
Natural Products Targeting Hsp90 for a Concurrent Strategy in Glioblastoma and Neurodegeneration

 ,
,  , and
, and 
Abstract
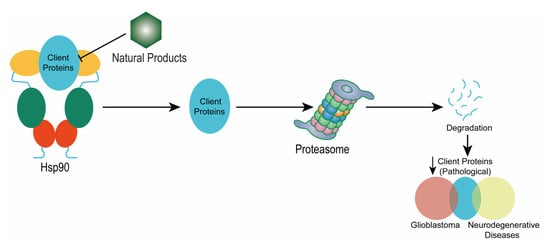
:
{kind=link}
{kind=link}
{kind=link}
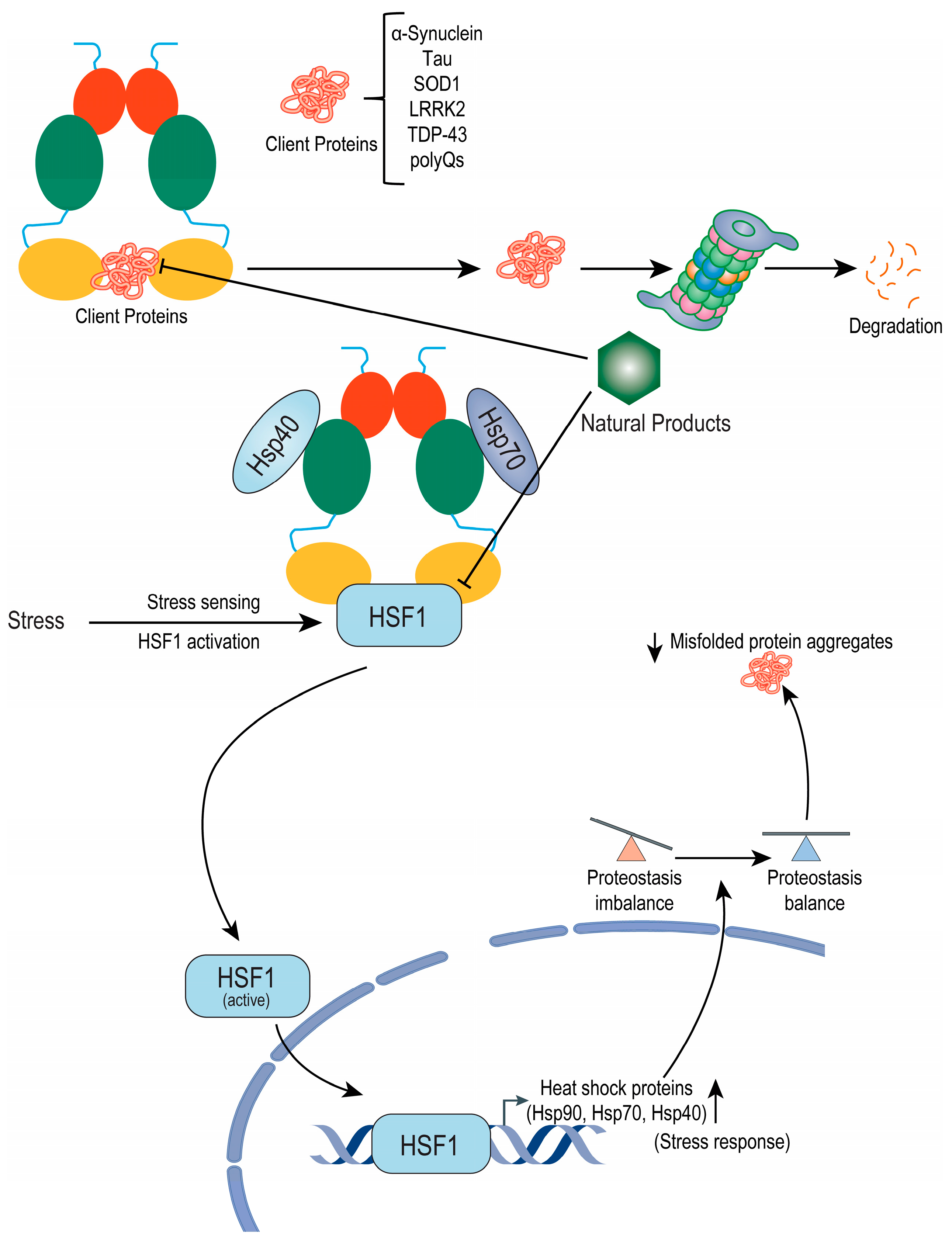{kind=link}
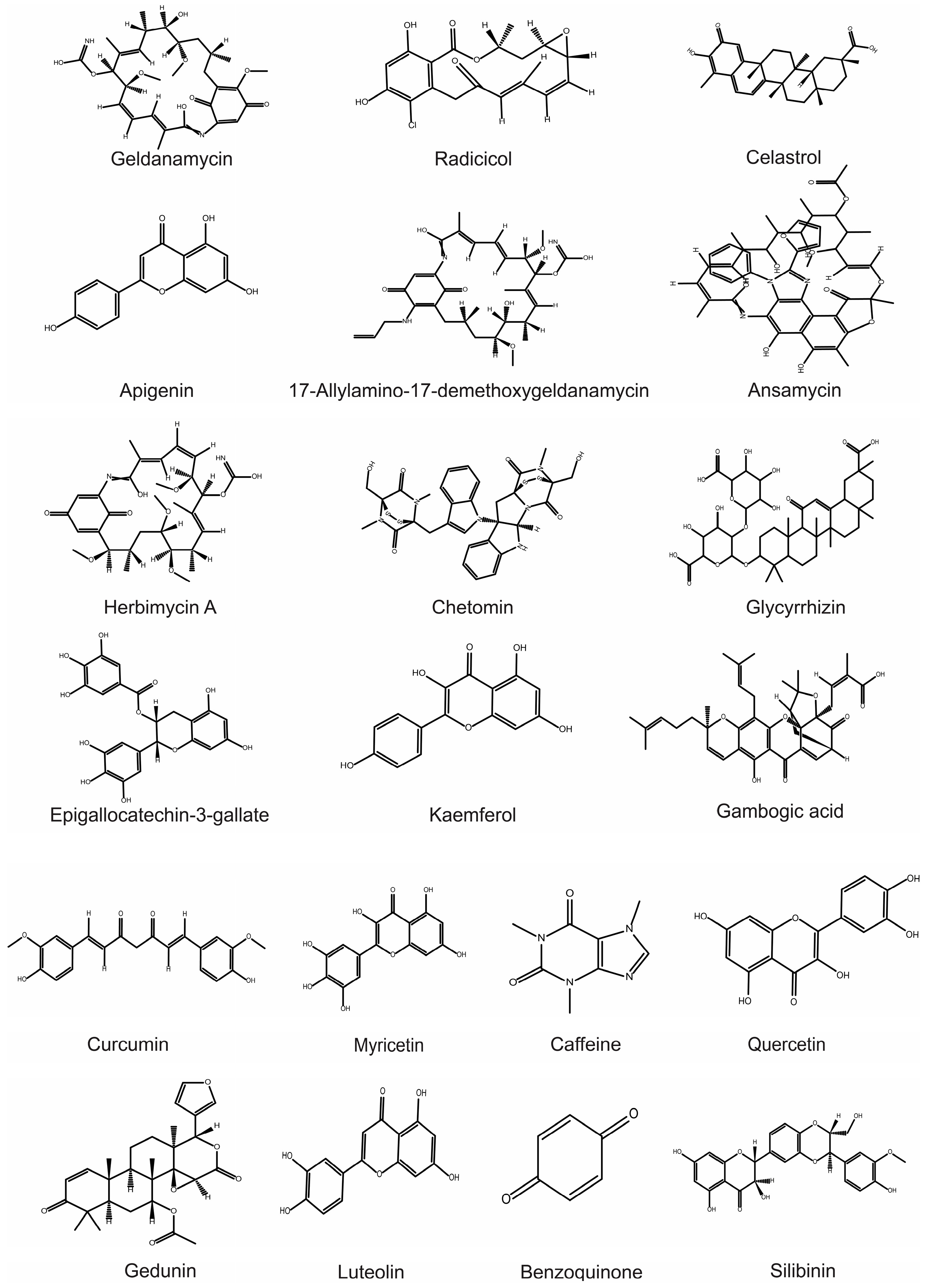{kind=link}
1. Introduction
2. An Overview of Glioblastoma Multiforme (GBM)
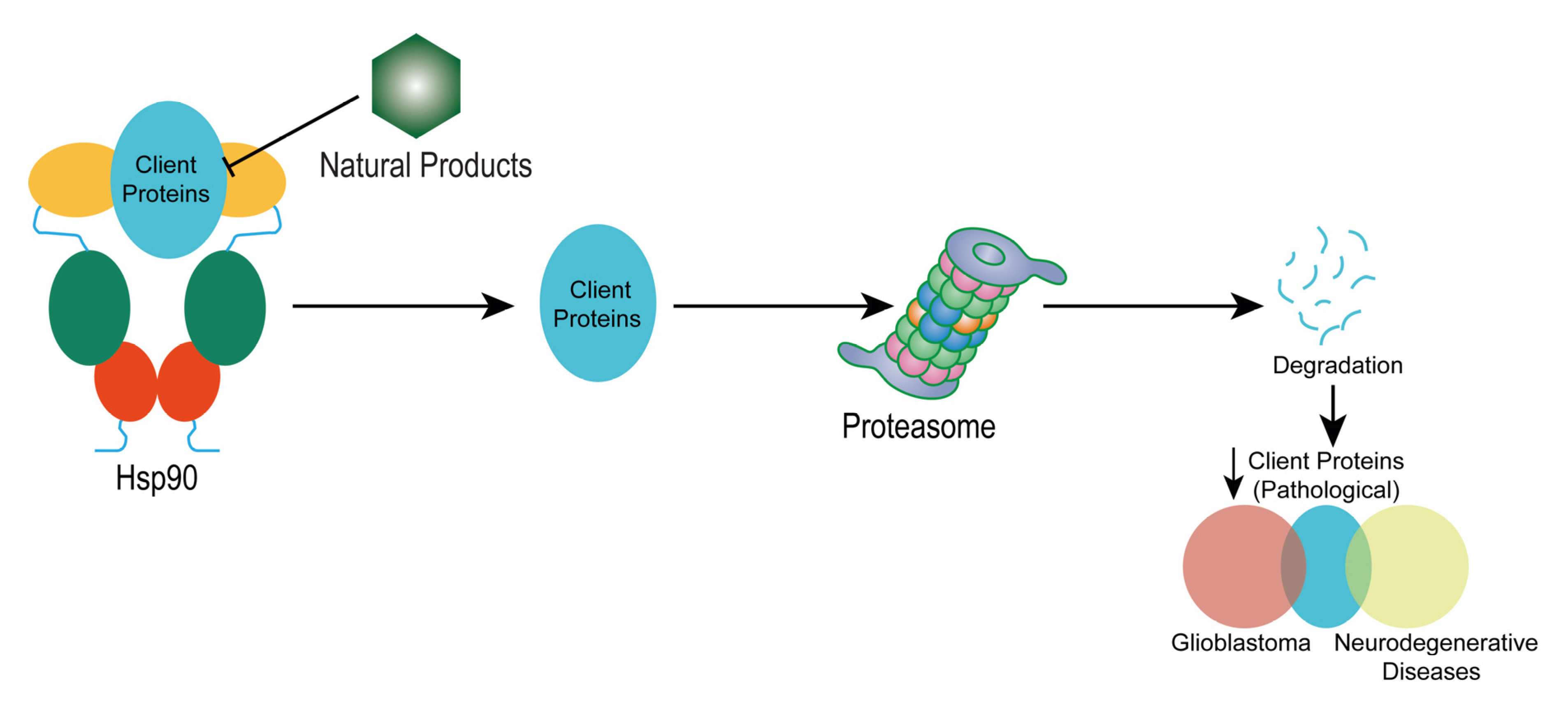
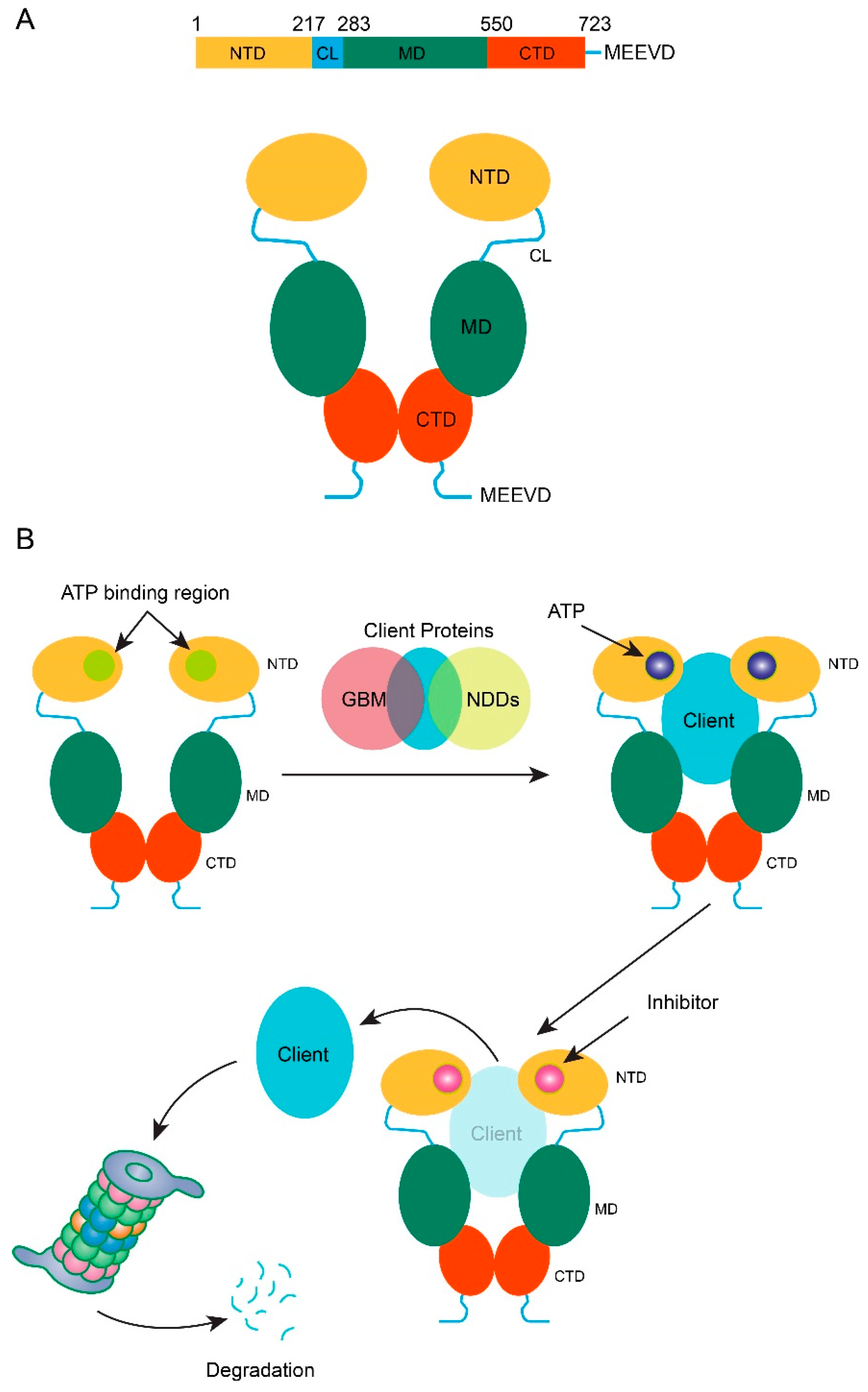
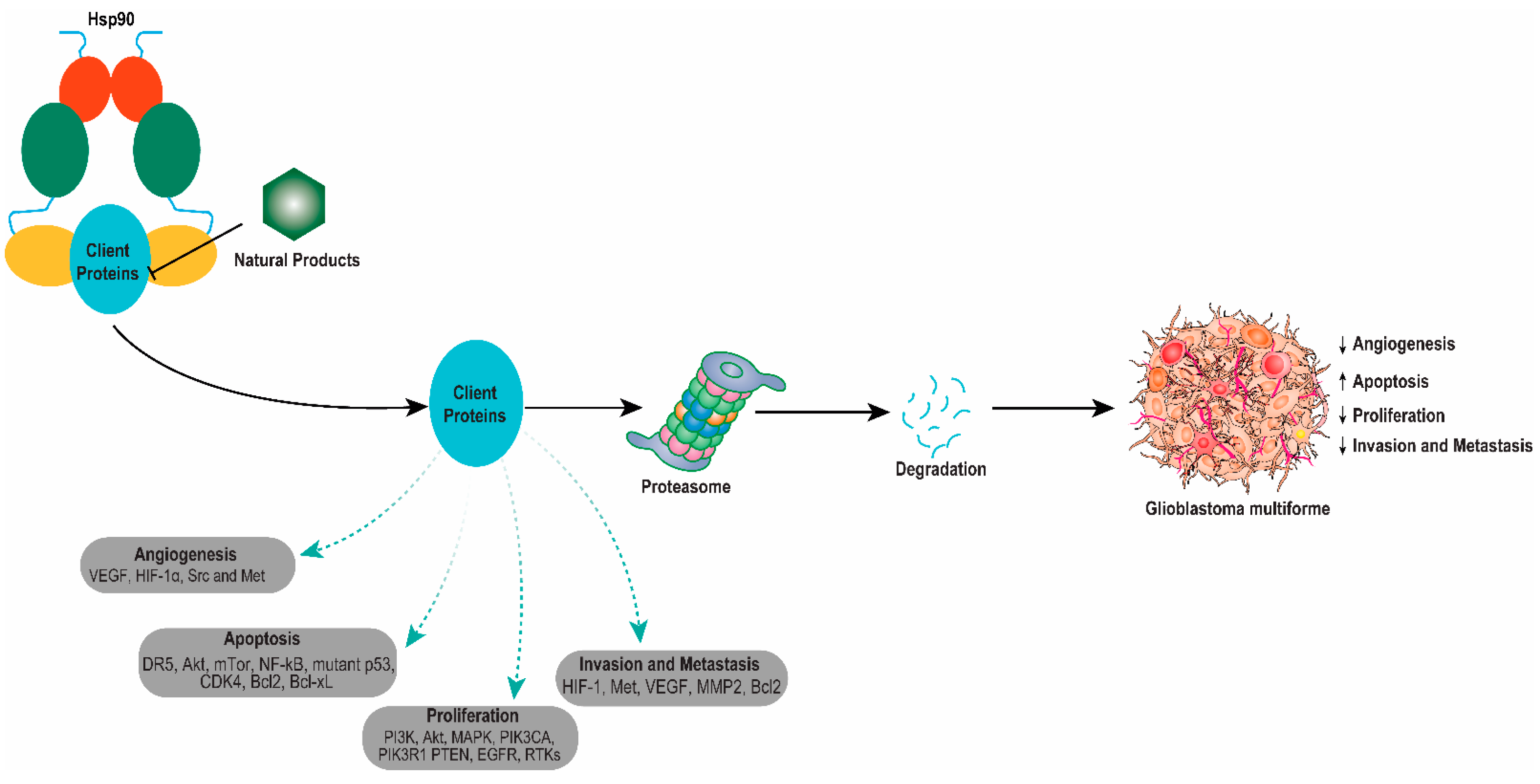
3. Involvement of Hsp90 in Glioblastoma Multiforme (GBM) Progression
4. HSP90 Inhibition in the Aggressiveness of Glioblastoma Multiforme (GBM)
5. Implication of Glioblastoma Multiforme (GBM) in Neurodegeneration
6. Inhibition of Hsp90 in Neurodegeneration
7. Natural Products Targeting Hsp90
7.1. Quinone
7.2. Polyphenol
7.3. Triterpenoid
7.4. Xanthone
7.5. Flavonoid
8. Conclusions and Future Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Squillaro, T.; Schettino, C.; Sampaolo, S.; Galderisi, U.; Di Iorio, G.; Giordano, A.; Melone, M.A.B. Adult-onset brain tumors and neurodegeneration: Are polyphenols protective? J. Cell. Physiol. 2018, 233, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, K.; Clavreul, A.; Lagarce, F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 2015, 20, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Kanu, O.O.; Mehta, A.; Di, C.; Lin, N.; Bortoff, K.; Bigner, D.D.; Yan, H.; Adamson, D.C. Glioblastoma multiforme: A review of therapeutic targets. Expert Opin Ther. Targets 2009, 13, 701–718. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N. Rooting out resistance. Nat. Rev. Cancer 2006, 6, 904. [Google Scholar] [CrossRef]
- Minniti, G.; Muni, R.; Lanzetta, G.; Marchetti, P.; Enrici, R.M. Chemotherapy for glioblastoma: Current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009, 29, 5171–5184. [Google Scholar] [PubMed]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2021, 7, 620677. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Dash, R.; Sohel, M.; Chowdhury, A.; Munni, Y.A.; Ali, M.C.; Hannan, M.A.; Islam, M.T.; Moon, I.S. Targeting estrogen signaling in the radiation-induced neurodegeneration: Possible role of phytoestrogens. Curr. Neuropharm. 2022. [Google Scholar] [CrossRef]
- Hoffermann, M.; Bruckmann, L.; Mahdy Ali, K.; Zaar, K.; Avian, A.; von Campe, G. Pre- and postoperative neurocognitive deficits in brain tumor patients assessed by a computer based screening test. J. Clin. Neurosci. 2017, 36, 31–36. [Google Scholar] [CrossRef]
- Gehrke, A.K.; Baisley, M.C.; Sonck, A.L.; Wronski, S.L.; Feuerstein, M. Neurocognitive deficits following primary brain tumor treatment: Systematic review of a decade of comparative studies. J. Neurooncol. 2013, 115, 135–142. [Google Scholar] [CrossRef]
- Bergo, E.; Lombardi, G.; Pambuku, A.; Della Puppa, A.; Bellu, L.; D’Avella, D.; Zagonel, V. Cognitive Rehabilitation in Patients with Gliomas and Other Brain Tumors: State of the Art. Biomed. Res. Int. 2016, 2016, 3041824. [Google Scholar] [CrossRef] [PubMed]
- Granholm, A.C.; Boger, H.; Emborg, M.E. Mood, memory and movement: An age-related neurodegenerative complex? Curr. Aging Sci. 2008, 1, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Levenson, R.W.; Sturm, V.E.; Haase, C.M. Emotional and behavioral symptoms in neurodegenerative disease: A model for studying the neural bases of psychopathology. Annu. Rev. Clin. Psychol. 2014, 10, 581–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, D.D. Neurocognitive function in brain tumors. Curr. Neurol. Neurosci. Rep. 2010, 10, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Embry, L. Neurocognitive dysfunction in survivors of childhood brain tumors. Semin. Pediatr. Neurol. 2012, 19, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zucchella, C.; Bartolo, M.; Di Lorenzo, C.; Villani, V.; Pace, A. Cognitive impairment in primary brain tumors outpatients: A prospective cross-sectional survey. J. Neurooncol. 