
scFASTCORMICS: A Contextualization Algorithm to Reconstruct Metabolic Multi-Cell Population Models from Single-Cell RNAseq Data

 ,
,
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.2. Equipment
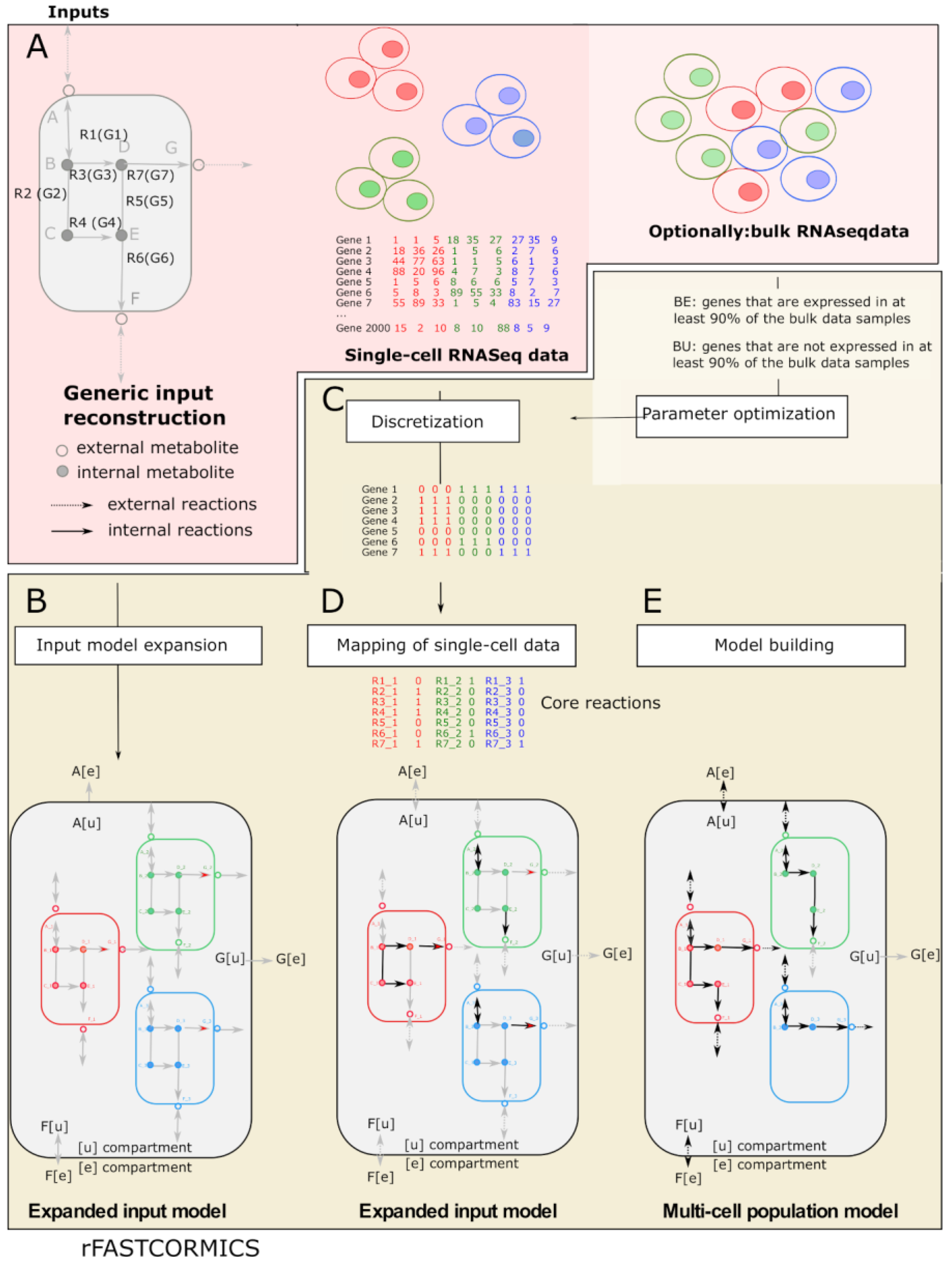
3. Procedure
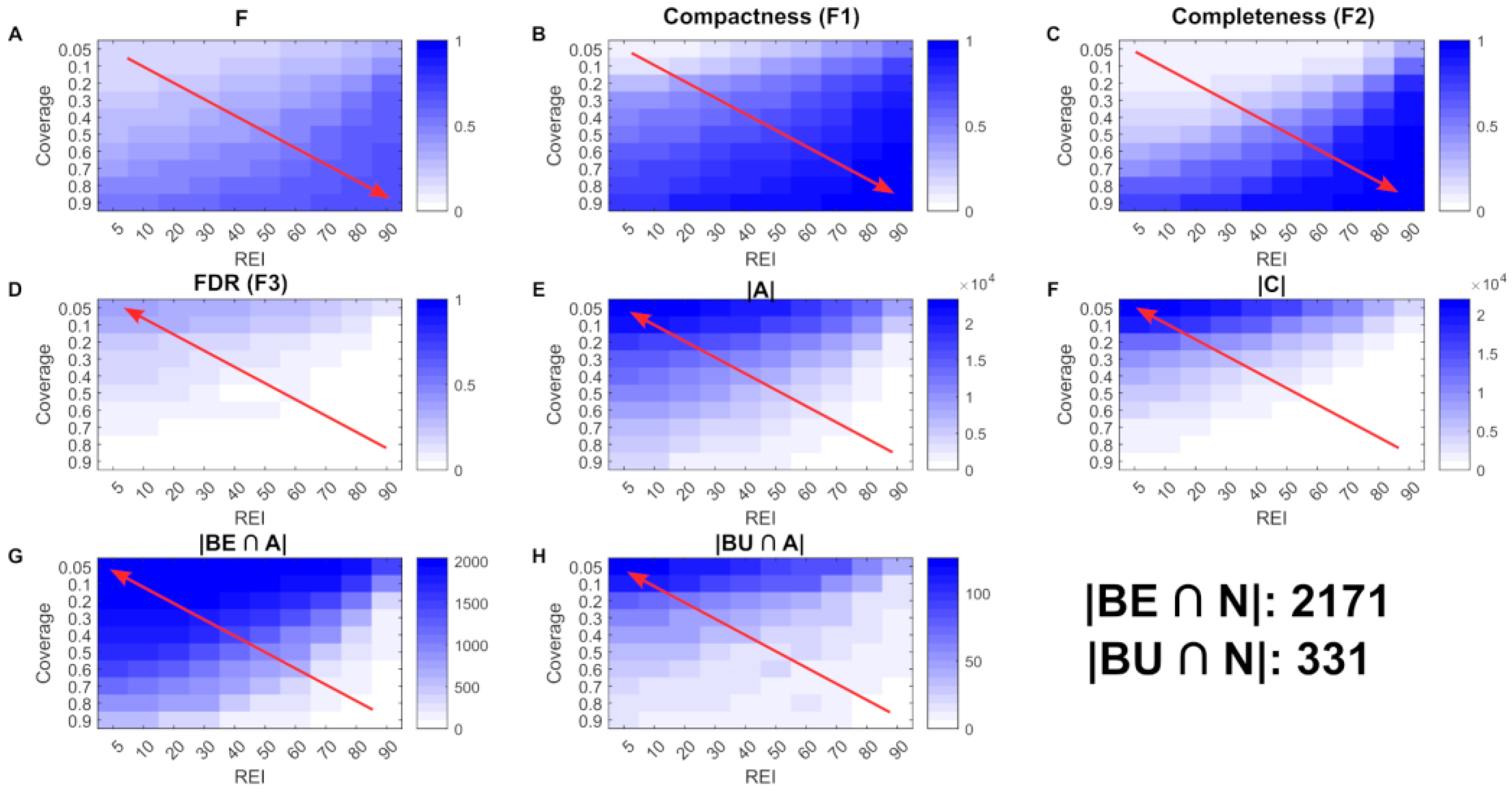
3.1. Parameter Optimization
3.2. Quality Check
3.3. Metabolite Exchange Prediction
4. Results
4.1. scFASTCORMICS Allows Building Compact, Complete and Specific Models Based on Single-Cell RNAseq Data
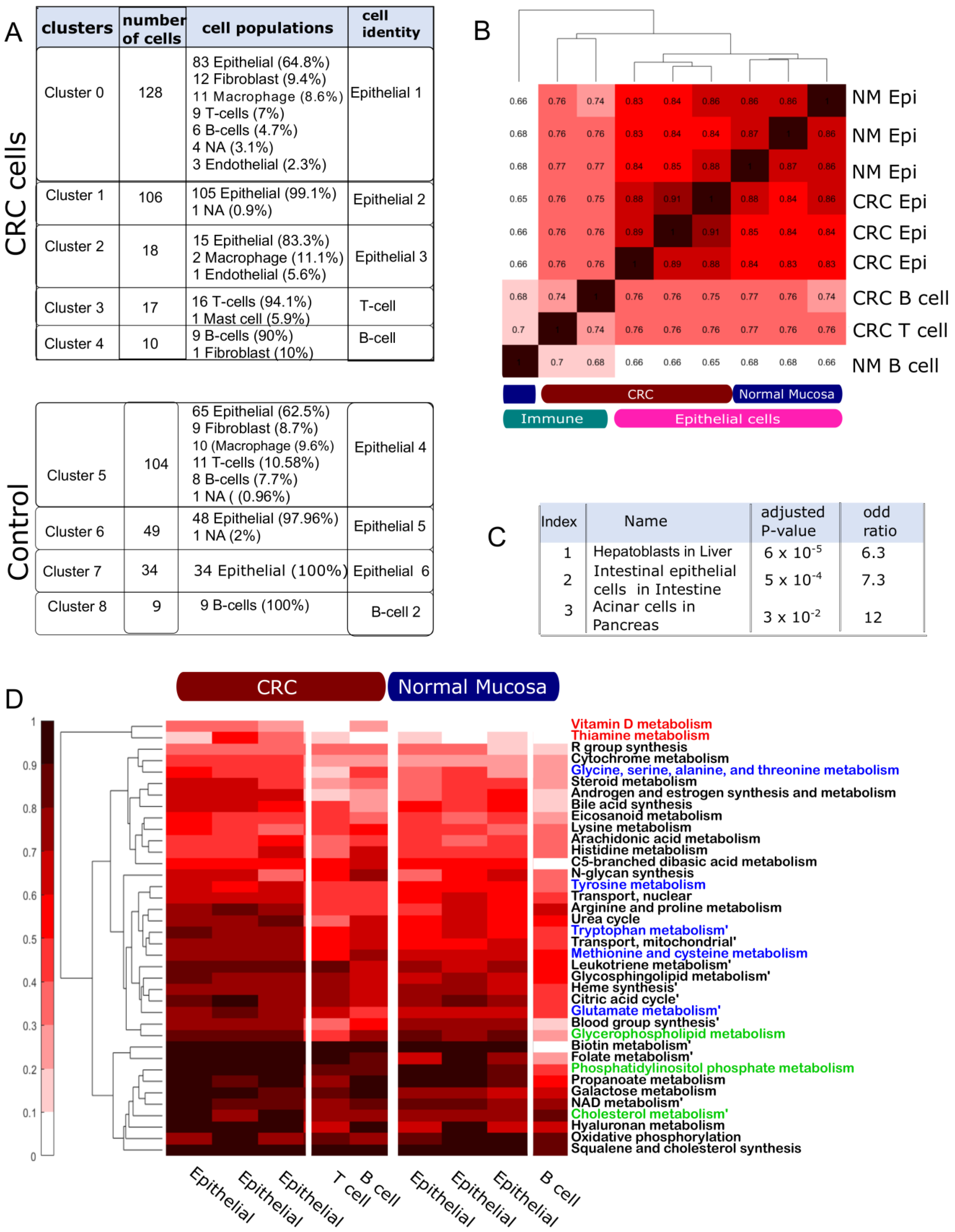
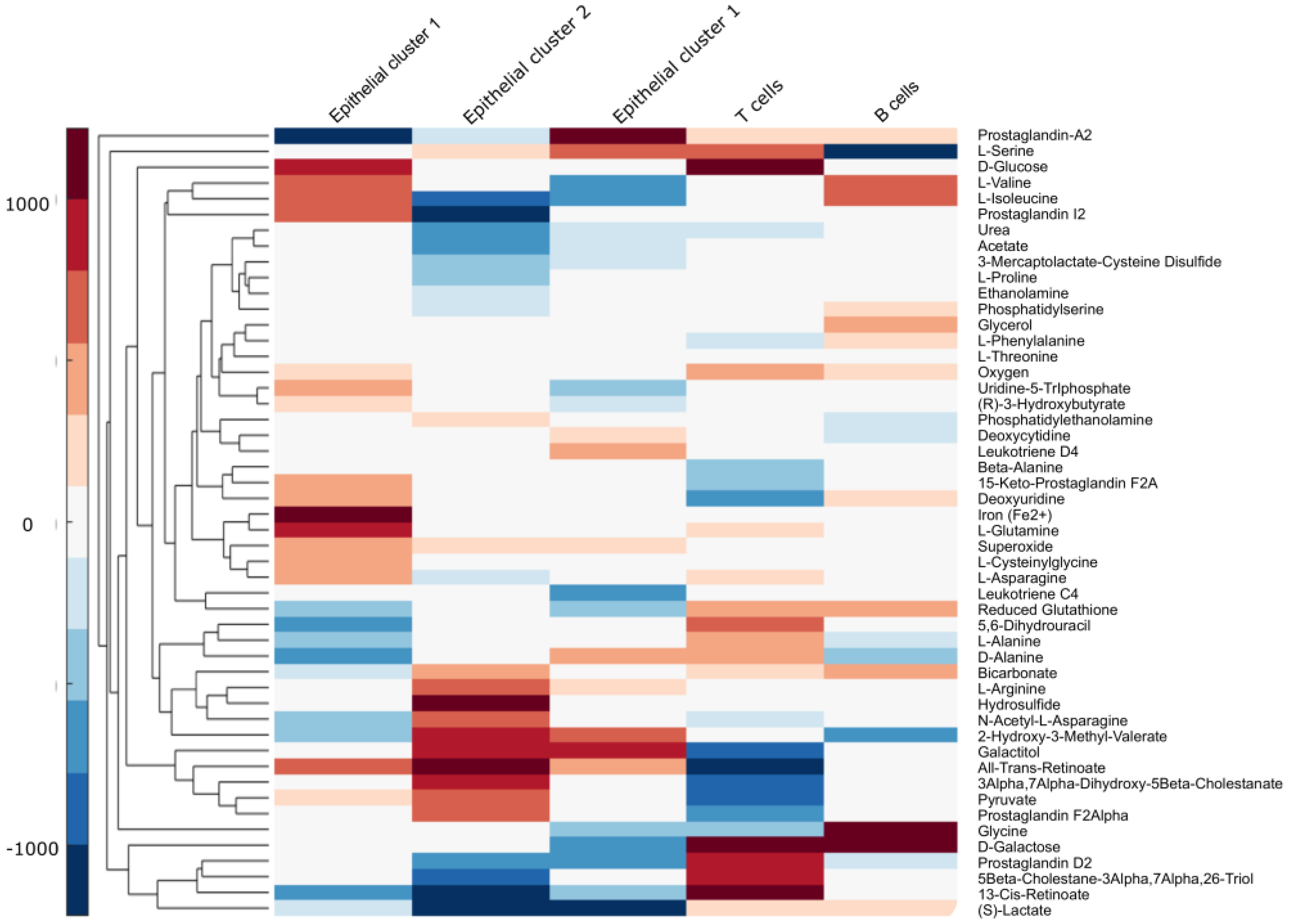
4.2. The Multi-Cell Population Model Captures Metabolic Variation among Cell Populations
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Tang, Y.Q.; Miao, H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm 2020, 1, 47–68. [Google Scholar] [CrossRef]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [Green Version]
- AlMusawi, S.; Ahmed, M.; Nateri, A.S. Understanding cell-cell communication and signaling in the colorectal cancer microenvironment. Clin. Transl. Med. 2021, 11, e308. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Tu, V.Y.; Ayari, A.; O’Connor, R.S. Beyond the lactate paradox: How lactate and acidity impact T cell therapies against cancer. Antibodies 2021, 10, 25. [Google Scholar] [CrossRef]
