; Freathy, R.M. Fetal Genotype and Maternal Glucose Have Independent and Additive Effects on Birth Weight. Diabetes 2018, 67, 1024–1029. [Google Scholar] [CrossRef] [Green Version]
- Urbanek, M.; Hayes, M.G.; Armstrong, L.L.; Morrison, J.; Lowe, L.P.; Badon, S.E.; Scheftner, D.A.; Pluzhnikov, A.; Levine, D.; Laurie, C.C.; et al. The chromosome 3q25 genomic region is associated with measures of adiposity in newborns in a multi-ethnic genome-wide association study. Hum. Mol. Genet. 2013, 22, 3583–3596. [Google Scholar] [CrossRef] [Green Version]
- Kadakia, R.; Scholtens, D.M.; Rouleau, G.W.; Talbot, O.; Ilkayeva, O.R.; George, T.; Josefson, J.L. Cord Blood Metabolites Associated with Newborn Adiposity and Hyperinsulinemia. J. Pediatr. 2018, 203, 144–149.e141. [Google Scholar] [CrossRef]
- Liu, Y.; Kuang, A.; Talbot, O.; Bain, J.R.; Muehlbauer, M.J.; Hayes, M.G.; Ilkayeva, O.R.; Lowe, L.P.; Metzger, B.E.; Newgard, C.B.; et al. Metabolomic and genetic associations with insulin resistance in pregnancy. Diabetologia 2020, 63, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N.; Linial, M.; Nachman, I.; Pe'er, D. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000, 7, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N.; Nachman, I.; Peer, D. Learning bayesian network structure from massive datasets: The Sparse Candidate algorithm. Proc. Fiftheenth Conf. Uncertain. Artif. Intell. 1999, 206–215. [Google Scholar] [CrossRef]
- Koch, M.; Broom, B.M.; Subramanian, D. Learning robust cell signalling models from high throughput proteomic data. Int. J. Bioinform. Res. Appl. 2009, 5, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Perez, D.; Lugo-Martinez, J.; Bourguignon, N.; Mathee, K.; Lerner, B.; Bar-Joseph, Z.; Narasimhan, G. Dynamic Bayesian Networks for Integrating Multi-omics Time Series Microbiome Data. mSystems 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, R.; Cheng, F.; Wei, Q.; Ji, Y.; Yang, H.; Zhong, X.; Tao, R.; Wen, Z.; Sutcliffe, J.S.; et al. A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data. Nat. Neurosci. 2019, 22, 691–699. [Google Scholar] [CrossRef]
- Aragram, B.; Zhou, Q. Concave Penalized Estimation of Sparse Gaussian Bayesian Networks. J. Mach. Learn. Res. 2015, 16, 2273–2328. [Google Scholar]
- Hartiala, J.A.; Wilson Tang, W.H.; Wang, Z.; Crow, A.L.; Stewart, A.F.R.; Roberts, R.; McPherson, R.; Erdmann, J.; Willenborg, C.; Hazen, S.L.; et al. Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat. Commun. 2016, 7, 10558. [Google Scholar] [CrossRef] [Green Version]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.-W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Suhre, K.; Shin, S.-Y.; Petersen, A.-K.; Mohney, R.P.; Meredith, D.; Wagele, B.; Altmaier, E.; CARDIoGRAM; Deloukas, P.; Erdmann, J.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. [Google Scholar] [CrossRef]
- Mittelstrass, K.; Ried, J.S.; Yu, Z.; Krumsiek, J.; Gieger, C.; Prehn, C.; Roemisch-Margl, W.; Polonikov, A.; Peters, A.; Theis, F.J.; et al. Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet. 2011, 7, e1002215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; Hrabe de Angelis, M.; Kronenberg, F.; Meitinger, T.; Mewes, H.-W.; Wichmann, H.-E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, C.; Petersen, A.-K.; Gieger, C.; Doring, A.; Illig, T.; Wang-Sattler, R.; Meisinger, C.; Peters, A.; Adamski, J.; Prehn, C.; et al. Body fat free mass is associated with the serum metabolite profile in a population-based study. PLoS ONE 2012, 7, e40009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffler, J.; Friedrich, N.; Arnold, M.; Kacprowski, T.; Rueedi, R.; Altmaier, E.; Bergmann, S.; Budde, K.; Gieger, C.; Homuth, G.; et al. Genome-wide association study with targeted and non-targeted NMR metabolomics identifies 15 novel loci of urinary human metabolic individuality. PLoS Genet. 2015, 11, e1005487. [Google Scholar] [CrossRef] [Green Version]
