
Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia

 , ,
, ,  , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Results
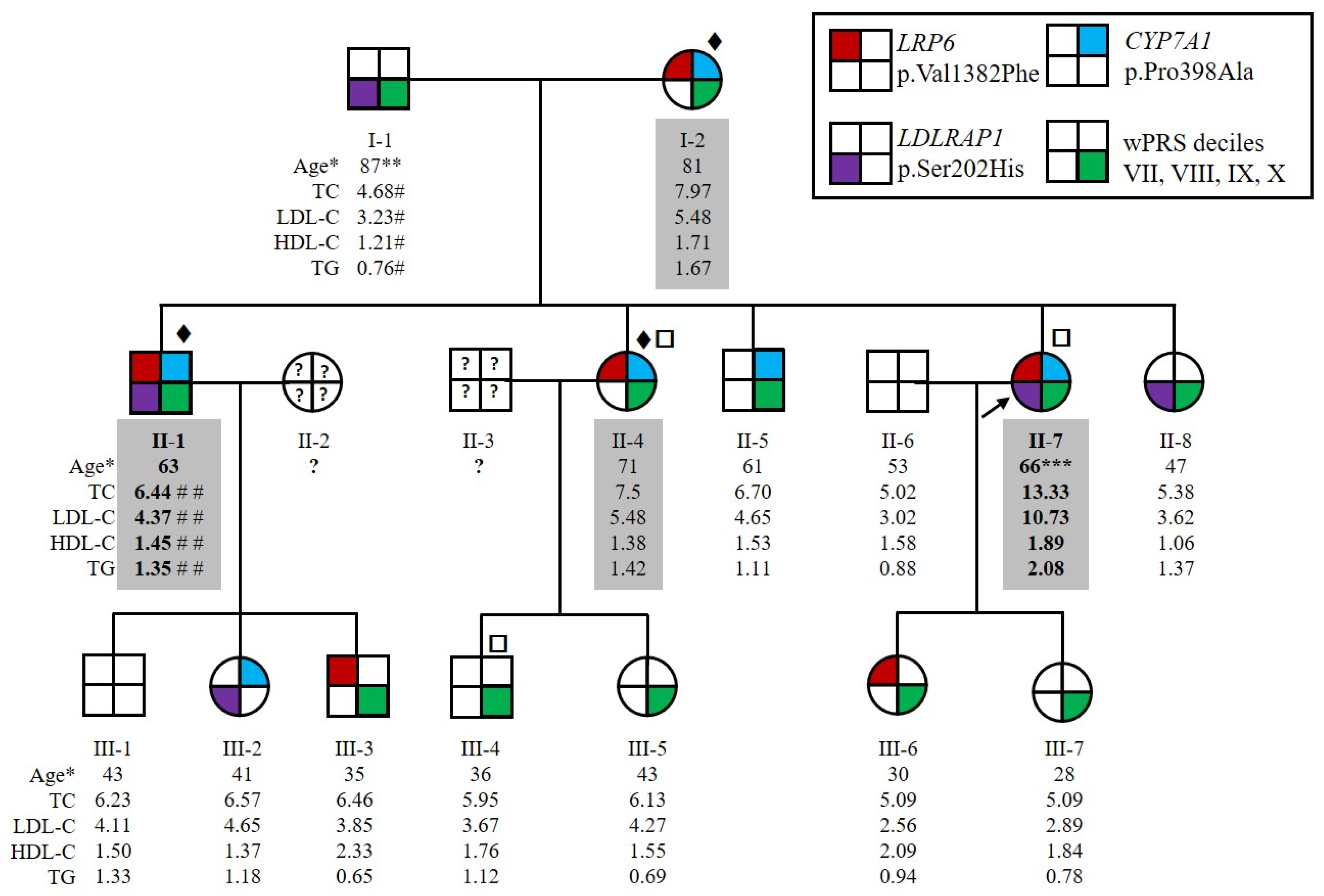
2.1. Patients Characteristics
2.2. Three Candidate Variants Were Identified by Whole Genome Sequencing, Whole Exome Sequencing and Positional Cloning
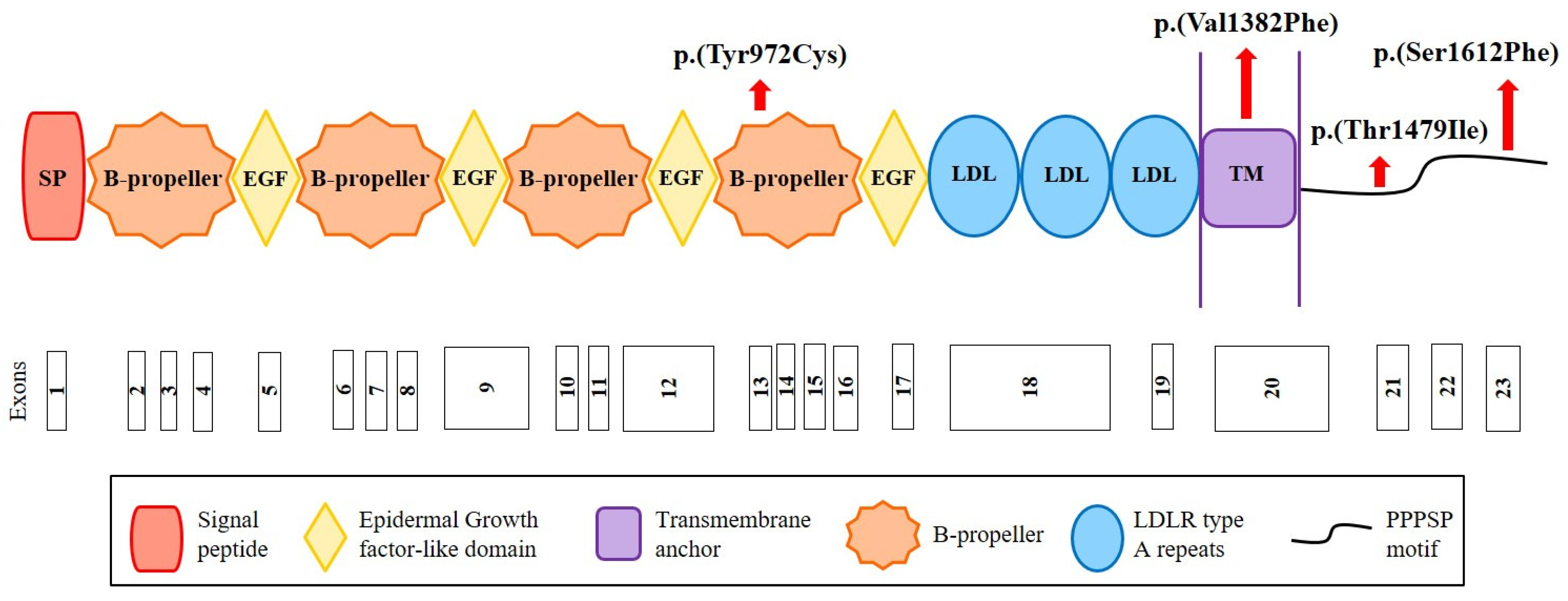
2.3. Identification of New Variants in CYP7A1, LDLRAP1 and LRP6
2.4. Bile Acid Synthesis Is Not Affected by the CYP7A1 Variant p.(Pro398Ala)
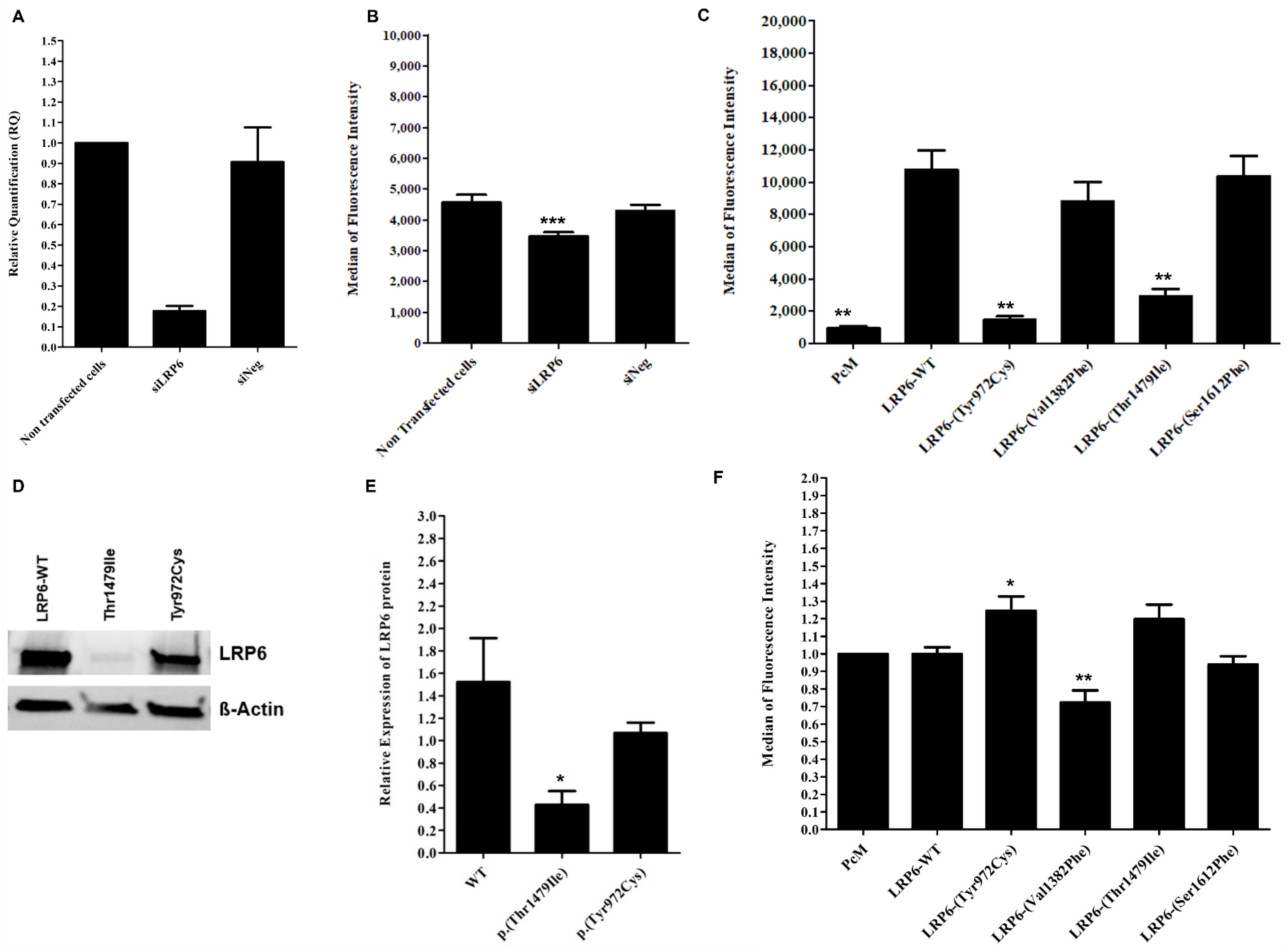
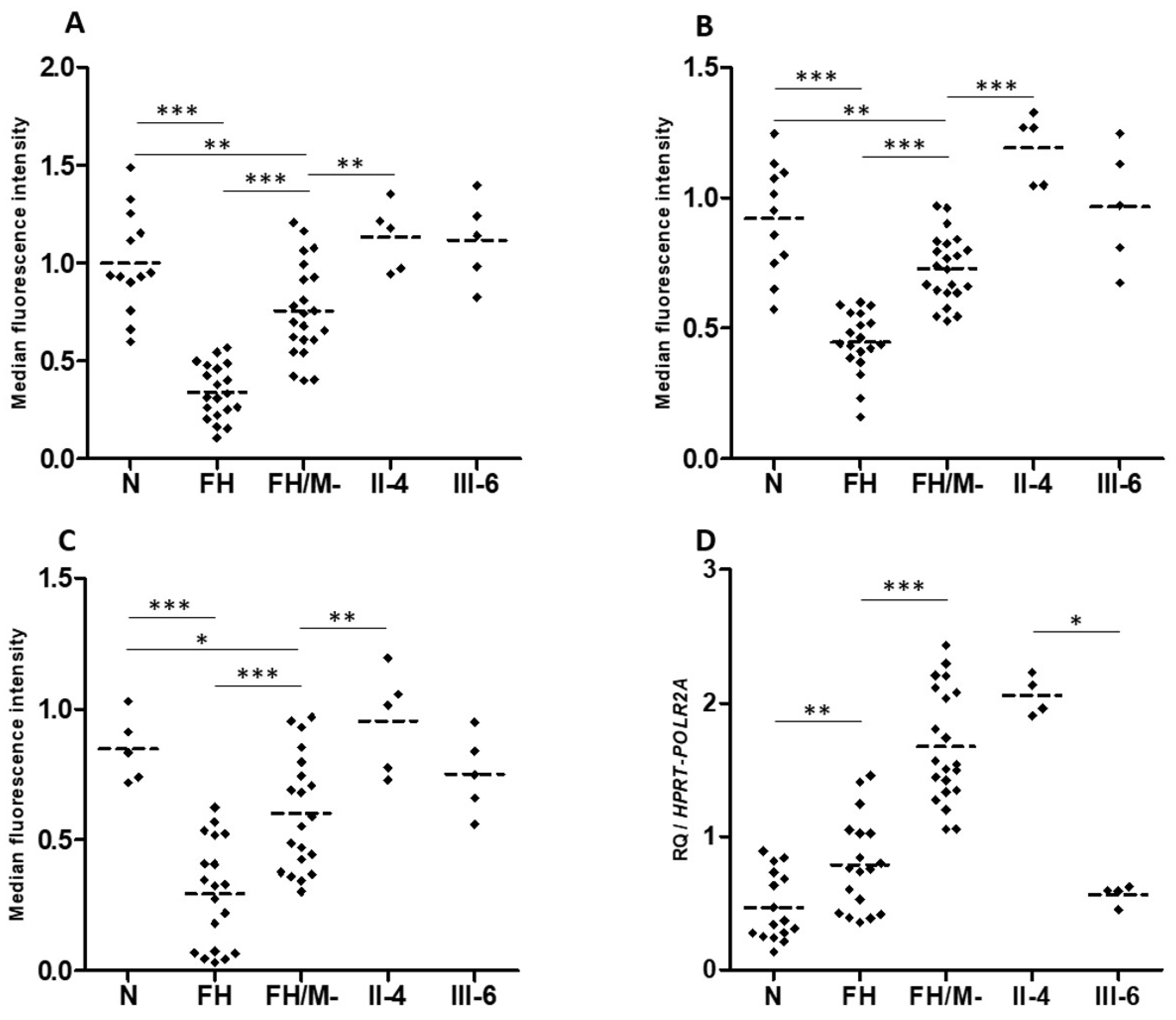
2.5. Contradictory Effects of LRP6 Variants on LRP6 Expression and LDL Binding and Uptake
2.6. Heterozygotes Carriers of p.(Val1382Phe) Did Not Present Altered LDL Uptake or LRP6 Gene Expression
2.7. Carriers of an LRP6 or LDLRAP1 Variant Present Lower LDL-C Levels than Non-Carriers Despite a Similar Polygenic Risk Score
3. Discussion
4. Materials and Methods
4.1. Probands and Family Recruitment
4.2. Whole Genome and Exome Sequencing and Data Analysis
4.3. Positional Cloning
4.4. Sequencing and In Silico Analysis of the Variants
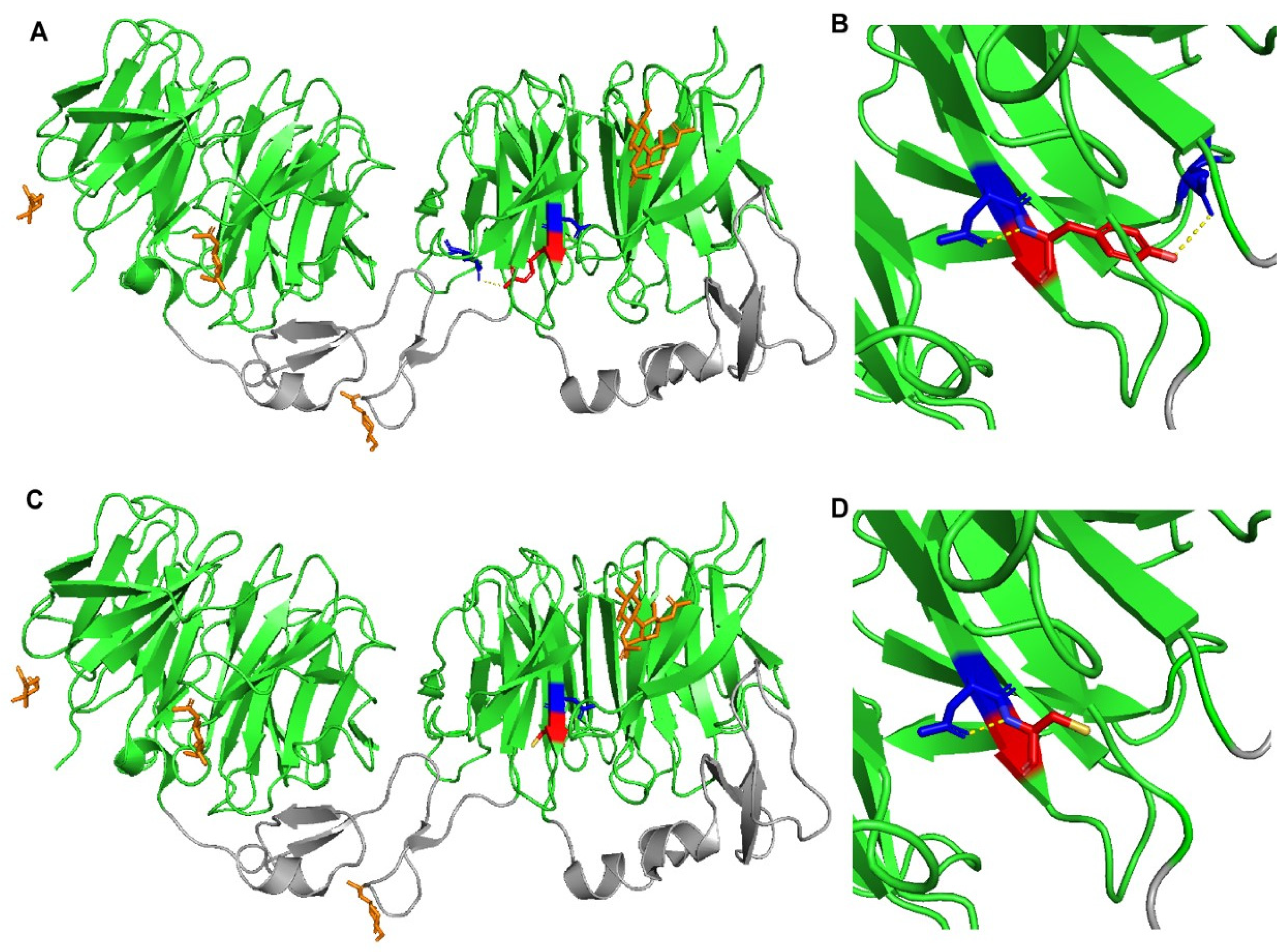
4.5. Construction of LRP6 Structural Models
4.6. Weighted Polygenic Risk Score (wPRS) Calculation
4.7. Sterol and Bile Acids Measurements
4.8. Site-Directed Mutagenesis
4.9. Cell Culture and Transfection
4.10. LRP6 and LDL Receptor Cell Surface Expression
4.11. LDL Uptake
4.12. Protein Extraction and Western Blot Assays
4.13. Total RNA Extraction, RT and Real-Time PCR Quantification
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béliard, S.; Boccara, F.; Cariou, B.; Carrié, A.; Collet, X.; Farnier, M.; Ferrières, J.; Krempf, M.; Peretti, N.; Rabès, J.-P.; et al. High burden of recurrent cardiovascular events in heterozygous familial hypercholesterolemia: The French Familial Hypercholesterolemia Registry. Atherosclerosis 2018, 277, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Schrott, H.G.; Hazzard, W.R.; Bierman, E.L.; Motulsky, A.G. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Investig. 