2013, 112, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Clare, D.K.; Saibil, H.R. ATP-driven molecular chaperone machines. Biopolymers 2013, 99, 846–859. [Google Scholar] [CrossRef]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, Á.S.T.; Erices, J.I.; Uribe, D.A.; Howden, J.; Niechi, I.; Muñoz, S.; Martín, R.S.; Monrás, C.A.Q. Current Therapeutic Alternatives and New Perspectives in Glioblastoma Multiforme. Curr. Med. Chem. 2017, 24, 2781–2795. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Di, C.; Mattox, A.K.; Wu, L.; Adamson, D.C. The future role of personalized medicine in the treatment of glioblastoma multiforme. Pharmgenom. Pers. Med. 2010, 3, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.s.; et al. Genetic Pathways to Glioblastoma: A Population-Based Study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovčevska, I.; Kočevar, N.; Komel, R. Glioma and glioblastoma-how much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Eckley, M.; Wargo, K.A. A review of glioblastoma multiforme. US Pharm. 2010, 35, 3–10. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro Oncol. 2002, 4, 278–299. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; de Ribaupierre, S.; Wöhrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, M.; Lee, M.; Miike, R.; Newman, B.; Bargar, G.; Davis, R.; Wiencke, J.; Neuhaus, J. Familial and Personal Medical History of Cancer and Nervous System Conditions among Adults with Glioma and Controls. Am. J. Epidemiol. 1997, 145, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.J.; Cha, S. Imaging glioblastoma multiforme. Cancer J. 2003, 9, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Frosch, M.P. Central Nervous System in Robbins Basic Pathology; Kumar, V., Abbas, A.K., Astor, J.C., Eds.; Elsevier Saunders: Philadelphia, PA, USA, 2013. [Google Scholar]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Chang, C.H.; Horton, J.; Schoenfeld, D.; Salazer, O.; Perez-Tamayo, R.; Kramer, S.; Weinstein, A.; Nelson, J.S.; Tsukada, Y. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint radiation therapy oncology group and eastern cooperative oncology group study. Cancer 1983, 52, 997–1007. [Google Scholar] [CrossRef]
- Simpson, J.R.; Horton, J.; Scott, C.; Curran, W.J.; Rubin, P.; Fischbach, J.; Isaacson, S.; Rotman, M.; Asbell, S.O.; Nelson, J.S.; et al. Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: Results of three consecutive radiation therapy oncology group (RTOG) clinical trials. Int. J. Radiat. Oncol. Biol. Phys. 1993, 26, 239–244. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Kristiansen, K.; Hagen, S.; Kollevold, T.; Torvik, A.; Holme, I.; Stat, M.; Nesbakken, R.; Hatlevoll, R.; Lindgren, M.; Brun, A.; et al. Combined modality therapy of operated astrocytomas grade III and IV. Confirmation of the value of postoperative irradiation and lack of potentiation of bleomycin on survival time: A prospective multicenter trial of the scandinavian glioblastoma study group. Cancer 1981, 47, 649–652. [Google Scholar] [CrossRef]
- Walker, M.D.; Alexander, E.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S.; Mealey, J.; Norrell, H.A.; Owens, G.; Ransohoff, J.; Wilson, C.B.; et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas: A cooperative clinical trial. J. Neurosurg. 1978, 49, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.J.; Fort, P.E. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Neurosci. 2018, 12, 821. [Google Scholar] [CrossRef] [Green Version]
- Subbarao Sreedhar, A.; Kalmár, É.; Csermely, P.; Shen, Y.-F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; You, Q.D.; Xu, X.L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Mollapour, M.; Prodromou, C.; Lee, C.-T.; Panaretou, B.; Yoshida, S.; Mayer, M.P.; Neckers, L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA 2012, 109, 2937–2942. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hsp90: Friends, clients and natural foes. Biochimie 2016, 127, 227–240. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Et Biophys. Acta. (BBA)-Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [Green Version]
