
Attenuation of Olanzapine-Induced Endoplasmic Reticulum Stress Improves Insulin Secretion in Pancreatic Beta Cells

 , and
, and
Abstract
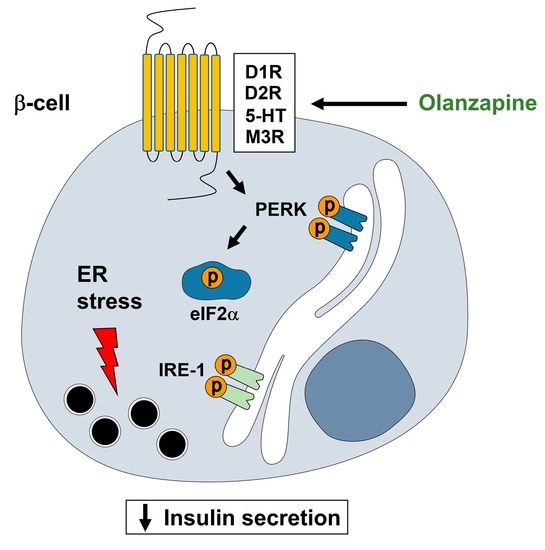
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
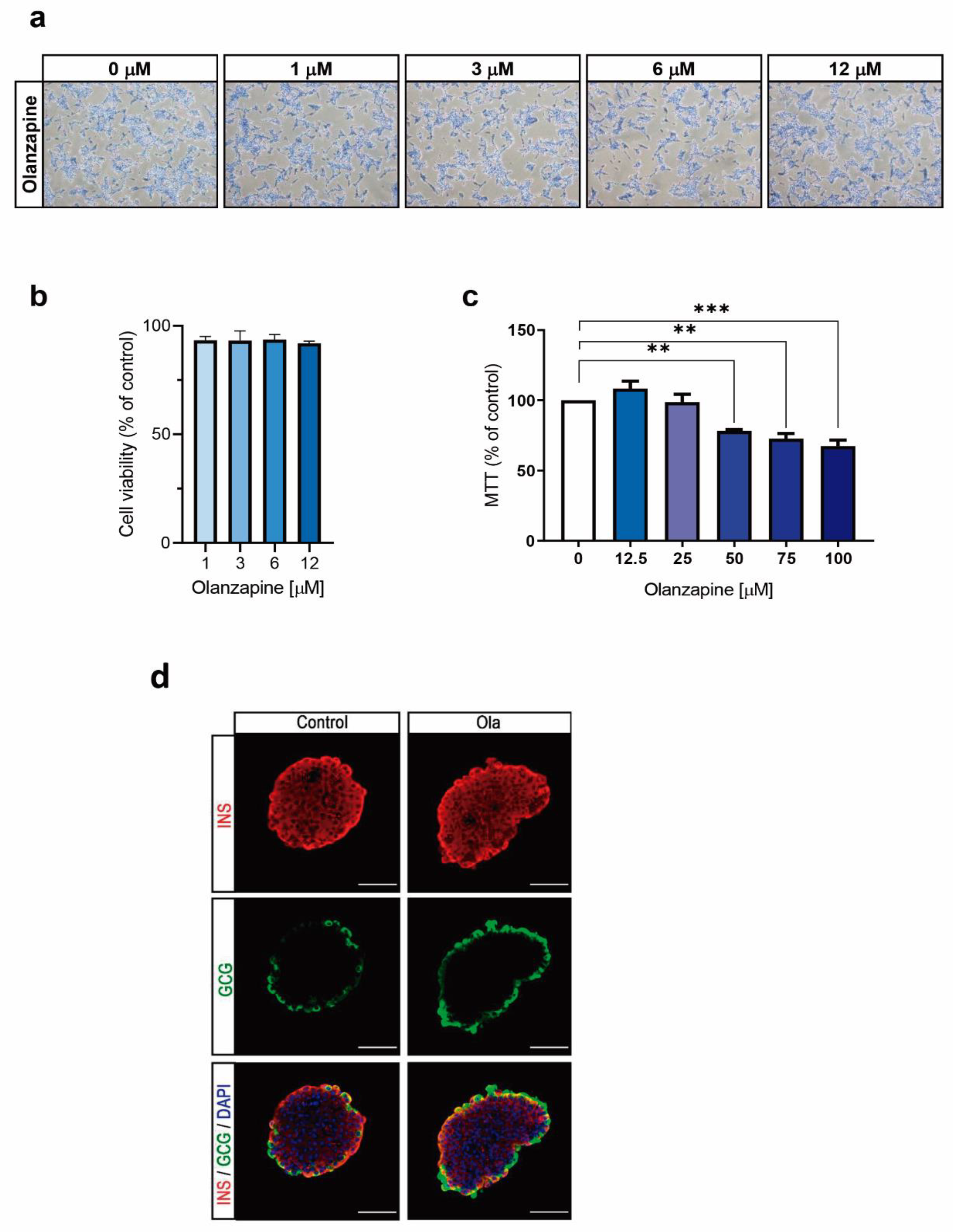
2.1. Treatment of INS-1 Beta Cells with Olanzapine Reduces Cell Viability in a Dose-Dependent Manner
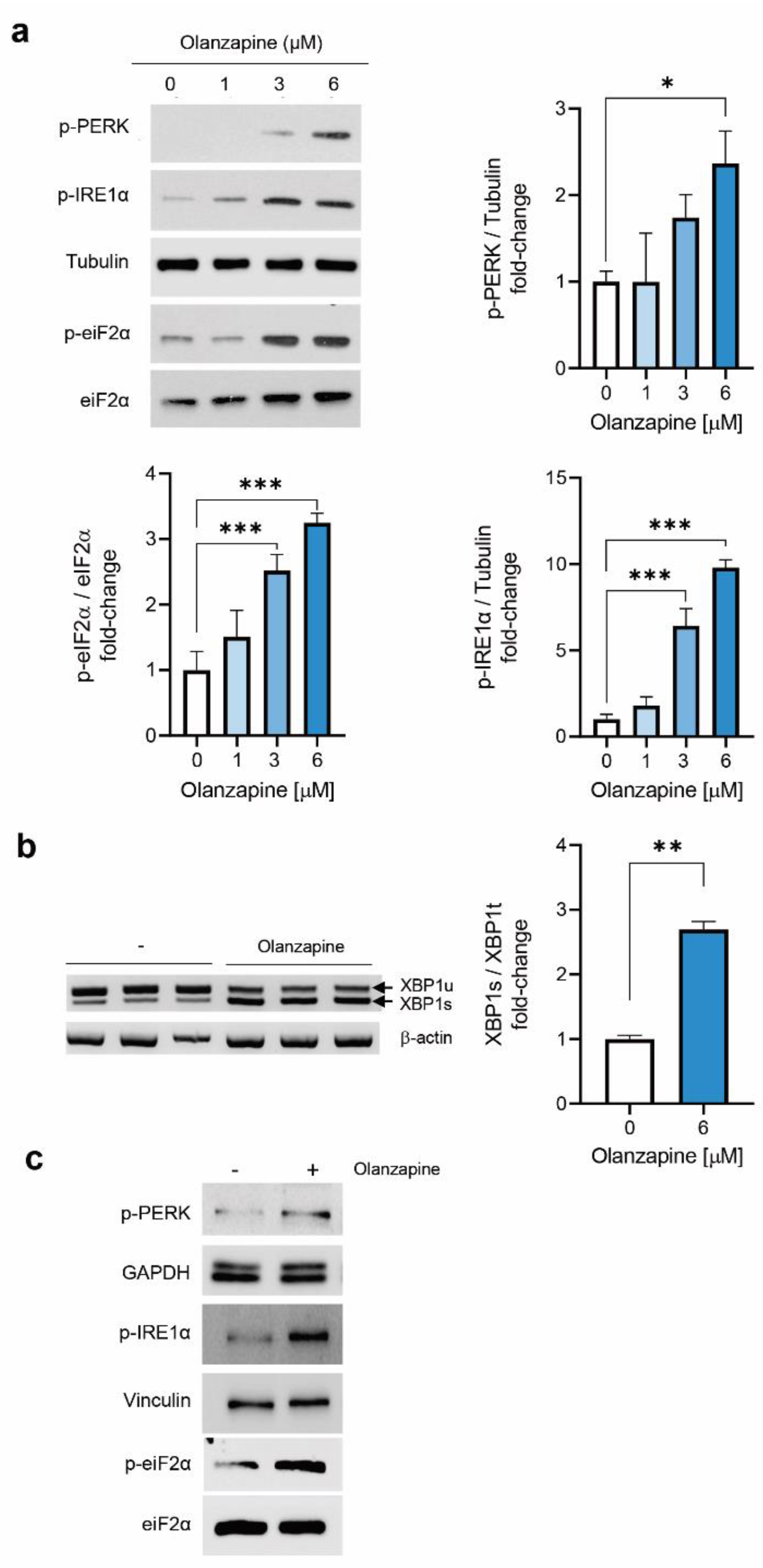
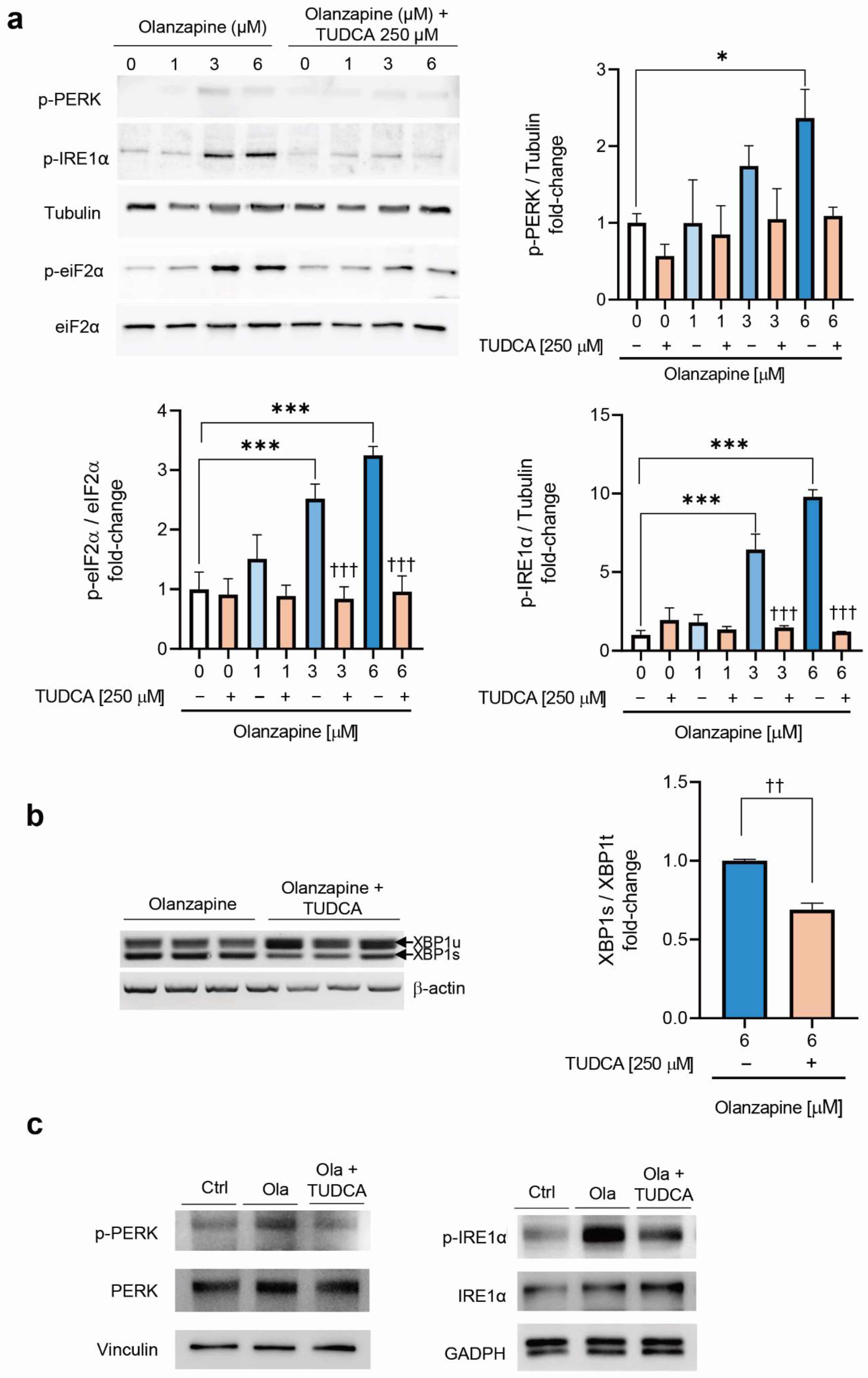
2.2. Short-Term Treatment with Olanzapine Induces Endoplasmic Reticulum (ER) Stress-Mediated Signaling in INS-1 Cells and Pancreatic Islets
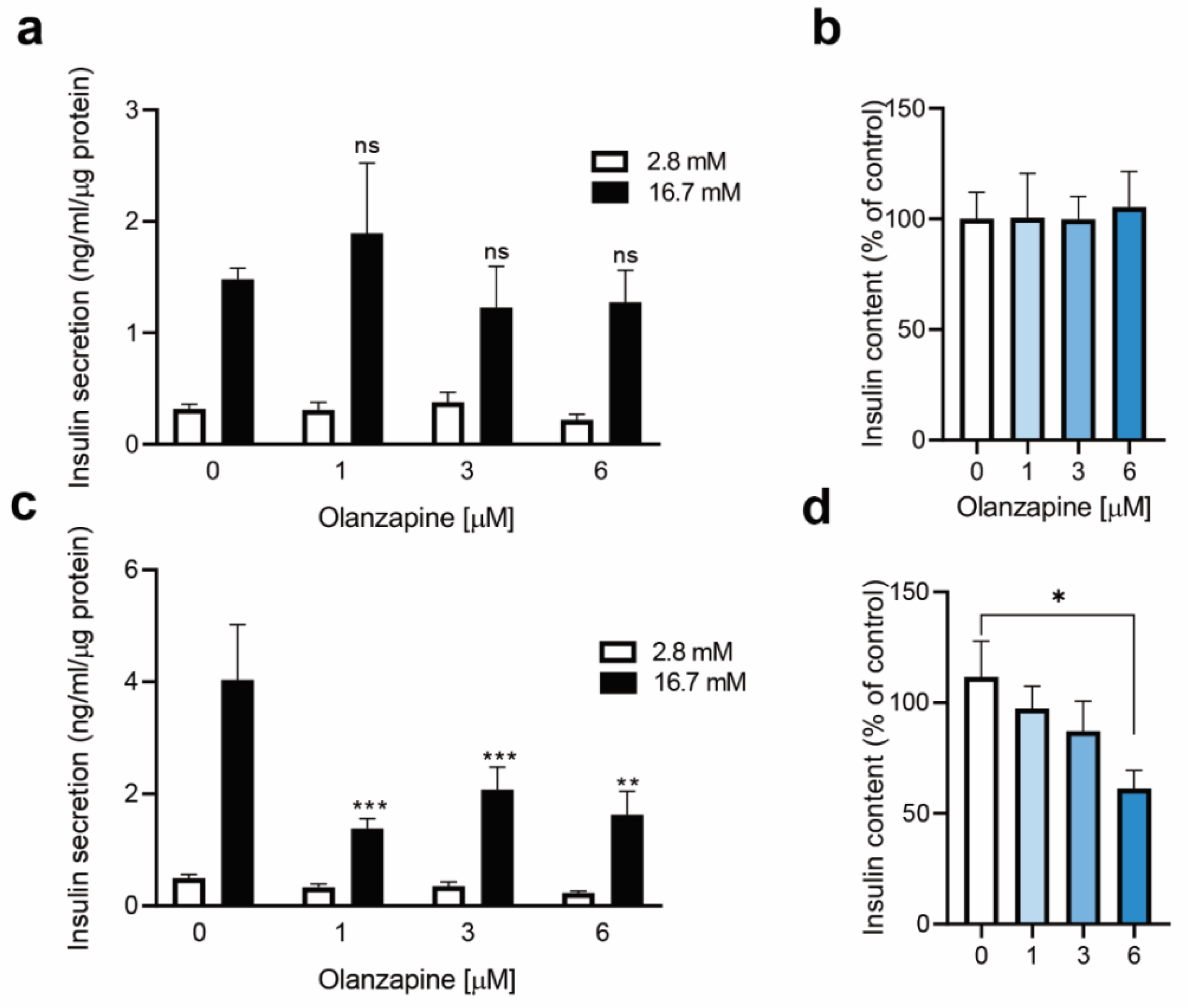
2.3. Twenty-Four Hour-Treatment with Non-Toxic Concentrations of Olanzapine Impaired Insulin Secretion in INS-1 Beta Cells
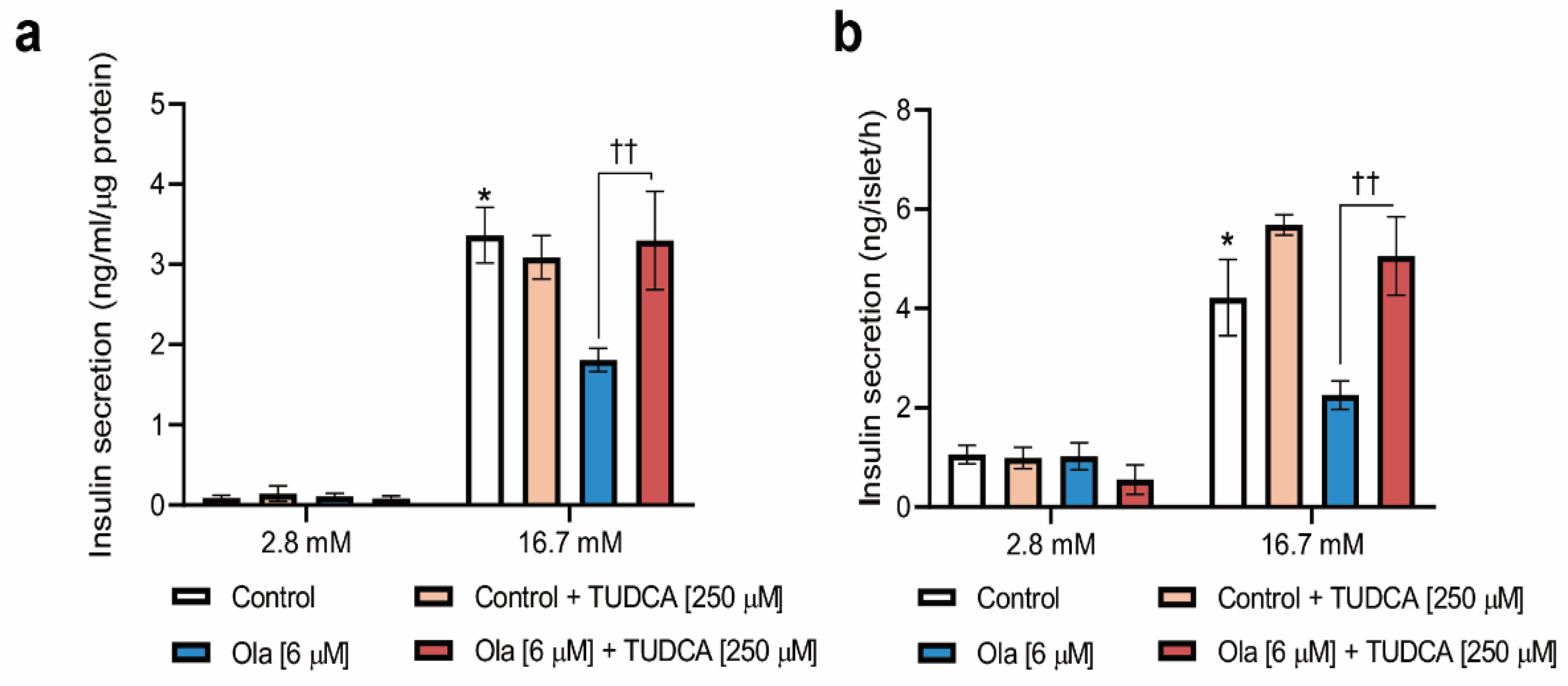
2.4. Co-Treatment of INS-1 Beta Cells or Pancreatic Islets with Olanzapine and the Bile Acid TUDCA Reduces ER Stress Signaling and Prevents the Impairment in Insulin Secretion
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Culture of INS-1 (821/13) Beta Cell Line
4.3. Cell Viability with Crystal Violet and MTT in INS-1 Cells
4.4. GSIS Measurement in INS-1 Cells
4.5. Analysis of Insulin Content in INS-1 Cells
4.6. Isolation of Pancreatic Islets of Langerhans from Female Mice