- Ried, J.S.; Shin, S.-Y.; Krumsiek, J.; Illig, T.; Theis, F.J.; Spector, T.D.; Adamski, J.; Wichmann, H.-E.; Strauch, K.; Soranzo, N.; et al. Novel genetic associations with serum level metabolites identified by phenotype set enrichment analyses. Hum. Mol. Genet. 2014, 23, 5847–5857. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-Y.; Fauman, E.B.; Petersen, A.-K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.-P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Raffler, J.; Pfeufer, A.; Suhre, K.; Kastenmuller, G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics 2015, 31, 1334–1336. [Google Scholar] [CrossRef]
- Kastenmuller, G.; Raffler, J.; Gieger, C.; Suhre, K. Genetics of human metabolism: An update. Hum. Mol. Genet. 2015, 24, R93–R101. [Google Scholar] [CrossRef] [Green Version]
- Feofanova, E.V.; Chen, H.; Dai, Y.; Jia, P.; Grove, M.L.; Morrison, A.C.; Qi, Q.; Daviglus, M.; Cai, J.; North, K.E.; et al. A Genome-wide Association Study Discovers 46 Loci of the Human Metabolome in the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 2020, 107, 849–863. [Google Scholar] [CrossRef]
- Herrera-Van Oostdam, A.S.; Salgado-Bustamante, M.; Lima-Rogel, V.; Oropeza-Valdez, J.J.; López, J.A.; Rodríguez, I.D.R.; Toro-Ortiz, J.C.; Herrera-Van Oostdam, D.A.; López-Hernández, Y.; Monárrez-Espino, J. Urinary Metabolomic Profile of Neonates Born to Women with Gestational Diabetes Mellitus. Metabolites 2021, 11, 723. [Google Scholar] [CrossRef]
- Procopciuc, L.M.; Caracostea, G.; Zaharie, G.; Stamatian, F. Newborn APOE genotype influences maternal lipid profile and the severity of high-risk pregnancy-preeclampsia: Interaction with maternal genotypes as a modulating risk factor in preeclampsia. Hypertens Pregnancy 2015, 34, 271–283. [Google Scholar] [CrossRef]
- Maria Procopciuc, L.; Caracostea, G.; Maria Hazi, G.; Nemeti, G.; Zaharie, G.; Stamatian, F. Maternal/fetal eNOS-Glu298Asp genotypes and their influence on the severity, prognosis, and lipid profile of preeclampsia. J. Matern.-Fetal Neonatal Med. 2018, 31, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, G.S.; Smye, M.; Sheridan, B.; Lappin, T.R.J.; Trimble, E.R.; HAPO Study Cooperative Research Group. Integration of local and central laboratory functions in a worldwide multicentre study: Experience from the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Clin. Trials 2006, 3, 397–407. [Google Scholar] [PubMed]
- HAPO Study Cooperative Research Group. The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study. Int. J. Gynaecol. Obstet. 2002, 78, 69–77. [Google Scholar] [CrossRef]
- HAPO Study Cooperative Research Group. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study: Associations with neonatal anthropometrics. Diabetes 2009, 58, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, S.; Nodzenski, M.; Reisetter, A.C.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Ilkayeva, O.R.; Lowe, L.P.; Metzger, B.E.; Newgard, C.B.; et al. Targeted Metabolomics Demonstrates Distinct and Overlapping Maternal Metabolites Associated With BMI, Glucose, and Insulin Sensitivity During Pregnancy Across Four Ancestry Groups. Diabetes Care 2017, 40, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadakia, R.; Talbot, O.; Kuang, A.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Ilkayeva, O.R.; Lowe, L.P.; Metzger, B.E.; Newgard, C.B.; et al. Cord Blood Metabolomics: Association With Newborn Anthropometrics and C-Peptide Across Ancestries. J. Clin. Endocrinol. Metab. 2019, 104, 4459–4472. [Google Scholar] [CrossRef]
- Scholtens, D.M.; Bain, J.R.; Reisetter, A.C.; Muehlbauer, M.J.; Nodzenski, M.; Stevens, R.D.; Ilkayeva, O.; Lowe, L.P.; Metzger, B.E.; Newgard, C.B.; et al. Metabolic Networks and Metabolites Underlie Associations Between Maternal Glucose During Pregnancy and Newborn Size at Birth. Diabetes 2016, 65, 2039–2050. [Google Scholar] [CrossRef] [Green Version]
- Halket, J.M.; Przyborowska, A.; Stein, S.E.; Mallard, W.G.; Down, S.; Chalmers, R.A. Deconvolution gas chromatography/mass spectrometry of urinary organic acids: Potential for pattern recognition and automated identification of metabolic disorders. Rapid Commun. Mass Spectrom. 1999, 13, 279–284. [Google Scholar] [CrossRef]
- AMDIS. Available online: http://www.amdis.net (accessed on 22 June 2021).