- Karta, J.; Bossicard, Y.; Kotzamanis, K.; Dolznig, H.; Letellier, E. Mapping the metabolic networks of tumor cells and cancer-associated fibroblasts. Cells 2021, 10, 304. [Google Scholar] [CrossRef]
- Ternes, D.; Tsenkova, M.; Pozdeev, V.I.; Meyers, M.; Koncina, E.; Atatri, S.; Schmitz, M.; Karta, J.; Schmoetten, M.; Heinken, A.; et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab. 2022, 4, 458–475. [Google Scholar] [CrossRef]
- Greenhalgh, K.; Ramiro-Garcia, J.; Heinken, A.; Ullmann, P.; Bintener, T.; Pacheco, M.P.; Baginska, J.; Shah, P.; Frachet, A.; Halder, R.; et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep. 2019, 27, 1621–1632.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Gatto, F.; Nielsen, J. Genome-scale modeling of human metabolism--a systems biology approach. Biotechnol. J. 2013, 8, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Goveia, J.; García-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.P.M.H.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [Green Version]
- Tourigny, D.S.; Goldberg, A.P.; Karr, J.R. Simulating single-cell metabolism using a stochastic flux-balance analysis algorithm. Biophys. J. 2021, 120, 5231–5242. [Google Scholar] [CrossRef]
- Wagner, A.; Wang, C.; Fessler, J.; DeTomaso, D.; Avila-Pacheco, J.; Kaminski, J.; Zaghouani, S.; Christian, E.; Thakore, P.; Schellhaass, B.; et al. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 2021, 184, 4168–4185.e21. [Google Scholar] [CrossRef]
- Alghamdi, N.; Chang, W.; Dang, P.; Lu, X.; Wan, C.; Gampala, S.; Huang, Z.; Wang, J.; Ma, Q.; Zang, Y.; et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021, 31, 1867–1884. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast reconstruction of compact context-specific metabolic network models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.P.; Sauter, T. The FASTCORE family: For the fast reconstruction of compact context-specific metabolic networks models. In Metabolic Network Reconstruction and Modeling; Springer: Berlin/Heidelberg, Germany, 2018; pp. 101–110. [Google Scholar]
- Pacheco, M.P.; John, E.; Kaoma, T.; Heinäniemi, M.; Nicot, N.; Vallar, L.; Bueb, J.L.; Sinkkonen, L.; Sauter, T. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genom. 2015, 16, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Reconstruction of genome-scale human metabolic models using omics data. Integr. Biol. 2015, 7, 859–868. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat, G.A.; Aurich, M.K.; Sastry, A.; et al. Recon3D: A resource enabling a three-dimensional view of gene variation in Human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Edgar, R. Gene Expression Omnibus: Microarray data storage, submission, retrieval, and analysis. Methods Enzym. 2006, 411, 352–369. [Google Scholar]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Maseda, D.; Ricciotti, E.; Crofford, L.J. Prostaglandin regulation of T cell biology. Pharmacol. Res. 2019, 149, 104456. [Google Scholar] [CrossRef]
- Bono, M.R.; Tejon, G.; Flores-Santibañez, F.; Fernandez, D.; Rosemblatt, M.; Sauma, D. Retinoic acid as a modulator of T cell immunity. Nutrients 2016, 8, 349. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, V.M.; Edwards, D.N.; Philip, M.; Chen, J. Microenvironmental metabolism regulates antitumor immunity. Cancer Res. 2019, 79, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Budinich, M.; Bourdon, J.; Larhlimi, A.; Eveillard, D. A multi-objective constraint-based approach for modeling genome-scale microbial ecosystems. PLoS ONE 2017, 12, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2021, 8, 603837. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, D.; Link, H.; Fuhrer, T.; Kochanowski, K.; Gerosa, L.; Sauer, U. Reserve Flux Capacity in the Pentose Phosphate Pathway Enables Escherichia coli’s Rapid Response to Oxidative Stress. Cell Syst. 2018, 6, 569–578.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Gu, I.; Gregory, E.; Atwood, C.; Lee, S.O.; Song, Y.H. Exploring the Role of Metabolites in Cancer and the Associated Nerve Crosstalk. Nutrients 2022, 14, 1722. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin D Deficiency: Effects on Oxidative Stress, Epigenetics, Gene Regulation, and Aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Quinn, W.J.; Jiao, J.; TeSlaa, T.; Stadanlick, J.; Wang, Z.; Wang, L.; Akimova, T.; Angelin, A.; Schäfer, P.M.; Cully, M.D.; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 2020, 33, 108500. [Google Scholar] [CrossRef]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef]
- Wang, W.; Guo, M.N.; Li, N.; Pang, D.Q.W.J.; Wu, J.-H. WJGO-14-1124.pdf 2022, 1124–1140. Available online: https://www.f6publishing.com/FeaturedArticleDetail?id=74252 (accessed on 21 September 2022).