- Holzbeierlein, J.M.; Windsperger, A.; Vielhauer, G. Hsp90: A Drug Target? Curr. Oncol. Rep. 2010, 12, 95–101. [Google Scholar] [CrossRef]
- Amolins, M.W.; Blagg, B.S. Natural product inhibitors of Hsp90: Potential leads for drug discovery. Mini Rev. Med. Chem. 2009, 9, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Graner, M.W.; Bigner, D.D. Chaperone proteins and brain tumors: Potential targets and possible therapeutics. Neuro-Oncol 2005, 7, 260–278. [Google Scholar] [CrossRef] [PubMed]
- Zohrabian, V.M.; Forzani, B.; Chau, Z.; Murali, R.A.J.; Jhanwar-Uniyal, M. Rho/ROCK and MAPK Signaling Pathways Are Involved in Glioblastoma Cell Migration and Proliferation. Anticancer Res. 2009, 29, 119. [Google Scholar] [PubMed]
- Waqas, M.; Enam, S.A.; Batool, M.; Rai, H.H. Basic mechanisms of glioblastoma multiforme cell invasion: A review article. J. Neurol. Neurosci. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Maity, A.; Pore, N.; Lee, J.; Solomon, D.; O’Rourke, D.M. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 2000, 60, 5879–5886. [Google Scholar]
- Sanderson, S.; Valenti, M.; Gowan, S.; Patterson, L.; Ahmad, Z.; Workman, P.; Eccles, S.A. Benzoquinone ansamycin heat shock protein 90 inhibitors modulate multiple functions required for tumor angiogenesis. Mol. Cancer Ther. 2006, 5, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Lauber, K.; Brix, N.; Ernst, A.; Hennel, R.; Krombach, J.; Anders, H.; Belka, C. Targeting the heat shock response in combination with radiotherapy: Sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity. Cancer Lett. 2015, 368, 209–229. [Google Scholar] [CrossRef]
- van Ommeren, R.; Staudt, M.D.; Xu, H.; Hebb, M.O. Advances in HSP27 and HSP90-targeting strategies for glioblastoma. J. Neurooncol. 2016, 127, 209–219. [Google Scholar] [CrossRef]
- Whitesell, L.; Sutphin, P.D.; Pulcini, E.J.; Martinez, J.D.; Cook, P.H. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol. Cell Biol. 1998, 18, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Saidon, C.; Carmi, I.; Keren, A.; Razin, E. Antiapoptotic function of Bcl-2 in mast cells is dependent on its association with heat shock protein 90β. Blood 2006, 107, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-C.; Liang, C.-M.; Lai, C.-Y.; Liang, S.-M. Involvement of Heat Shock Protein (Hsp)90β but Not Hsp90α in Antiapoptotic Effect of CpG-B Oligodeoxynucleotide. J. Immunol. 2007, 178, 6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matts, R.L.; Manjarrez, J.R. Assays for identification of Hsp90 inhibitors and biochemical methods for discriminating their mechanism of action. Curr. Top Med. Chem. 2009, 9, 1462–1478. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Alarcon, S.V.; Lee, S.; Lee, M.J.; Giaccone, G.; Neckers, L.; Trepel, J.B. Update on Hsp90 inhibitors in clinical trial. Curr. Top Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.Y.; Ham, S.W.; Kim, J.K.; Jin, X.; Lee, S.Y.; Shin, Y.J.; Choi, C.Y.; Sa, J.K.; Kim, S.H.; Chun, T.; et al. Ly6G(+) inflammatory cells enable the conversion of cancer cells to cancer stem cells in an irradiated glioblastoma model. Cell Death Differ. 2019, 26, 2139–2156. [Google Scholar] [CrossRef]
- Otomo, T.; Hishii, M.; Arai, H.; Sato, K.; Sasai, K. Microarray analysis of temporal gene responses to ionizing radiation in two glioblastoma cell lines: Up-regulation of DNA repair genes. J. Radiat. Res. 2004, 45, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Albrecht, V.; Seidl, K.; Kinzel, L.; Unger, K.; Hess, J.; Kreutzer, L.; Sun, N.; Stegen, B.; Nieto, A.; et al. Inhibition of HSP90 as a Strategy to Radiosensitize Glioblastoma: Targeting the DNA Damage Response and Beyond. Front. Oncol. 2021, 11, 612354. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gong, Y.; Ma, Y.; Thompson, R.C.; Wang, J.; Cheng, Z.; Xue, L. A Brain-Penetrating Hsp90 Inhibitor NXD30001 Inhibits Glioblastoma as a Monotherapy or in Combination With Radiation. Front. Pharm. 2020, 11, 974. [Google Scholar] [CrossRef]
