
Molecular Screening via Sanger Sequencing of the Genetic Variants in Non-Alcoholic Fatty Liver Disease Subjects in the Saudi Population: A Hospital-Based Study
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Design
2.3. Anthropometric Measurements
2.4. Histological Specimen
2.5. Molecular Screening
2.6. Sanger Sequencing Analysis
2.7. Statistical Analysis
3. Results
3.1. HWE Analysis
3.2. Clinical Characteristics between NAFLD and Non-NAFLD Subjects
3.3. Genotyping of the Five SNPs in Patients with and without NAFLD
3.4. Clinical Characteristics of Patients with NASH and without NAFLD
3.5. Genotyping in Patients with NASH and without NAFLD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a premature change in terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Akkiz, H.; Taskin, E.; Karaogullarindan, U.; Delik, A.; Kuran, S.; Kutlu, O. The influence of RS738409 I148M polymorphism of patatin-like phospholipase domain containing 3 gene on the susceptibility of non-alcoholic fatty liver disease. Medicine 2021, 100, e25893. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; McPherson, S.; Day, C.P. How big a problem is non-alcoholic fatty liver disease? BMJ 2011, 343, d3897. [Google Scholar] [CrossRef] [Green Version]
- Margini, C.; Dufour, J.F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. Off. J. Int. Assoc. Study Liver 2016, 36, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2020, 110, 921–937. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, Y.; Liu, S.; Chen, G.; Cao, L.; Xin, Y. Association of LDLR rs1433099 with the Risk of NAFLD and CVD in Chinese Han Population. J. Clin. Transl. Hepatol. 2021, 9, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and nonalcoholic fatty liver disease: Current perspectives. Diabetes Metab. Syndr. Obes. Targets Ther. 2018, 11, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, E.; Aburizaiza, O.S.; Siddique, A.; Khwaja, H.; Carpenter, D.O. Obesity and public health in the Kingdom of Saudi Arabia. Rev. Environ. Health 2015, 30, 191–205. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Ratziu, V.; Oren, R. Nutrition and physical activity in NAFLD: An overview of the epidemiological evidence. World J. Gastroenterol. 2011, 17, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.Y.; Fan, J.G. What are the clinical settings and outcomes of lean NAFLD? Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 289–290. [Google Scholar] [CrossRef]
- Cai, W.; Weng, D.-H.; Yan, P.; Lin, Y.-T.; Dong, Z.-H.; Yao, H. Genetic polymorphisms associated with nonalcoholic fatty liver disease in Uyghur population: A case-control study and meta-analysis. Lipids Health Dis. 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struben, V.M.; Hespenheide, E.E.; Caldwell, S.H. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am. J. Med. 2000, 108, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.R.; Waters, B.; Patil, S.R.; Reuben, A.; Morelli, J.; Riely, C.A. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am. J. Gastroenterol. 2001, 96, 2957–2961. [Google Scholar] [CrossRef] [PubMed]