4.7. Ex Vivo GSIS in Islets
4.8. Analysis of XBP-1 Splicing
4.9. Protein Extraction and Western Blotting
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lauriello, J.; Perkins, D.O. Enhancing the treatment of patients with schizophrenia through continuous care. J. Clin. Psychiatry 2019, 80, 3721. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too much of a good thing is bad. BMC Med. 2020, 18, 224. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Skadhede, S.; Correll, C.U. Antipsychotics associated with the development of type 2 diabetes in antipsychotic-naïve schizophrenia patients. Neuropsychopharmacology 2010, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef]
- Kowalchuk, C.; Castellani, L.N.; Chintoh, A.; Remington, G.; Giacca, A.; Hahn, M.K. Antipsychotics and glucose metabolism: How brain and body collide. Am. J. Physiol. Endocrinol. Metab. 2019, 316, e1–e15. [Google Scholar] [CrossRef] [Green Version]
- Grajales, D.; Ferreira, V.; Valverde, A.M. Second-generation antipsychotics and dysregulation of glucose metabolism: Beyond weight gain. Cells 2019, 8, 1336. [Google Scholar] [CrossRef] [Green Version]
- Fulton, B.; Goa, K.L. Olanzapine: A review of its pharmacological properties and therapeutic efficacy in the management of schizophrenia and related psychoses. Drugs 1997, 53, 281–298. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology 2006, 66, S102–S109. [Google Scholar] [CrossRef]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, s60–s68. [Google Scholar] [CrossRef]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Ozasa, R.; Okada, T.; Nadanaka, S.; Nagamine, T.; Zyryanova, A.; Harding, H.; Ron, D.; Mori, K. The antipsychotic olanzapine induces apoptosis in insulin-secreting pancreatic beta cells by blocking PERK-mediated translational attenuation. Cell Struct. Funct. 2013, 38, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauressergues, E.; Bert, E.; Duriez, P.; Hum, D.; Majd, Z.; Staels, B.; Cussac, D. Does endoplasmic reticulum stress participate in APD-induced hepatic metabolic dysregulation? Neuropharmacology 2012, 62, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Weston-Green, K.; Babic, I.; de Santis, M.; Pan, B.; Montgomery, M.K.; Mitchell, T.; Huang, X.F.; Nealon, J. Disrupted sphingolipid metabolism following acute clozapine and olanzapine administration. J. Biomed. Sci. 2018, 25, 40. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Huang, X.F.; Gao, G.; Zhou, T.; Li, W.; Hu, J.; Chen, J.; Li, J.; Sun, T. Olanzapine-induced endoplasmic reticulum stress and inflammation in