- Choi, E.J.; Cho, B.J.; Lee, D.J.; Hwang, Y.H.; Chun, S.H.; Kim, H.H.; Kim, I.A. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Sasame, J.; Ikegaya, N.; Kawazu, M.; Natsumeda, M.; Hayashi, T.; Isoda, M.; Satomi, K.; Tomiyama, A.; Oshima, A.; Honma, H.; et al. HSP90 Inhibition Overcomes Resistance to Molecular Targeted Therapy in BRAFV600E-mutant High-grade Glioma. Clin. Cancer Res. 2022, 28, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Sharp, S.Y.; Eccles, S.A.; Gowan, S.; Popov, S.; Jones, C.; Pearson, A.; Vassal, G.; Workman, P. Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and pediatric glioblastoma. Mol. Cancer Ther. 2010, 9, 1219–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Yang, S.; Chi, G.; Jin, X. Hsp90 inhibitor NMS-E973 exerts the anticancer effect against glioblastoma via induction of PUMA-mediated apoptosis. Onco. Targets Ther. 2018, 11, 1583–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachsberger, P.R.; Lawrence, Y.R.; Liu, Y.; Rice, B.; Feo, N.; Leiby, B.; Dicker, A.P. Hsp90 inhibition enhances PI-3 kinase inhibition and radiosensitivity in glioblastoma. J. Cancer Res. Clin. Oncol. 2014, 140, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yin, D.; Yu, S.; Lin, X.; Savani, M.R.; Du, K.; Ku, Y.; Wu, D.; Li, S.; Liu, H.; et al. Antitumor Activity of a Mitochondrial-Targeted HSP90 Inhibitor in Gliomas. Clin. Cancer Res. 2022, 28, 2180–2195. [Google Scholar] [CrossRef]
- Ho, K.T.; Chen, P.F.; Chuang, J.Y.; Gean, P.W.; Hsueh, Y.S. A heat shock protein 90 inhibitor reduces oncoprotein expression and induces cell death in heterogeneous glioblastoma cells with EGFR, PDGFRA, CDK4, and NF1 aberrations. Life Sci. 2022, 288, 120176. [Google Scholar] [CrossRef]
- Li, T.; Mehraein-Ghomi, F.; Forbes, M.E.; Namjoshi, S.V.; Ballard, E.A.; Song, Q.; Chou, P.-C.; Wang, X.; Parker Kerrigan, B.C.; Lang, F.F.; et al. HSP90-CDC37 functions as a chaperone for the oncogenic FGFR3-TACC3 fusion. Mol. Ther. 2022, 30, 1610–1627. [Google Scholar] [CrossRef]
- Memmel, S.; Sisario, D.; Zöller, C.; Fiedler, V.; Katzer, A.; Heiden, R.; Becker, N.; Eing, L.; Ferreira, F.L.R.; Zimmermann, H.; et al. Migration pattern, actin cytoskeleton organization and response to PI3K-, mTOR-, and Hsp90-inhibition of glioblastoma cells with different invasive capacities. Oncotarget 2017, 8, 45298–45310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, T.; Tojo, N.; Ohnishi, K. Preferential radiosensitization to glioblastoma cancer stem cell-like cells by a Hsp90 inhibitor, N-vinylpyrrolidone-AUY922. Oncol. Lett. 2022, 23, 102. [Google Scholar] [CrossRef]
- Xu, J.; Wu, P.J.; Lai, T.H.; Sharma, P.; Canella, A.; Welker, A.M.; Beattie, C.E.; Elder, J.B.; Easley, M.; Lonser, R.; et al. Disruption of DNA Repair and Survival Pathways through Heat Shock Protein Inhibition by Onalespib to Sensitize Malignant Gliomas to Chemoradiation Therapy. Clin. Cancer Res. 2022, 28, 1979–1990. [Google Scholar] [CrossRef]
- Canella, A.; Welker, A.M.; Yoo, J.Y.; Xu, J.; Abas, F.S.; Kesanakurti, D.; Nagarajan, P.; Beattie, C.E.; Sulman, E.P.; Liu, J.; et al. Efficacy of Onalespib, a Long-Acting Second-Generation HSP90 Inhibitor, as a Single Agent and in Combination with Temozolomide against Malignant Gliomas. Clin. Cancer Res. 2017, 23, 6215–6226. [Google Scholar] [CrossRef]
- Mummudi, N.; Jalali, R. Palliative care and quality of life in neuro-oncology. F1000Prime Rep. 2014, 6, 71. [Google Scholar] [CrossRef]
- Villa, C.; Miquel, C.; Mosses, D.; Bernier, M.; Di Stefano, A.L. The 2016 World Health Organization classification of tumours of the central nervous system. La Presse Médicale 2018, 47, e187–e200. [Google Scholar] [CrossRef] [PubMed]