- Makkonen, J.; Pietilainen, K.H.; Rissanen, A.; Kaprio, J.; Yki-Jarvinen, H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: A study in monozygotic and dizygotic twins. J. Hepatol. 2009, 50, 1035–1042. [Google Scholar] [CrossRef]
- Bambha, K.; Belt, P.; Abraham, M.; Wilson, L.A.; Pabst, M.; Ferrell, L.; Unalp-Arida, A.; Bass, N. Ethnicity and nonalcoholic fatty liver disease. Hepatology 2012, 55, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; Valenti, L. Genetics of nonalcoholic fatty liver disease. Metabolism 2016, 65, 1026–1037. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Österreicher, C.H.; Brenner, D.A. The genetics of nonalcoholic fatty liver disease. Ann. Hepatol. 2007, 6, 83–88. [Google Scholar] [CrossRef]
- Al-hamoudi, W.; El-Sabbah, M.; Ali, S.; Altuwaijri, M.; Bedewi, M.; Adam, M.; Alhammad, A.; Sanai, F.; Alswat, K.; Abdo, A. Epidemiological, clinical, and biochemical characteristics of Saudi patients with nonalcoholic fatty liver disease: A hospital-based study. Ann. Saudi Med. 2012, 32, 288–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallatah, H.I.; Al-Dabbagh, A.; Hiejazi, M.T.; Hanbazazah, S.A.; Hussein, A.O.; Al-Sahafi, M.A.; Akbar, H.O. Prevalence and clinical characteristics of NAFLD in chronic liver disease patients from King Abdulaziz University Hospital, Jeddah. Saudi J. Med. Med. Sci. 2020, 8, 118. [Google Scholar] [PubMed]
- Goud, K.I.; Kavitha, M.; Mahalakshmi, A.; Vempati, R.; Alodhayani, A.A.; Mohammed, A.A.; Khan, I.A. Molecular detection of Mycobacterium tuberculosis in pulmonary and extrapulmonary samples in a hospital-based study. Afr. Health Sci. 2020, 20, 1617–1623. [Google Scholar] [CrossRef]
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease: Definitions, risk factors, and workup. Clin. Liver Dis. 2012, 1, 99. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Althumiri, N.A.; Basyouni, M.H.; AlMousa, N.; AlJuwaysim, M.F.; Almubark, R.A.; BinDhim, N.F.; Alkhamaali, Z.; Alqahtani, S.A. Obesity in Saudi Arabia in 2020: Prevalence, distribution, and its current association with various health conditions. Healthcare 2021, 9, 311. [Google Scholar] [CrossRef]
- Alsuliman, M.A.; Alotaibi, S.A.; Zhang, Q.; Durgampudi, P.K. A systematic review of factors associated with uncontrolled diabetes and meta-analysis of its prevalence in Saudi Arabia since 2006. Diabetes/Metab. Res. Rev. 2021, 37, e3395. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; M Anstee, Q.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvnjak, M.; Baršić, N.; Tomašić, V.; Lerotić, I. Genetic polymorphisms in non-alcoholic fatty liver disease: Clues to pathogenesis and disease progression. World J. Gastroenterol. WJG 2009, 15, 6023. [Google Scholar] [CrossRef] [PubMed]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2012, 5, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; Del Menico, B. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Mazo, D.F.; Malta, F.M.; Stefano, J.T.; Salles, A.P.M.; Gomes-Gouvea, M.S.; Nastri, A.C.S.; Almeida, J.R.; Pinho, J.R.R.; Carrilho, F.J.; Oliveira, C.P. Validation of PNPLA3 polymorphisms as risk factor for NAFLD and liver fibrosis in an admixed population. Ann. Hepatol. 2019, 18, 466–471. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Gao, Y.; Ma, H.; Zhan, S.; Yang, Y.; Xin, Y.; Xuan, S. Association of TM6SF2 rs58542926 gene polymorphism with the risk of non-alcoholic fatty liver disease and colorectal adenoma in Chinese Han population. BMC Biochem. 2019, 20, 3. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, J.; Mei, T.-T.; Guo, H.-Q.; Wei, X.-H.; Zhang, W.-Y.; Liu, Y.-L.; Liang, S.; Fan, Z.-P.; Ma, L.-X. Association of TM6SF2 rs58542926 T/C gene polymorphism with hepatocellular carcinoma: A meta-analysis. BMC Cancer 2019, 19, 1128. [Google Scholar] [CrossRef]
- Musso, G.; Cipolla, U.; Cassader, M.; Pinach, S.; Saba, F.; De Michieli, F.; Paschetta, E.; Bongiovanni, D.; Framarin, L.; Leone, N. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J. Lipid Res. 2017, 58, 1221–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferder, J.; Gnirke, A.; Thomas, W. A novel MHC class I-like gene is mutated in patients with hereditary hemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Britton, L.J.; Subramaniam, V.N.; Crawford, D.H. Iron and non-alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 8112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saremi, L.; Lotfipanah, S.; Mohammadi, M.; Hosseinzadeh, H.; Sayad, A.; Saltanatpour, Z. Association of HFE gene mutations with nonalcoholic fatty liver disease in the Iranian population. Cell. Mol. Biol. 2016, 62, 123–128. [Google Scholar] [PubMed]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Ruscica, M.; Dongiovanni, P.; Steffani, L.; Motta, B.M.; Canavesi, E.; Fracanzani, A.L.; Mozzi, E.; Roviaro, G. The I148M PNPLA3 polymorphism influences serum adiponectin in patients with fatty liver and healthy controls. BMC Gastroenterol. 2012, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Goran, M.I.; Walker, R.; Le, K.-A.; Mahurkar, S.; Vikman, S.; Davis, J.N.; Spruijt-Metz, D.; Weigensberg, M.J.; Allayee, H. Effects of PNPLA3 on liver fat and metabolic profile in Hispanic children and adolescents. Diabetes 2010, 59, 3127–3130. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Yoneda, M.; Hyogo, H.; Ochi, H.; Mizusawa, S.; Ueno, T.; Chayama, K.; Nakajima, A.; Nakao, K.; Sekine, A. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med. Genet. 2010, 11, 172. [Google Scholar] [CrossRef]