1973, 52, 1544–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innerarity, T.L.; Mahley, R.W.; Weisgraber, K.H.; Bersot, T.P.; Krauss, R.M.; Vega, G.L.; Grundy, S.M.; Friedl, W.; Davignon, J.; McCarthy, B.J. Familial defective apolipoprotein B-100: A mutation of apolipoprotein B that causes hypercholesterolemia. J. Lipid Res. 1990, 31, 1337–1349. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Marduel, M.; Carrié, A.; Sassolas, A.; Devillers, M.; Carreau, V.; Filippo, M.D.; Erlich, D.; Abifadel, M.; Marques-Pinheiro, A.; Munnich, A.; et al. Molecular Spectrum of Autosomal Dominant Hypercholesterolemia in France. Hum. Mutat. 2010, 31, E1811–E1824. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Higuero, J.A.; Benito-Vicente, A.; Etxebarria, A.; Milicua, J.C.G.; Ostolaza, H.; Arrondo, J.L.R.; Martín, C. Structural changes induced by acidic pH in human apolipoprotein B-100. Sci. Rep. 2016, 6, 36324. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, P.; Elbitar, S.; Ghaleb, Y.; Khalil, Y.A.; Varret, M.; Boileau, C.; Abifadel, M. PCSK9 Mutations in Familial Hypercholesterolemia: From a Groundbreaking Discovery to Anti-PCSK9 Therapies. Curr. Atheroscler. Rep. 2017, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Khalil, Y.A.; Rabès, J.-P.; Boileau, C.; Varret, M. APOE gene variants in primary dyslipidemia. Atherosclerosis 2021, 328, 11–22. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.-A.; Nohara, A.; Inazu, A.; Mabuchi, H.; Yamagishi, M. Impact of clinical signs and genetic diagnosis of familial hypercholesterolaemia on the prevalence of coronary artery disease in patients with severe hypercholesterolaemia. Eur. Heart J. 2017, 38, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Reeskamp, L.F.; Volta, A.; Zuurbier, L.; Defesche, J.C.; Hovingh, G.K.; Grefhorst, A. ABCG5 and ABCG8 genetic variants in familial hypercholesterolemia. J. Clin. Lipidol. 2020, 14, 207–217.e7. [Google Scholar] [CrossRef]
- Chora, J.R.; Alves, A.C.; Medeiros, A.M.; Mariano, C.; Lobarinhas, G.; Guerra, A.; Mansilha, H.; Cortez-Pinto, H.; Bourbon, M. Lysosomal acid lipase deficiency: A hidden disease among cohorts of familial hypercholesterolemia? J. Clin. Lipidol. 2017, 11, 477–484.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Investig. 2002, 110, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Go, G. Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis. Nutrients 2015, 7, 4453–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Tomaszewski, M.; Charchar, F.J.; Barnes, T.; Gawron-Kiszka, M.; Sedkowska, A.; Podolecka, E.; Kowalczyk, J.; Rathbone, W.; Kalarus, Z.; Grzeszczak, W.; et al. A common variant in low-density lipoprotein receptor-related protein 6 gene (LRP6) is associated with LDL-cholesterol. Arter. Thromb. Vasc. Biol. 2009, 29, 1316–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, A.; Mani, M.-A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montazeri-Najafabady, N.; Dabbaghmanesh, M.H.; Mohammadian Amiri, R. The association of LRP6 rs2302685 (V1062I) polymorphism with the risk of hyperlipidemia in Iranian children and adolescents. Ann. Hum. Genet. 2018, 82, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.A.; Hu, Y.; Zhang, H.; Xue, C.; Schmidt, E.M.; Tang, Z.-Z.; Bizon, C.; Lange, E.M.; Smith, J.D.; Turner, E.H.; et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 2014, 94, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic Versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arter. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef] [Green Version]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Rabès, J.-P.; Béliard, S.; Carrié, A. Familial hypercholesterolemia: Experience from France. Curr. Opin. Lipidol. 2018, 29, 65–71. [Google Scholar] [CrossRef]
- Varret, M.; Abifadel, M.; Rabès, J.-P.; Boileau, C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin. Genet. 2008, 73, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sjouke, B.; Defesche, J.C.; Hartgers, M.L.; Wiegman, A.; van Lennep, J.E.R.; Kastelein, J.J.; Hovingh, G.K. Double-heterozygous autosomal dominant hypercholesterolemia: Clinical characterization of an underreported disease. J. Clin. Lipidol. 2016, 10, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Elbitar, S.; Susan-Resiga, D.; Ghaleb, Y.; El Khoury, P.; Peloso, G.; Stitziel, N.; Rabès, J.-P.; Carreau, V.; Hamelin, J.; Ben-Djoudi-Ouadda, A.; et al. New Sequencing technologies help revealing unexpected mutations in Autosomal Dominant Hypercholesterolemia. Sci. Rep. 2018, 8, 1943. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.-A.; Nomura, A.; Teramoto, R.; Hosomichi, K.; Nohara, A.; Inazu, A.; Mabuchi, H.; Tajima, A.; Yamagishi, M. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J. Clin. Lipidol. 2018, 12, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Haralambos, K.; Whatley, S.D.; Edwards, R.; Gingell, R.; Townsend, D.; Ashfield-Watt, P.; Lansberg, P.; Datta, D.B.N.; McDowell, I.F.W. Clinical experience of scoring criteria for Familial Hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis 2015, 240, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, H.; Einarsson, K.; Johansson, G. Effect of biliary drainage on individual reactions in the conversion of cholesterol to taurochlic acid. Bile acids and steroids 180. Eur. J. Biochem. 1967, 2, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Shefer, S.; Hauser, S.; Bekersky, I.; Mosbach, E.H. Biochemical site of regulation of bile acid biosynthesis in the rat. J. Lipid Res. 1970, 11, 404–411. [Google Scholar] [CrossRef]
- Couture, P.; Otvos, J.D.; Cupples, L.A.; Wilson, P.W.; Schaefer, E.J.; Ordovas, J.M. Association of the A-204C polymorphism in the cholesterol 7alpha-hydroxylase gene with variations in plasma low density lipoprotein cholesterol levels in the Framingham Offspring Study. J. Lipid Res. 1999, 40, 1883–1889. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Ye, Z.; Go, G.-W.; Singh, R.; Liu, W.; Keramati, A.R.; Mani, A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J. Biol. Chem. 2012, 287, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. TEM 2013, 24, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc. Res. 2020, 116, 1254–1274. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Mani, S.; Davis, N.R.; Sarrafzadegan, N.; Kavathas, P.B.; Mani, A. Mutation in EGFP domain of LDL receptor-related protein 6 impairs cellular LDL clearance. Circ. Res. 2008, 103, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Siest, G.; Visvikis, S.; Herbeth, B.; Gueguen, R.; Vincent-Viry, M.; Sass, C.; Beaud, B.; Lecomte, E.; Steinmetz, J.; Locuty, J.; et al. Objectives, design and recruitment of a familial and longitudinal cohort for studying gene-environment interactions in the field of cardiovascular risk: The Stanislas cohort. Clin. Chem. Lab. Med. 1998, 36, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Peloso, G.M.; Abifadel, M.; Cefalu, A.B.; Fouchier, S.; Motazacker, M.M.; Tada, H.; Larach, D.B.; Awan, Z.; Haller, J.F.; et al. Exome sequencing in suspected monogenic dyslipidemias. Circ. Cardiovasc. Genet. 