- Kind, T.; Wohlgemuth, G.; Lee Do, Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [Green Version]
- Nodzenski, M.; Muehlbauer, M.J.; Bain, J.R.; Reisetter, A.C.; Lowe, W.L., Jr.; Scholtens, D.M. Metabomxtr: An R package for mixture-model analysis of non-targeted metabolomics data. Bioinformatics 2014, 30, 3287–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisetter, A.C.; Muehlbauer, M.J.; Bain, J.R.; Nodzenski, M.; Stevens, R.D.; Ilkayeva, O.; Metzger, B.E.; Newgard, C.B.; Lowe, W.L., Jr.; Scholtens, D.M. Mixture model normalization for non-targeted gas chromatography/mass spectrometry metabolomics data. BMC Bioinform. 2017, 18, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrrell, J.; Richmond, R.C.; Palmer, T.M.; Feenstra, B.; Rangarajan, J.; Metrustry, S.; Cavadino, A.; Paternoster, L.; Armstrong, L.L.; De Silva, N.M.; et al. Genetic Evidence for Causal Relationships Between Maternal Obesity-Related Traits and Birth Weight. JAMA 2016, 315, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, M.C.; Agrawal, A.; Cole, J.W.; Hansel, N.N.; Barnes, K.C.; Beaty, T.H.; Bennett, S.N.; Bierut, L.J.; Boerwinkle, E.; Doheny, K.F.; et al. The Gene, Environment Association Studies consortium (GENEVA): Maximizing the knowledge obtained from GWAS by collaboration across studies of multiple conditions. Genet. Epidemiol. 2010, 34, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Rosseel, Y. lavaan: An R Package for Structural Equation Modeling. J. Stat. Softw. 2012, 48, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.F. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression-Based Approach; Guilford: New York, NY, USA, 2013. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bojanowski, M. Intergraph: Coercion Routines for Network Data Objects; R package version 2.0-2. 2015. Available online: http://mbojan.github.io/intergraph/ (accessed on 28 April 2022).
- Butts, C.T.; Leslie-Cook, A.; Krivitsky, P.N.; Bender-deMoll, S. NetworkDynamic: Dynamic Extensions for Network Objects; R package version 0.10.0. 2019. Available online: https://cran.r-project.org/web/packages/networkDynamic/index.html (accessed on 28 April 2022).
- Butts, C.T. network: A Package for Managing Relational Data in R. J. Stat. Softw. 2008, 24, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Butts, C.T. network: Classes for Relational Data. The Statnet Project; R package version 1.13.0.1. 2015. Available online: https://cran.r-project.org/web/packages/network/index.html (accessed on 28 April 2022).