- Peng, X.-E.; Wu, Y.-L.; Lin, S.-W.; Lu, Q.-Q.; Hu, Z.-J.; Lin, X. Genetic variants in PNPLA3 and risk of non-alcoholic fatty liver disease in a Han Chinese population. PLoS ONE 2012, 7, e50256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xing, C.; Cohen, J.C.; Hobbs, H.H. Genetic variant in PNPLA3 associated with nonalcoholic fatty liver disease in China. Hepatology 2012, 55, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uygun, A.; Ozturk, K.; Demirci, H.; Oztuna, A.; Eren, F.; Kozan, S.; Yilmaz, Y.; Kurt, O.; Turker, T.; Vatansever, S. The association of nonalcoholic fatty liver disease with genetic polymorphisms: A multicenter study. Eur. J. Gastroenterol. Hepatol. 2017, 29, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, G.S.; Chu, X.; Wood, G.C.; Gerhard, G.M.; Benotti, P.; Petrick, A.T.; Gabrielsen, J.; Strodel, W.E.; Still, C.D.; Argyropoulos, G. Next-generation sequence analysis of genes associated with obesity and nonalcoholic fatty liver disease-related cirrhosis in extreme obesity. Hum. Hered. 2013, 75, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.-H.; Li, Y.-L.; Li, D.; Wang, N.-N.; Jing, L.; Huang, Y.-H. The rs738409 (I148M) variant of the PNPLA3 gene and cirrhosis: A meta-analysis. J. Lipid Res. 2015, 56, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, G.; Liu, P.; Li, X.; Zhou, X.; He, S. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: A meta-analysis. Medicine 2019, 98, e14324. [Google Scholar] [CrossRef]
- Xu, R.; Tao, A.; Zhang, S.; Deng, Y.; Chen, G. Association between patatin-like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: A HuGE review and meta-analysis. Sci. Rep. 2015, 5, 9284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.-C.; Anty, R. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Z.; Peng, Z.; Liu, W. The TM6SF2 rs58542926 T allele is significantly associated with non-alcoholic fatty liver disease in Chinese. J. Hepatol. 2015, 62, 1438–1439. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.; Wong, G.; Tse, C.-H.; Chan, H. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J. Hepatol. 2014, 61, 708–709. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Llauradó, G.; Orešič, M.; Hyötyläinen, T.; Orho-Melander, M.; Yki-Järvinen, H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J. Hepatol. 2015, 62, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirola, C.J.; Sookoian, S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology 2015, 62, 1742–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, T.-T.; Zhang, J.; Tang, S.; Guo, H.-Q.; Wei, X.-H.; Zhang, W.-Y.; Liu, Y.-L.; Liang, S.; Fan, Z.-P.; Ma, L.-X. Association between TM6SF2 rs58542926 T/C gene polymorphism and significant liver fibrosis: A meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Z.; Que, S.; Zhou, L.; Zheng, S.; Romeo, S.; Mardinoglu, A.; Valenti, L. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: A meta-analysis. Sci. Rep. 2017, 7, 9273. [Google Scholar] [CrossRef] [Green Version]