2015, 8, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Schäffer, A.A.; Lemire, M.; Ott, J.; Lathrop, G.M.; Weeks, D.E. Coordinated conditional simulation with SLINK and SUP of many markers linked or associated to a trait in large pedigrees. Hum. Hered. 2011, 71, 126–134. [Google Scholar] [CrossRef]
- O’Connell, J.R.; Weeks, D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998, 63, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishelson, M.; Geiger, D. Exact genetic linkage computations for general pedigrees. Bioinforma. Oxf. Engl. 2002, 18 (Suppl. S1), S189–S198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, D.E. A likelihood-based analysis of consistent linkage of a disease locus to two nonsyntenic marker loci: Osteogenesis imperfecta versus COL1A1 and COL1A2. Am. J. Hum. Genet. 1990, 47, 592–594. [Google Scholar]
- Lindner, T.H.; Hoffmann, K. easyLINKAGE: A PERL script for easy and automated two-/multi-point linkage analyses. Bioinforma. Oxf. Engl. 2005, 21, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Marmontel, O.; Rollat-Farnier, P.A.; Wozny, A.-S.; Charrière, S.; Vanhoye, X.; Simonet, T.; Chatron, N.; Collin-Chavagnac, D.; Nony, S.; Dumont, S.; et al. Development of a new expanded next-generation sequencing panel for genetic diseases involved in dyslipidemia. Clin. Genet. 2020, 98, 589–594. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinform. Oxf. Engl. 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, Y.; Elbitar, S.; El Khoury, P.; Bruckert, E.; Carreau, V.; Carrié, A.; Moulin, P.; Di-Filippo, M.; Charriere, S.; Iliozer, H.; et al. Usefulness of the genetic risk score to identify phenocopies in families with familial hypercholesterolemia? Eur. J. Hum. Genet. EJHG 2018, 26, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lütjohann, D.; Hahn, C.; Prange, W.; Sudhop, T.; Axelson, M.; Sauerbruch, T.; von Bergmann, K.; Reichel, C. Influence of rifampin on serum markers of cholesterol and bile acid synthesis in