- Bender-deMoll, S. ndtv: Network Dynamic Temporal Visualizations, R package version 0.13.0. 2019. Available online: https://cran.r-project.org/web/packages/ndtv/index.html (accessed on 28 April 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BNMs Involving Fasting Metabolites and Fasting Maternal Glucose | |||||
| Numbers of edges—N (% of total) | |||||
| Types of nodes in each edge pair | λ = 15 | λ = 10 | λ = 7 | λ = 2 | λ = 1 |
| MG-MG | 8 (6%) | 9 (4%) | 11 (3%) | 16 (1%) | 16 (1%) |
| MG-MM | 0 (0%) | 1 (0%) | 3 (1%) | 293 (10%) | 292 (10%) |
| MG-MP | 0 (0%) | 0 (0%) | 0 (0%) | 8 (0%) | 22 (1%) |
| MG-NG | 26 (19%) | 37 (15%) | 38 (9%) | 45 (2%) | 45 (1%) |
| MG-NM | 0 (0%) | 0 (0%) | 0 (0%) | 114 (4%) | 113 (4%) |
| MG-NP | 0 (0%) | 0 (0%) | 0 (0%) | 8 (0%) | 8 (0%) |
| MM-MM | 49 (36%) | 84 (34%) | 159 (37%) | 904 (31%) | 901 (29%) |
| MM-MP | 0 (0%) | 0 (0%) | 3 (1%) | 8 (0%) | 20 (1%) |
| MM-NM | 4 (3%) | 18 (7%) | 44 (10%) | 629 (22%) | 627 (20%) |
| MM-NP | 0 (0%) | 0 (0%) | 0 (0%) | 30 (1%) | 44 (1%) |
| MP-MM | 0 (0%) | 0 (0%) | 1 (0%) | 31 (1%) | 92 (3%) |
| MP-MP | 0 (0%) | 0 (0%) | 1 (0%) | 1 (0%) | 1 (0%) |
| MP-NM | 0 (0%) | 0 (0%) | 0 (0%) | 13 (0%) | 60 (2%) |
| MP-NP | 0 (0%) | 0 (0%) | 2 (0%) | 4 (0%) | 5 (0%) |
| NG-NG | 8 (6%) | 9 (4%) | 11 (3%) | 11 (0%) | 11 (0%) |
| NG-NM | 0 (0%) | 0 (0%) | 0 (0%) | 95 (3%) | 95 (3%) |
| NG-NP | 0 (0%) | 0 (0%) | 1 (0%) | 6 (0%) | 6 (0%) |
| NM-NM | 41 (30%) | 90 (36%) | 155 (36%) | 662 (23%) | 661 (22%) |
| NM-NP | 0 (0%) | 1 (0%) | 1 (0%) | 16 (1%) | 16 (1%) |
| NP-NM | 0 (0%) | 0 (0%) | 0 (0%) | 30 (1%) | 30 (1%) |
| NP-NP | 1 (0%) | 1 (0%) | 1 (0%) | 4 (0%) | 5 (0%) |
| λ = 15 most stringent penalty, λ = 1 least stringent penalty λ = 10 first occurrence of genotype to non-genotype edge λ = 7 first occurrence of maternal exposure phenotype to offspring outcome phenotype λ = 2 first occurrence of SMM pathway evaluated from maternal fasting AC C2 to cord glucose outcome | |||||
| BNMs involving 1-hr Metabolites and 1-hr Maternal Glucose | |||||
| Numbers of edges—N (% of total) | |||||
| Types of nodes in each edge pair | λ = 15 | λ = 9 | λ = 7 | λ = 3 | λ = 1 |
| MG-MG | 8 (6.06%) | 10 (3.38%) | 11 (2.54%) | 16 (1.08%) | 16 (0.53%) |
| MG-MM | 0 (0%) | 1 (0.34%) | 3 (0.69%) | 89 (6.02%) | 272 (8.97%) |
| MG-MP | 0 (0%) | 0 (0%) | 0 (0%) | 8 (0.54%) | 34 (1.12%) |
| MG-NG | 26 (19.7%) | 37 (12.5%) | 38 (8.78%) | 45 (3.04%) | 45 (1.48%) |
| MG-NM | 0 (0%) | 0 (0%) | 0 (0%) | 21 (1.42%) | 93 (3.07%) |
| MG-NP | 0 (0%) | 0 (0%) | 0 (0%) | 1 (0.07%) | 10 (0.33%) |
| MM-MM | 43 (32.57%) | 105 (35.47%) | 156 (36.03%) | 507 (34.28%) | 893 (29.44%) |
| MM-MP | 0 (0%) | 1 (0.33%) | 4 (0.92%) | 12 (0.81%) | 40 (1.32%) |
| MM-NM | 5 (3.79%) | 27 (9.12%) | 41 (9.47%) | 242 (16.36%) | 628 (20.71%) |
| MM-NP | 0 (0%) | 0 (0%) | 1 (0.23%) | 14 (0.95%) | 47 (1.55%) |
| MP-MM | 0 (0%) | 1 (0.34%) | 4 (0.93%) | 16 (1.08%) | 59 (1.95%) |
| MP-MP | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| MP-NM | 0 (0%) | 0 (0%) | 0 (0%) | 4 (0.27%) | 55 (1.81%) |
| MP-NP | 0 (0%) | 0 (0%) | 1 (0.23%) | 4 (0.27%) | 4 (0.13%) |