- Hawcroft, G.; Gardner, S.H.; Hull, M.A. Expression of prostaglandin D2 receptors DP1 and DP2 by human colorectal cancer cells. Cancer Lett. 2004, 210, 81–84. [Google Scholar] [CrossRef]
- Devalaraja, S.; To, T.K.J.; Folkert, I.W.; Natesan, R.; Alam, M.Z.; Li, M.; Tada, Y.; Budagyan, K.; Dang, M.T.; Zhai, L.; et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell 2020, 180, 1098–1114.e16. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, L.; Zhang, L.; Hu, X.; Ren, X.; Zhang, Z. Deep single-cell RNA sequencing data of individual T cells from treatment-naïve colorectal cancer patients. Sci. Data 2019, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Zhou, F.; Tang, D.; Lin, L.; Zou, C.; Tan, W.; Dai, Y.; Yi, K. Single-cell transcriptional profiling reveals the heterogenicity in colorectal cancer. Medicine 2019, 98, e16916. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, S.; Yang, S. An Effcient and Flexible Method for Deconvoluting Bulk RNA-Seq Data with Single-Cell RNA-Seq Data. Cells 2019, 8, 1161. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Number of Reactions | Number of Metabolites | Number of Genes |
|---|---|---|---|
| Input generic model (Recon 3D) | 10,600 | 5835 | 1883 |
| Expanded input model CRC | 48,320 | 24,495 | 11,240 |
| Expanded input model NM | 39,280 | 20,220 | 8992 |
| Multi-cell population CRC | 22,961 | 16,292 | 9231 |
| Multi-cell population NM | 17,429 | 12,758 | 7327 |
| Cluster 0 sub-model (CRC epithelium) | 4731 | 3165 | 1896 |
| Cluster 1 sub-model (CRC epithelium) | 4678 | 3157 | 1951 |
| Cluster 2 sub-model (CRC epithelium) | 4542 | 3078 | 1844 |
| Cluster 3 sub-model (CRC T cell) | 3902 | 2777 | 1777 |
| Cluster 4 sub-model (CRC B cell) | 3988 | 2835 | 1763 |
| Cluster 5 sub-model (NM epithelium) | 4224 | 2993 | 1841 |
| Cluster 6 sub-model (NM epithelium) | 4344 | 3032 | 1916 |
| Cluster 7 sub-model (NM epithelium) | 4340 | 3012 | 1871 |
| Cluster 8 sub-model (NM B-cell) | 3457 | 2513 | 1699 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco, M.P.; Ji, J.; Prohaska, T.; García, M.M.; Sauter, T. scFASTCORMICS: A Contextualization Algorithm to Reconstruct Metabolic Multi-Cell Population Models from Single-Cell RNAseq Data. Metabolites 2022, 12, 1211. https://doi.org/10.3390/metabo12121211
Pacheco MP, Ji J, Prohaska T, García MM, Sauter T. scFASTCORMICS: A Contextualization Algorithm to Reconstruct Metabolic Multi-Cell Population Models from Single-Cell RNAseq Data. Metabolites. 2022; 12(12):1211. https://doi.org/10.3390/metabo12121211
Chicago/Turabian StylePacheco, Maria Pires, Jimmy Ji, Tessy Prohaska, María Moscardó García, and Thomas Sauter. 2022. "scFASTCORMICS: A Contextualization Algorithm to Reconstruct Metabolic Multi-Cell Population Models from Single-Cell RNAseq Data" Metabolites 12, no. 12: 1211. https://doi.org/10.3390/metabo12121211