men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Šošić-Jurjević, B.; Lütjohann, D.; Renko, K.; Filipović, B.; Radulović, N.; Ajdžanović, V.; Trifunović, S.; Nestorović, N.; Živanović, J.; Manojlović Stojanoski, M.; et al. The isoflavones genistein and daidzein increase hepatic concentration of thyroid hormones and affect cholesterol metabolism in middle-aged male rats. J. Steroid Biochem. Mol. Biol. 2019, 190, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | c.notation p.notation | rs Number | Pathway | GTEx-TPM_Liver # | gnomAD (Total) * | gnomAD (ENF) * | FREX ** | Varsome *** | PolyPhen2 | Provean † | ClinVar | CADD Score ‡ | Splice AI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP7A1 (NM_000780) | c.38C > T p.(Ala13Val) | rs147162838 | Bile acid and bile salt metabolism | 2.612 | 0.181% (512/282,726) | 0.35% (451/129,062) | 0. | LB | B | N (−0.189) | LB/VUS | 7.125 | No-consq (0) |
| c.1039G > A p.(Asp347Asn) | rs8192875 | 0.274% (776/282,802) | 0.019% (25/129,150) | … | LB | PD | D (−0.2990) | … | 33 | Donor gain (0.48) | |||
| c.1192C > G p.(Pro398Ala) | rs142708991 | 0.336% (951/282,868) | 0.43% (555/129,184) | 0.0871% | LB | PD | D (−7.559) | LB | 25.1 | No-consq (0) | |||
| LDLRAP1 (NM_015627) | c.603dupC p.(Ser202LeufsTer19) | rs781585299 | Clathrin-mediated endocytosis | 112.23 | … | … | … | P | … | … | P | … | … |
| c.604_605delTCinsCA p.(Ser202His) | rs386629678 | … | … | … | LB | PD | N (−2.072) | LB | … | … | |||
| LRP6 (NM_002336) | c.2915A > G p.(Tyr972Cys) | rs772441071 | Vesicle-mediated transport | 9.662 | 0.001193% (3/251,364) | 0.002640% (3/113,656) | … | VUS | PD | D (−7.227) | … | 26.9 | Acceptor gain (0.04) |
| c.4144G > T p.(Val1382Phe) | rs139480047 | 0.08379% (237/282,856) | 0.1061% (137/129,164) | 0.261% | B | B | N (−1.246) | LB | 22.3 | Donor gain (0.02) | |||
| c.4436C > T p.(Thr1479Ile) | rs144175121 | 0.02263% (64/282,836) | 0.04335% (56/129,168) | … | B | B | N (−1.906) | … | 23.3 | No-consq (0) | |||
| c.4835C > T p.(Ser1612Phe) | … | 0.0008097% (2/247,016) | 0% (0/111,624) | … | VUS | PD | D (−2.879) | … | 29.4 | Donor gain (0.02) |
| Gene | Variant | Sex | Age * | TC ** | LDL-C ** | HDL-C ** | TG ** | wPRS | Decile | Clinic | Family History |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP7A1 (NM_000780) | c.38C > T p.(Ala13Val) | M | 59 | … | … | … | … | 0.571 | IV | … | … |