| NG-NG | 8 (6.06%) | 10 (3.38%) | 11 (2.54%) | 11 (0.74%) | 11 (0.36%) |
| NG-NM | 0 (0%) | 0 (0%) | 0 (0%) | 23 (1.56%) | 101 (3.33%) |
| NG-NP | 0 (0%) | 0 (0%) | 1 (0.23%) | 1 (0.07%) | 8 (0.26%) |
| NM-NM | 41 (31.06%) | 102 (34.46%) | 160 (36.95%) | 442 (29.88%) | 664 (21.89%) |
| NM-NP | 0 (0%) | 1 (0.34%) | 1 (0.23%) | 10 (0.68%) | 18 (0.59%) |
| NP-NM | 0 (0%) | 0 (0%) | 0 (0%) | 9 (0.61%) | 30 (0.99%) |
| NP-NP | 1 (0.76%) | 1 (0.34%) | 1 (0.23%) | 4 (0.27%) | 5 (0.17%) |
| λ = 15 most stringent penalty, λ = 1 least stringent penalty λ = 9 first occurrence of genotype to non-genotype edge λ = 7 first occurrence of maternal exposure phenotype to offspring outcome phenotype λ = 3 first occurrence of all SMM pathways involving maternal 1-hr metabolites MG = maternal genotype MM = maternal metabolite MP = maternal phenotype NG = newborn genotype NM = newborn metabolite NP = newborn phenotype | |||||
| PATHWAY MEMBERS | Training BNM | Validation Data |
|---|---|---|
| IDE (95% Confidence Interval); FDR-Adjusted P, PM (%) | IDE (95% Confidence Interval); Nominal P, PM (%) | |
| Maternal rs1171619 SLC16A9 -> Cord blood AC C2 -> Cord blood AC C16:1 -> Cord C-peptide | −1.60 × 10−2 (−2.42 × 10−2–−7.78 × 10−3); 0.0086, 25.51% | −1.36 × 10−2 (−2.41 × 10−2–−3.20 × 10−3); 0.0120, 173.98% |
| Maternal fasting AC C2 -> Cord blood AC C2 -> Cord blood Alanine -> Cord glucose | 1.26 × 10−2 (6.12 × 10−3–1.90 × 10−2); 0.0202, 129.82% | 1.21 × 10−2 (1.38 × 10−3–2.29 × 10−2); 0.0151, 24.17% |
| Maternal 1-hr AC C5 -> Maternal 1-hr AC C4/Ci4 -> Cord blood AC C4/Ci4 -> Cord blood AC C3 -> Cord glucose | −1.21 × 10−2 (−1.72 × 10−2–−6.94 × 10−3); 0.0018, 28.58% | −8.86 × 10−3 (−1.45 × 10−2–−3.23 × 10−3); 0.0101, 17.63% |
| Maternal 1-hr Tyrosine -> Cord blood Tyrosine -> Cord blood AC C4/Ci4 -> Cord blood AC C3 -> Cord glucose | −8.06 × 10−3 (−1.20 × 10−2–−4.13 × 10−3); 0.0053, 55.5% | −1.13 × 10−2 (−1.83 × 10−2–−4.35 × 10−3); 0.0067, 64.19% |
| Maternal 1-hr Tyrosine -> Cord blood Tyrosine -> Cord blood Phenylalanine -> Cord blood AC C3 -> Cord glucose | −7.55 × 10−3 (−1.16 × 10−2–−3.48 × 10−3); 0.0106, 52.02% | −8.04 × 10−3 (−1.41 × 10−2–−2.03 × 10−3); 0.0261, 45.56% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuang, A.; Hayes, M.G.; Hivert, M.-F.; Balasubramanian, R.; Lowe, W.L., Jr.; Scholtens, D.M. Network Approaches to Integrate Analyses of Genetics and Metabolomics Data with Applications to Fetal Programming Studies. Metabolites 2022, 12, 512. https://doi.org/10.3390/metabo12060512
Kuang A, Hayes MG, Hivert M-F, Balasubramanian R, Lowe WL Jr., Scholtens DM. Network Approaches to Integrate Analyses of Genetics and Metabolomics Data with Applications to Fetal Programming Studies. Metabolites. 2022; 12(6):512. https://doi.org/10.3390/metabo12060512
Chicago/Turabian StyleKuang, Alan, M. Geoffrey Hayes, Marie-France Hivert, Raji Balasubramanian, William L. Lowe, Jr., and Denise M. Scholtens. 2022. "Network Approaches to Integrate Analyses of Genetics and Metabolomics Data with Applications to Fetal Programming Studies" Metabolites 12, no. 6: 512. https://doi.org/10.3390/metabo12060512