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tintó, S. Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol. 2019, 17, e3000545. [Google Scholar] [CrossRef] [PubMed]
- Herting, C.J.; Chen, Z.; Maximov, V.; Duffy, A.; Szulzewsky, F.; Shayakhmetov, D.M.; Hambardzumyan, D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 2019, 142, 3834–3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.S. Alzheimer pathology in elderly patients with glioblastoma multiforme. Arch. Pathol. Lab. Med. 2002, 126, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- Klotz, S.; Ricken, G.; Preusser, M.; Dieckmann, K.; Widhalm, G.; Rössler, K.; Fischer, P.; Kalev, O.; Wöhrer, A.; Kovacs, G.G.; et al. Enhanced expression of autophagy-related p62 without increased deposits of neurodegeneration-associated proteins in glioblastoma and surrounding tissue—An autopsy-based study. Brain Pathol. 2022, 32, e13058. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.; Garcia-Rendueles, M.E.R. Oncogenic Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 3223. [Google Scholar] [CrossRef]
- Brat, D.J.; Gearing, M.; Goldthwaite, P.T.; Wainer, B.H.; Burger, P.C. Tau-associated neuropathology in ganglion cell tumours increases with patient age but appears unrelated to ApoE genotype. Neuropathol. Appl. Neurobiol. 2001, 27, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Soffer, D.; Umansky, F.; Goldman, J.E. Ganglioglioma with neurofibrillary tangles (NFTs): Neoplastic NFTs share antigenic determinants with NFTs of Alzheimer’s disease. Acta Neuropathol. 1995, 89, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Oberc-Greenwood, M.A.; McKeever, P.E.; Kornblith, P.L.; Smith, B.H. A human ganglioglioma containing paired helical filaments. Hum. Pathol. 1984, 15, 834–838. [Google Scholar] [CrossRef]
- Goates, J.J.; Dickson, D.W.; Horoupian, D.S. Meningioangiomatosis: An immunocytochemical study. Acta Neuropathol. 1991, 82, 527–532. [Google Scholar] [CrossRef]
- Halper, J.; Scheithauer, B.W.; Okazaki, H.; Laws, E.R., Jr. Meningio-angiomatosis: A report of six cases with special reference to the occurrence of neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1986, 45, 426–446. [Google Scholar] [CrossRef]
- Jarabo, P.; de Pablo, C.; Herranz, H.; Martín, F.A.; Casas-Tintó, S. Insulin signaling mediates neurodegeneration in glioma. Life Sci. Alliance 2021, 4, e202000693. [Google Scholar] [CrossRef]
- Loh, K.M.; van Amerongen, R.; Nusse, R. Generating Cellular Diversity and Spatial Form: Wnt Signaling and the Evolution of Multicellular Animals. Dev. Cell 2016, 38, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Oliva, C.A.; Vargas, J.Y.; Inestrosa, N.C. Wnts in adult brain: From synaptic plasticity to cognitive deficiencies. Front. Cell. Neurosci. 2013, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Packard, M.; Koo, E.S.; Gorczyca, M.; Sharpe, J.; Cumberledge, S.; Budnik, V. The Drosophila Wnt, Wingless, Provides an Essential Signal for Pre- and Postsynaptic Differentiation. Cell 2002, 111, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Rojo, A.I.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R.; et al. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry 2018, 8, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the Wings of Glioblastoma: Modulation of WNT as a Novel Therapeutic Strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, R.; Hundelt, M.; Shahani, N. Tau alteration and neuronal degeneration in tauopathies: Mechanisms and models. Biochim. Et Biophys Acta (BBA)-Mol. Basis Dis. 2005, 1739, 331–354. [Google Scholar] [CrossRef]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, I.; Tsuiki, H.; Kenyon, L.C.; Godwin, A.K.; Emlet, D.R.; Holgado-Madruga, M.; Lanham, I.S.; Joynes, C.J.; Vo, K.T.; Guha, A.; et al. Proteolytic Cleavage of the CD44 Adhesion Molecule in Multiple Human Tumors. Am. J. Pathol. 2002, 160, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kim, D.; Ju, S.; Shin, S.; Cho, I.-j.; Park, S.-H.; Grailhe, R.; Lee, C.; Kim, Y.K. Glioblastoma-secreted soluble CD44 activates tau pathology in the brain. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [CrossRef]