| c.1039G > A p.(Asp347Asn) | F | 71 | 7.92 | 6.24 | … | 0.98 | 0.25 | I | … | … | |
| c.1192C > G p.(Pro398Ala) | M | 71 | 7.52 | 5.69 | … | … | … | … | … | … | |
| LDLRAP1 (NM_015627) | c.603dupC p.(Ser202LeufsTer19) | F | 37 | 7.75 | 5.56 | 1.37 | 1.81 | 0.752 | VII | … | … |
| c.604_605delTCinsCA p.(Ser202His) | F | 39 | … | 5.44 | … | … | 0.622 | V | … | … | |
| LRP6 (NM_002336) | c.2915A > G p.(Tyr972Cys) | M † | 40 | 6.35 | 4.70 | 1.21 | 0.89 | … | … | CAD | Yes Yes |
| M ‡ | 32 | 7.47 | 5.30 | 1.34 | 1.83 | 0.371 | II | No | |||
| c.4436C > T p.(Thr1479Ile) | F | 48 | 8.15 | 5.57 | 2.12 | 1.01 | 0.581 | IV | No | Yes | |
| c.4835C > T p.(Ser1612Phe) | M | 51 | 8.04 | 5.89 | 1.71 | 0.94 | 0.542 | III | CAD | Yes |
| II-1 *,† | II-4 **,† | II-5 † | III-4 | III-6 | III-7 | Carriers *** | Non-Carriers *** | Marker of | p-Value | |
|---|---|---|---|---|---|---|---|---|---|---|
| Serum total cholesterol (mmol/L) | 5.08 | 7.84 | 6.95 | 5.73 | 5.46 | 3.67 | 6.62 ± 1.41 | 4.95 ± 1.12 | 0.306 | |
| R_campesterol (μg/mg) ‡ | 1.77 | 1.13 | 1.41 | 1.66 | 1.98 | 2.02 | 1.44 ± 0.32 | 0.32 ± 1.89 | Cholesterol absorption | 0.061 |
| R_sitosterol (μg/mg) ‡ | 1.65 | 0.87 | 0.97 | 0.94 | 1.26 | 1.30 | 1.16 ± 0.43 | 1.17 ± 0.20 | Cholesterol absorption | 0.495 |
| R_cholestanol (μg/mg) ‡ | 1.05 | 1.00 | 0.79 | 1.32 | 1.25 | 1.38 | 0.95 ± 0.14 | 1.32 ± 0.07 | Cholesterol absorption | 0.015 |
| R_lathosterol (μg/mg) ‡ | 0.68 | 1.46 | 1.39 | 2.02 | 1.22 | 2.01 | 1.18 ± 0.43 | 1.75 ± 0.46 | Cholesterol synthesis | 0.094 |
| R_lanosterol (µg/mg) ‡ | 0.10 | 0.14 | 0.12 | 0.17 | 0.11 | 0.20 | 0.12 ± 0.02 | 0.16 ± 0.05 | Cholesterol synthesis | 0.126 |
| R_desmosterol (μg/mg) ‡ | 0.65 | 0.78 | 0.77 | 0.91 | 0.58 | 0.77 | 0.74 ± 0.07 | 0.75 ± 0.17 | Cholesterol synthesis | 0.433 |
| R_7αOH-cholesterol (ng/mg) ‡ | 23 | 25 | 39 | 41 | 44 | 52 | 29 ± 9 | 46 ± 6 | Degradation to bile acids | 0.032 |
| R_27OH-cholesterol (ng/mg) ‡ | 113 | 108 | 95 | 107 | 117 | 128 | 105 ± 9 | 118 ± 10 | Degradation to bile acids | 0.104 |
| Chenodeoxycholic acid (μmol/L) | 3.05 | 2.76 | 1.34 | 2.11 | 0.01 | 3.49 | 2.38 ± 0.91 | 1.87 ± 1.75 | Bile acids | 0.342 |