the hypothalamus were inhibited by an ER stress inhibitor 4-phenylbutyrate. Psychoneuroendocrinology 2019, 104, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Huang, X.F.; Deng, C.; Zhang, B.H.; Qian, K.; He, M.; Sun, T.L. Olanzapine Induces inflammation and immune response via activating ER stress in the rat prefrontal cortex. Curr. Med. Sci. 2021, 41, 788–802. [Google Scholar] [CrossRef]
- Ninagawa, S.; Tada, S.; Okumura, M.; Inoguchi, K.; Kinoshita, M.; Kanemura, S.; Imami, K.; Umezawa, H.; Ishikawa, T.; Mackin, R.B.; et al. Antipsychotic olanzapine-induced misfolding of proinsulin in the endoplasmic reticulum accounts for atypical development of diabetes. eLife 2020, 9, e60970. [Google Scholar] [CrossRef]
- Melkersson, K. Clozapine and olanzapine, but not conventional antipsychotics, increase insulin release in vitro. Eur. Neuropsychopharmacol. 2004, 14, 115–119. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhong, J.J.; Jin, J.F.; Yin, X.M.; Miao, H. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic β-cells by reducing ER stress. Exp. Clin. Endocrinol. Diabetes 2013, 121, 43–47. [Google Scholar] [CrossRef]
- Liu, M.Z.; He, H.Y.; Luo, J.Q.; He, F.Z.; Chen, Z.R.; Liu, Y.P.; Xiang, D.X.; Zhou, H.H.; Zhang, W. Drug-induced hyperglycaemia and diabetes: Pharmacogenomics perspectives. Arch. Pharmacal Res. 2018, 41, 725–736. [Google Scholar] [CrossRef]