- Ryskalin, L.; Ferese, R.; Morucci, G.; Biagioni, F.; Busceti, C.L.; Michetti, F.; Lenzi, P.; Frati, A.; Fornai, F. Occurrence of Total and Proteinase K-Resistant Alpha-Synuclein in Glioblastoma Cells Depends on mTOR Activity. Cancers 2022, 14, 1382. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.K.; Fang, K.M.; Huang, H.T.; Chang, W.R.; Chuang, C.C.; Tzeng, S.F. Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency. Life 2021, 11, 961. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Barros, L.F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb. Perspect. Biol. 2015, 7, a020396. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.; Ferreyra Vega, S.; Kling, T.; Bontell, T.O.; Jakola, A.S.; Carén, H. Intratumor DNA methylation heterogeneity in glioblastoma: Implications for DNA methylation-based classification. Neuro Oncol. 2019, 21, 616–627. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fike, J.R.; Rola, R.; Limoli, C.L. Radiation response of neural precursor cells. Neurosurg. Clin. N. Am. 2007, 18, 115–127. [Google Scholar] [CrossRef]
- Fike, J.R.; Rosi, S.; Limoli, C.L. Neural precursor cells and central nervous system radiation sensitivity. Semin. Radiat. Oncol. 2009, 19, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, B.P.; Giedzinski, E.; Izadi, A.; Suarez, T.; Lan, M.L.; Tran, K.K.; Acharya, M.M.; Nelson, G.A.; Raber, J.; Parihar, V.K.; et al. Functional consequences of radiation-induced oxidative stress in cultured neural stem cells and the brain exposed to charged particle irradiation. Antioxid. Redox Signal. 2014, 20, 1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Prithivirajsingh, S.; Story, M.D.; Bergh, S.A.; Geara, F.B.; Ang, K.K.; Ismail, S.M.; Stevens, C.W.; Buchholz, T.A.; Brock, W.A. Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett. 2004, 571, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malakhova, L.; Bezlepkin, V.G.; Antipova, V.; Ushakova, T.; Fomenko, L.; Sirota, N.; Gaziev, A.I. The increase in mitochondrial DNA copy number in the tissues of gamma-irradiated mice. Cell. Mol. Biol. Lett. 2005, 10, 721–732. [Google Scholar] [PubMed]
- Kobashigawa, S.; Suzuki, K.; Yamashita, S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun. 2011, 414, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Kim, J.-H.; Kim, T.; Lee, S.B. Pharmacological intervention of early neuropathy in neurodegenerative diseases. Pharm. Res. 2017, 119, 169–177. [Google Scholar] [CrossRef]
- Wileman, T.; Kane, L.P.; Carson, G.R.; Terhorst, C. Depletion of cellular calcium accelerates protein degradation in the endoplasmic reticulum. J. Biol. Chem. 1991, 266, 4500–4507. [Google Scholar] [CrossRef]
- Torres, M.; Encina, G.; Soto, C.; Hetz, C. Abnormal calcium homeostasis and protein folding stress at the ER: A common factor in familial and infectious prion disorders. Commun. Integr. Biol. 2011, 4, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Grzybowska, E.A. Calcium-Binding Proteins with Disordered Structure and Their Role in Secretion, Storage, and Cellular Signaling. Biomolecules 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Ivy, S.P. Heat shock protein 90. Curr. Opin. Oncol. 2003, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Sõti, C.; Nagy, E.; Giricz, Z.; Vígh, L.; Csermely, P.; Ferdinandy, P. Heat shock proteins as emerging therapeutic targets. Br. J. Pharm. 2005, 146, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Nollen, E.A.; Morimoto, R.I. Chaperoning signaling pathways: Molecular chaperones as stress-sensing ‘heat shock’ proteins. J. Cell Sci. 2002, 115, 2809–2816. [Google Scholar] [CrossRef]