| Cholic acid (µmol/L) | 1.76 | 1.92 | 0.44 | 1.74 | 0.01 | 1.28 | 1.38 ± 0.81 | 1.01 ± 0.89 | Bile acids | 0.314 |
| Lithocholic acid (µmol/L) | 0.06 | 0.16 | 0.21 | 0.20 | 0.31 | 0.11 | 0.14 ± 0.08 | 0.21 ± 0.10 | Bile acids | 0.224 |
| Deoxycholic acid (µmol/L) | 0.27 | 2.16 | 2.30 | 2.99 | 1.95 | 1.06 | 1.58 ± 1.13 | 2.00 ± 0.96 | Bile acids | 0.324 |
| Non-ADH Subjects * | ADH Probands ** | CYP7A1 Variant Carriers *** | LRP6 Variant Carriers # | LDLRAP1 Variant Carriers ## | CYP7A1 and LRP6 Variants Carriers ### | CYP7A1, LRP6 and LDLRAP1 Variants Carriers † | |
|---|---|---|---|---|---|---|---|
| N | 13 | 152 | 4 | 6 | 3 | 2 | 2 |
| Sex (% of women) | 53.8 | 60.4 | 25 | 33.3 | 66.7 | 100 | 50 |
| Age (years) | 50 ± 15 | 48 ± 18 | 66 ± 6 | 39 ± 9 | 54 ± 28 | 81–71 | 6366 |
| p-value vs. non-ADH subjects | 0.4897 | 0.0266 | 0.0520 | 0.3185 | |||
| p-value vs. ADH probands | 0.0094 | 0.0453 | 0.4945 | ||||
| LDL-C (mmol/L) | 3.4 ± 0.5 | 6.1 ± 1.5 | 5.5 ± 0.8 | 4.6 ± 1.3 | 4.7 ± 1.3 | 5.5–5.5 | 7.9–10.7 |
| p-value vs. non-ADH subjects | <0.0001 | 0.0053 | 0.0242 | 0.0693 | |||
| p-value vs. ADH probands | 0.2364 | 0.0088 | 0.0542 | ||||
| wPRS | 0.665 ± 0.165 | 0.700 ± 0.187 | 0.574 ± 0.326 | 0.655 ± 0.243 | 0.792 ± 0.193 | 0.731–0.831 | 0.831–0.902 |
| p-value vs. non-ADH subjects | 0.2024 | 0.3428 | 0.500 | 0.2091 | |||
| p-value vs. ADH probands | 0.2091 | 0.2548 | 0.2676 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghaleb, Y.; Elbitar, S.; Philippi, A.; El Khoury, P.; Azar, Y.; Andrianirina, M.; Loste, A.; Abou-Khalil, Y.; Nicolas, G.; Le Borgne, M.; et al. Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia. Metabolites 2022, 12, 262. https://doi.org/10.3390/metabo12030262
Ghaleb Y, Elbitar S, Philippi A, El Khoury P, Azar Y, Andrianirina M, Loste A, Abou-Khalil Y, Nicolas G, Le Borgne M, et al. Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia. Metabolites. 2022; 12(3):262. https://doi.org/10.3390/metabo12030262
Chicago/Turabian StyleGhaleb, Youmna, Sandy Elbitar, Anne Philippi, Petra El Khoury, Yara Azar, Miangaly Andrianirina, Alexia Loste, Yara Abou-Khalil, Gaël Nicolas, Marie Le Borgne, and et al. 2022. "Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia" Metabolites 12, no. 3: 262. https://doi.org/10.3390/metabo12030262