- Vancampfort, D.; Correll, C.U.; Galling, B.; Probst, M.; de Hert, M.; Ward, P.B.; Rosenbaum, S.; Gaughran, F.; Lally, J.; Stubbs, B. Diabetes mellitus in people with schizophrenia, bipolar disorder and major depressive disorder: A systematic review and large scale meta-analysis. World Psychiatry 2016, 15, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Burghardt, K.J.; Seyoum, B.; Mallisho, A.; Burghardt, P.R.; Kowluru, R.A.; Yi, Z. Atypical antipsychotics, insulin resistance and weight; a meta-analysis of healthy volunteer studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 83, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Grajales, D.; Vázquez, P.; Ruíz-Rosario, M.; Tudurí, E.; Mirasierra, M.; Ferreira, V.; Hitos, A.B.; Koller, D.; Zubiaur, P.; Cigudosa, J.C.; et al. The second-generation antipsychotic drug aripiprazole modulates the serotonergic system in pancreatic islets and induces beta cell dysfunction in female mice. Diabetologia 2022, 65, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Chintoh, A.F.; Mann, S.W.; Lam, L.; Lam, C.; Cohn, T.A.; Fletcher, P.J.; Nobrega, J.N.; Giacca, A.; Remington, G. Insulin resistance and decreased glucose-stimulated insulin secretion after acute olanzapine administration. J. Clin. Psychopharmacol. 