- Raszeja-Wyszomirska, J.; Kurzawski, G.; Lawniczak, M.; Miezynska-Kurtycz, J.; Lubinski, J. Nonalcoholic fatty liver disease and HFE gene mutations: A Polish study. World J. Gastroenterol. WJG 2010, 16, 2531. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Qian, B.-X.; Yin, W.-L.; Wang, F.-M.; Han, T. Association between the HFE C282Y, H63D polymorphisms and the risks of non-alcoholic fatty liver disease, liver cirrhosis and hepatocellular carcinoma: An updated systematic review and meta-analysis of 5758 cases and 14,741 controls. PLoS ONE 2016, 11, e0163423. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef]


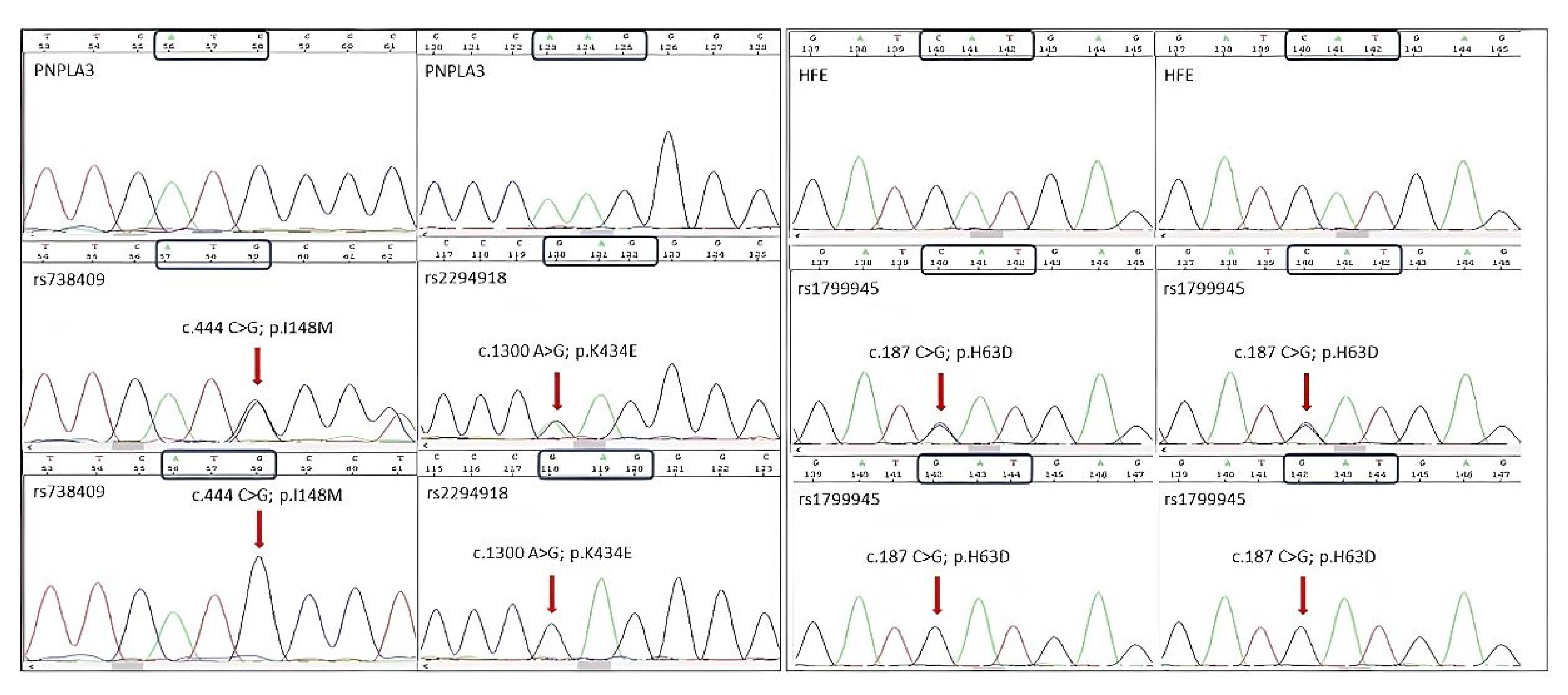{kind=link}
| S. No | Gene | SNP | Rsnumber | Mutation | Amino Acid Substitution |
|---|---|---|---|---|---|
| 1 | HFE | H63D | rs1799945 | C-G | Histidine-63-Aspartic acid |
| 2 | HFE | C282Y | rs1800562 | G-A | Cysteine-282-Tyrosine |
| 3 | PNPLA3 | I148M | rs738409 | C-G | Isoleucine-148-Methionine |
| 4 | PNPLA3 | K434E | rs2294918 | G-A | Lysine-434-Glutamic acid |
| 5 | TM6SF2 | E167K | rs58542926 | G-A | Glutamic acid-167-Lysine |
| Anthropometric | NAFLD (n = 95) | Non-NAFLD (n = 78) | p Value |
|---|---|---|---|
| Age (Years) | 43.6 ± 11.67 | 34.9 ± 11.05 | 0.0001 |
| Gender (Male; Female) | 31 (31.7%): 64 (65.3%) | 15 (19.2%): 63 (80.8%) | 0.26 |
| Weight (Kgs) | 83.9 ± 15.35 | 75.7 ± 15.79 | 0.0007 |
| Height (Cms) | 153.2 ± 0.08 | 151.9 ± 0.09 | 0.46 |
| BMI (kg/m2) | 32.5 ± 6.01 | 30.0 ± 5.79 | 0.006 |
| Genes | Rsnumber | Genotype/Alleles | NAFLD (%) | Non-NAFLD (%) | OR | 95% CIs | p Value |
|---|---|---|---|---|---|---|---|
| PNPLA3 (I148M) | rs738409 | CC | 45 (47.4%) | 45 (57.7%) | Reference | Reference | Reference |
| CG | 39 (41.0%) | 28 (35.9%) | 1.393 | 0.733–2.649 | 0.309 | ||
| GG | 11 (11.6%) | 05 (6.4%) | 2.200 | 0.707–6.844 | 0.166 | ||
| CG + GG vs. CC | 45 (47.4%) | 45 (57.7%) | 1.515 | 0.828–2.770 | 0.176 | ||
| CG vs. CC + GG | 56 (59.0%) | 51 (64.1%) | 1.244 | 0.670–2.306 | 0.488 | ||
| CC + CG vs. GG | 84 (88.4%) | 73 (93.6%) | 1.912 | 0.634–5.759 | 0.243 | ||
| C | 129 (67.8%) | 118 (75.6%) | Reference | Reference | Reference | ||
| G | 61 (32.2%) | 38 (24.4%) | 1.468 | 0.912–2.363 | 0.112 | ||
| PNPLA3(K434E) | rs2294918 | GG | 21 (22.1%) | 18 (23.1%) | Reference | Reference | Reference |
| GA | 40 (42.1%) | 44 (56.4%) | 0.779 | 0.363–1.668 | 0.520 | ||
| AA | 34 (35.8%) | 16 (20.5%) | 1.821 | 0.766–4.329 | 0.179 | ||
| AA vs. GA + AA | 21 (22.1%) | 18 (23.1%) | 1.057 | 0.516–2.162 | 0.870 | ||
| GA vs. GG + AA | 55 (57.9%) | 34 (43.6%) | 0.562 | 0.306–1.029 | 0.061 | ||
| GG + GA vs. AA | 61 (64.2%) | 62 (79.5%) | 2.160 | 1.082–4.312 | 0.027 | ||
| G | 82 (43.1%) | 80 (51.2%) | Reference | Reference | Reference | ||
| A | 108 (56.9%) | 76 (48.8%) | 1.386 | 0.906–2.121 | 0.131 | ||
| TM6SF2 (E167K) | rs58542926 | GG | 89 (93.7%) | 73 (93.6%) | Reference | Reference | Reference |