- Carman, A.; Kishinevsky, S.; Koren, J., 3rd; Lou, W.; Chiosis, G. Chaperone-dependent Neurodegeneration: A Molecular Perspective on Therapeutic Intervention. J. Alzheimers Dis. Park. 2013, 2013 (Suppl. S10), S10-007. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Cells in stress: Transcriptional activation of heat shock genes. Science 1993, 259, 1409–1410. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Murray, J.I.; Hartman, S.J.; Botstein, D.; Myers, R.M. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell 2004, 15, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Vihervaara, A.; Sistonen, L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Batista-Nascimento, L.; Neef, D.W.; Liu, P.C.; Rodrigues-Pousada, C.; Thiele, D.J. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. PLoS ONE 2011, 6, e15976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, S.; Madorsky, I.; Pileggi, J.G.; Kamal, A.; Notterpek, L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol. Dis. 2008, 32, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Koren, J., 3rd; Jinwal, U.K.; Jin, Y.; O’Leary, J.; Jones, J.R.; Johnson, A.G.; Blair, L.J.; Abisambra, J.F.; Chang, L.; Miyata, Y.; et al. Facilitating Akt clearance via manipulation of Hsp70 activity and levels. J. Biol. Chem. 2010, 285, 2498–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Ma, J.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-Jun. ASN Neuro 2012, 4, e00102. [Google Scholar] [CrossRef] [Green Version]
- Hentze, N.; Le Breton, L.; Wiesner, J.; Kempf, G.; Mayer, M.P. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. eLife 2016, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Kijima, T.; Prince, T.L.; Tigue, M.L.; Yim, K.H.; Schwartz, H.; Beebe, K.; Lee, S.; Budzynski, M.A.; Williams, H.; Trepel, J.B.; et al. HSP90 inhibitors disrupt a transient HSP90-HSF1 interaction and identify a noncanonical model of HSP90-mediated HSF1 regulation. Sci. Rep. 2018, 8, 6976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhury, S.; Keegan, B.M.; Blagg, B.S.J. The role and therapeutic potential of Hsp90, Hsp70, and smaller heat shock proteins in peripheral and central neuropathies. Med. Res. Rev. 2021, 41, 202–222. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, D.; Yang, F.; Pang, G.; Sun, Z.; Chang, J.; Dou, Y. Hsp90 co-chaperone degradation combined with antioxidation nanostrategy to rescue tauopathy-induced Alzheimer’s disease. Chem. Eng. J. 2022, 432, 134352. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology 2006, 66, S24–S36. [Google Scholar] [CrossRef]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T.; et al. Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J. Pharm. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzer, K.M.; Ruf, W.P.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.T.; McLean, P.J. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Riedel, M.; Goldbaum, O.; Schwarz, L.; Schmitt, S.; Richter-Landsberg, C. 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PLoS ONE 2010, 5, e8753. [Google Scholar] [CrossRef] [PubMed]
- Batulan, Z.; Taylor, D.M.; Aarons, R.J.; Minotti, S.; Doroudchi, M.M.; Nalbantoglu, J.; Durham, H.D. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 24, 213–225. [Google Scholar] [CrossRef]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [Green Version]
- Tokui, K.; Adachi, H.; Waza, M.; Katsuno, M.; Minamiyama, M.; Doi, H.; Tanaka, K.; Hamazaki, J.; Murata, S.; Tanaka, F.; et al. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum. Mol. Genet. 2008, 18, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Costa Mdo, C.; Paulson, H.L. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 2012, 97, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Fernandes, A.; Duarte-Silva, S.; Neves-Carvalho, A.; Amorim, M.; Soares-Cunha, C.; Oliveira, P.; Thirstrup, K.; Teixeira-Castro, A.; Maciel, P. Chronic Treatment with 17-DMAG Improves Balance and Coordination in A New Mouse Model of Machado-Joseph Disease. Neurotherapeutics 2014, 11, 433–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [Green Version]