2008, 28, 494–499. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic reticulum chaperone glucose-regulated protein 94 is essential for proinsulin handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, G.; Bachar, E.; Shaked, M.; Sinai, A.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N. Glucose regulation of β-cell stress in type 2 diabetes. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 66–75. [Google Scholar] [CrossRef]
- Stone, S.I.; Abreu, D.; McGill, J.B.; Urano, F. Monogenic and syndromic diabetes due to endoplasmic reticulum stress. J. Diabetes Complicat. 2020, 35, 107618. [Google Scholar] [CrossRef]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Nagata, M.; Yokooji, T.; Nakai, T.; Miura, Y.; Tomita, T.; Taogoshi, T.; Sugimoto, Y.; Matsuo, H. Blockade of multiple monoamines receptors reduce insulin secretion from pancreatic β-cells. Sci. Rep. 2019, 9, 16438. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.H.; Lau, Y.S.; Miller, A.A.; Ku, J.M.; Potocnik, S.; Ye, J.M.; Woodman, O.L.; Herbert, T.P. Angiotensin II causes β-cell dysfunction through an ER stress-induced proinflammatory response. Endocrinology 2017, 158, 3162–3173. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Tomer, Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J. Autoimmun. 2017, 80, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forno, F.; Maatuf, Y.; Boukeileh, S.; Dipta, P.; Mahameed, M.; Darawshi, O.; Ferreira, V.; Rada, P.; García-Martinez, I.; Gross, E.; et al. Aripiprazole cytotoxicity coincides with activation of the unfolded protein response in human hepatic cells. J. Pharmacol. Exp. Ther. 2020, 374, 452–461. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grajales, D.; Vázquez, P.; Alén, R.; Hitos, A.B.; Valverde, Á.M. Attenuation of Olanzapine-Induced Endoplasmic Reticulum Stress Improves Insulin Secretion in Pancreatic Beta Cells. Metabolites 2022, 12, 443. https://doi.org/10.3390/metabo12050443
Grajales D, Vázquez P, Alén R, Hitos AB, Valverde ÁM. Attenuation of Olanzapine-Induced Endoplasmic Reticulum Stress Improves Insulin Secretion in Pancreatic Beta Cells. Metabolites. 2022; 12(5):443. https://doi.org/10.3390/metabo12050443
Chicago/Turabian StyleGrajales, Diana, Patricia Vázquez, Rosa Alén, Ana B. Hitos, and Ángela M. Valverde. 2022. "Attenuation of Olanzapine-Induced Endoplasmic Reticulum Stress Improves Insulin Secretion in Pancreatic Beta Cells" Metabolites 12, no. 5: 443. https://doi.org/10.3390/metabo12050443