| GA | 06 (6.3%) | 04 (5.1%) | 1.230 | 0.334–4.525 | 0.754 | ||
| AA | 00 (0%) | 01 (1.3%) | 0.273 | 0.010–6.819 | 0.398 * | ||
| GA + AA vs. GG | 89 (93.7%) | 73 (93.6%) | 0.984 | 0.288–3.355 | 0.979 | ||
| GA vs. GG + AA | 89 (93.7%) | 74 (94.9%) | 1.202 | 0.347–4.157 | 0.770 * | ||
| GG + GA vs. AA | 95 (100%) | 77 (98.7%) | 0.270 | 0.010–6.733 | 0.393 * | ||
| G | 184 (96.8%) | 150 (96.1%) | Reference | Reference | Reference | ||
| A | 06 (3.2%) | 06 (3.9%) | 0.81 | 0.257–2.579 | 0.72 | ||
| HFE (H63D) | rs1799945 | CC | 73 (76.8%) | 54 (69.2%) | Reference | Reference | Reference |
| CG | 20 (21.1%) | 21 (26.9%) | 0.704 | 0.347–1.427 | 0.329 | ||
| GG | 02 (2.10%) | 03 (3.90%) | 0.493 | 0.079–3.054 | 0.438 | ||
| CG + GG vs. CC | 73 (76.8%) | 54 (69.2%) | 0.671 | 0.344–1.334 | 0.260 | ||
| CG vs. CC + GG | 75 (78.9%) | 57 (73.1%) | 0.723 | 0.358–1.461 | 0.366 | ||
| CC + CG vs. GG | 93 (97.9%) | 75 (96.1%) | 0.537 | 0.087–3.301 | 0.496 | ||
| C | 166 (87.3%) | 129 (82.6%) | Reference | Reference | Reference | ||
| G | 24 (12.7%) | 27 (17.4%) | 0.690 | 0.380–1.254 | 0.222 | ||
| HFE (C282Y) | rs1800562 | GG | 95 (100%) | 78 (100%) | Reference | Reference | Reference |
| GA | 00 (0%) | 00 (0%) | 3.697 | 0.148–92.01 | 0.393 * | ||
| AA | 00 (0%) | 00 (0%) | 3.697 | 0.148–92.01 | 0.393 * | ||
| G | 190 (100%) | 156 (100%) | Reference | Reference | Reference | ||
| A | 00 (0%) | 00 (0%) | 0.781 | 0.015–39.79 | 0.901 * |
| Anthropometric | NASH (n = 25) | Non-NAFLD (n = 78) | p Value |
|---|---|---|---|
| Age (Years) | 47.42 ± 10.95 | 34.9 ± 11.05 | 0.0003 |
| Gender (Male; Female) | 19 (76%): 06 (24%) | 15 (19.2%): 63 (80.8%) | 0.24 |
| Weight (Kgs) | 85.57 ± 17.23 | 75.7 ± 15.79 | 0.009 |
| Height (Cms) | 152.8 ± 0.08 | 151.9 ± 0.09 | 0.06 |
| BMI (kg/m2) | 33.48 ± 6.12 | 30.0 ± 5.79 | 0.01 |
| Genes | Rsnumber | Genotype/Alleles | NASH (%) | Non-NAFLD (%) | OR | 95% CIs | p Value |
|---|---|---|---|---|---|---|---|
| PNPLA3 (I148M) | rs738409 | CC | 12 (48%) | 45 (57.7%) | Reference | Reference | Reference |
| CG | 10 (40%) | 28 (35.9%) | 0.730 | 0.301–1.768 | 0.485 | ||
| GG | 03 (12%) | 05 (6.4%) | 1.227 | 0.268–5.608 | 0.791 | ||
| CG + GG vs. CC | 12 (48%) | 45 (57.7%) | 1.477 | 0.598–3.648 | 0.396 | ||
| CG vs. CC + GG | 15 (60%) | 51 (64.1%) | 1.190 | 0.472–3.000 | 0.711 | ||
| CC + CG vs. GG | 22 (88%) | 73 (93.6%) | 1.991 | 0.440–8.999 | 0.363 | ||
| C | 34 (68%) | 118 (75.6%) | Reference | Reference | Reference | ||
| G | 16 (32%) | 38 (24.4%) | 1.461 | 0.727–2.936 | 0.286 | ||
| PNPLA3(K434E) | rs2294918 | GG | 09 (36%) | 18 (23.1%) | Reference | Reference | Reference |
| GA | 13 (52%) | 44 (56.4%) | 0.590 | 0.214–1.625 | 0.307 | ||
| AA | 03 (12%) | 16 (20.5%) | 0.375 | 0.086–1.631 | 0.182 | ||
| GA + AA vs. GG | 09 (36%) | 18 (23.1%) | 0.533 | 0.201–1.409 | 0.201 | ||
| GA vs. GG + AA | 12 (48%) | 34 (43.6%) | 0.837 | 0.339–2.066 | 0.699 | ||
| GG + GA vs. AA | 22 (88%) | 62 (79.5%) | 0.528 | 0.140–1.989 | 0.339 | ||
| G | 31 (62%) | 80 (51.2%) | Reference | Reference | Reference | ||
| A | 19 (38%) | 76 (48.8%) | 0.645 | 0.336–1.238 | 0.186 | ||
| TM6SF2 (E167K) | rs58542926 | GG | 24 (96%) | 73 (93.6%) | Reference | Reference | Reference |
| GA | 01 (04%) | 04 (5.1%) | 0.760 | 0.081–7.137 | 0.810 | ||
| AA | 00 (00%) | 01 (1.3%) | 1.000 | 0.039–25.35 | 0.999 * | ||
| GA + AA vs. GG | 24 (96%) | 73 (93.6%) | 0.608 | 0.067–5.468 | 0.654 | ||
| GA vs. GG + AA | 24 (96%) | 74 (94.9%) | 0.770 | 0.082–7.234 | 0.819 | ||
| GG + GA vs. AA | 25 (100%) | 77 (98.7%) | 1.013 | 0.040–25.65 | 0.993 * | ||
| G | 49 (98%) | 150 (96.1%) | Reference | Reference | Reference | ||
| A | 01 (02%) | 06 (3.9%) | 0.510 | 0.059–4.342 | 0.530 | ||
| HFE (H63D) | rs1799945 | CC | 24 (96%) | 54 (69.2%) | Reference | Reference | Reference |
| CG | 00 (00%) | 21 (26.9%) | 0.051 | 0.003–0.889 | 0.005 * | ||
| GG | 01 (04%) | 03 (3.90%) | 0.750 | 0.074–7.583 | 0.806 | ||
| CG + GG vs. CC | 24 (96%) | 54 (69.2%) | 0.093 | 0.011–0.733 | 0.006 | ||
| CG vs. CC + GG | 25 (100%) | 57 (73.1%) | 0.052 | 0.003–0.899 | 0.005 * | ||
| CC + CG vs. GG | 24 (06%) | 75 (96.1%) | 1.042 | 0.103–10.49 | 0.972 | ||
| C | 48 (96%) | 129 (82.6%) | Reference | Reference | Reference | ||
| G | 02 (04%) | 27 (17.4%) | 0.199 | 0.045–0.869 | 0.01 | ||
| HFE (C282Y) | rs1800562 | GG | 25 (100%) | 78 (100%) | Reference | Reference | Reference |
| GA | 00 (0%) | 00 (0%) | 3.078 | 0.059–159.1 | 0.556 * | ||
| AA | 00 (0%) | 00 (0%) | 3.078 | 0.059–159.1 | 0.556 * | ||
| G | 50 (100%) | 156 (100%) | Reference | Reference | Reference | ||
| A | 00 (0%) | 00 (0%) | 3.099 | 0.060–158.2 | 0.552 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsaif, F.; Al-hamoudi, W.; Alotaiby, M.; Alsadoon, A.; Almayouf, M.; Almadany, H.; Abuhaimed, J.; Ghufran, N.; Merajuddin, A.; Ali Khan, I. Molecular Screening via Sanger Sequencing of the Genetic Variants in Non-Alcoholic Fatty Liver Disease Subjects in the Saudi Population: A Hospital-Based Study. Metabolites 2022, 12, 1240. https://doi.org/10.3390/metabo12121240
Alsaif F, Al-hamoudi W, Alotaiby M, Alsadoon A, Almayouf M, Almadany H, Abuhaimed J, Ghufran N, Merajuddin A, Ali Khan I. Molecular Screening via Sanger Sequencing of the Genetic Variants in Non-Alcoholic Fatty Liver Disease Subjects in the Saudi Population: A Hospital-Based Study. Metabolites. 2022; 12(12):1240. https://doi.org/10.3390/metabo12121240
Chicago/Turabian StyleAlsaif, Faisal, Waleed Al-hamoudi, Maram Alotaiby, Amani Alsadoon, Mohammed Almayouf, Hadeel Almadany, Jawahir Abuhaimed, Noman Ghufran, Ahmed Merajuddin, and Imran Ali Khan. 2022. "Molecular Screening via Sanger Sequencing of the Genetic Variants in Non-Alcoholic Fatty Liver Disease Subjects in the Saudi Population: A Hospital-Based Study" Metabolites 12, no. 12: 1240. https://doi.org/10.3390/metabo12121240