- Rickner, H.; Jiang, L.; Hong, R.; O’Neill, N.; Wolozin, B.; Cheng, C. Single cell transcriptomic profiling of neurodegeneration mediated by tau propagation in a novel 3D neuron-astrocyte coculture model. Alzheimer’s Dement. 2021, 17, e058551. [Google Scholar] [CrossRef]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Tidwell, J.L.; Houenou, L.J.; Tytell, M. Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress Chaperones 2004, 9, 88–98. [Google Scholar] [CrossRef]
- Elgendy, A.; Abbas, A. Effect of heat Shock protein 90 Inhibition on Peripheral Neuropathy In Rats with Streptozocin-Induced Diabetes. 2020; Unpublished manuscript. [Google Scholar]
- Zhang, L.; Zhao, H.; Blagg, B.S.; Dobrowsky, R.T. C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome. Res. 2012, 11, 2581–2593. [Google Scholar] [CrossRef]
- Sofis, M.J.; Jarmolowicz, D.P.; Kaplan, S.V.; Gehringer, R.C.; Lemley, S.M.; Garg, G.; Blagg, B.S.; Johnson, M.A. KU32 prevents 5-fluorouracil induced cognitive impairment. Behav. Brain Res. 2017, 329, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.B.; Genilloud, O.; Peláez, F. 2.05-Terrestrial Microorganisms—Filamentous Bacteria. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 109–140. [Google Scholar]
- Hadden, M.K.; Galam, L.; Gestwicki, J.E.; Matts, R.L.; Blagg, B.S.J. Derrubone, an Inhibitor of the Hsp90 Protein Folding Machinery. J. Nat. Prod. 2007, 70, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Liew, H.Y.; Tan, X.Y.; Chan, H.H.; Khaw, K.Y.; Ong, Y.S. Natural HSP90 inhibitors as a potential therapeutic intervention in treating cancers: A comprehensive review. Pharm. Res 2022, 181, 106260. [Google Scholar] [CrossRef]
- Li, L.; Chen, N.-N.; You, Q.-D.; Xu, X.-L. An updated patent review of anticancer Hsp90 inhibitors (2013-present). Expert Opin. Ther. Pat. 2021, 31, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.A.; Moody, C.J. Learning from Nature: Advances in Geldanamycin- and Radicicol-Based Inhibitors of Hsp90. J. Org. Chem. 2013, 78, 5117–5141. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Jahan, I.; Ali, M.C.; Mitra, S.; Munni, Y.A.; Timalsina, B.; Hannan, M.A.; Moon, I.S. Potential roles of natural products in the targeting of proteinopathic neurodegenerative diseases. Neurochem. Int. 2021, 145, 105011. [Google Scholar] [CrossRef] [PubMed]
- Ngo, L.T.; Okogun, J.I.; Folk, W.R. 21st century natural product research and drug development and traditional medicines. Nat. Prod. Rep. 2013, 30, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Pastvova, N.; Dolezel, P.; Mlejnek, P. Heat Shock Protein Inhibitor 17-Allyamino-17-Demethoxygeldanamycin, a Potent Inductor of Apoptosis in Human Glioma Tumor Cell Lines, Is a Weak Substrate for ABCB1 and ABCG2 Transporters. Pharmaceuticals 2021, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Chiba, T.; Tanaka, K. CHIP: A quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 2003, 35, 572–578. [Google Scholar] [CrossRef]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, Y.; Kasahara, T.; Yokota-Aizu, E.; Ueno, M.; Watanabe, S. A Suppressive Role of p125FAK Protein Tyrosine Kinase in Hydrogen Peroxide-Induced Apoptosis of T98G Cells. Biochem. Biophys. Res. Commun. 1997, 241, 769–774. [Google Scholar] [CrossRef]
- Hamasuna, R.; Kataoka, H.; Moriyama, T.; Itoh, H.; Seiki, M.; Koono, M. Regulation of matrix metalloproteinase-2 (MMP-2) by hepatocyte growth factor/scatter factor (HGF/SF) in human glioma cells: HGF/SF enhances MMP-2 expression and activation accompanying up-regulation of membrane type-1 MMP. Int. J. Cancer 1999, 82, 274–281. [Google Scholar] [CrossRef]
- Pratt, W.B.; Morishima, Y.; Peng, H.M.; Osawa, Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp. Biol. Med. 2010, 235, 278–289. [Google Scholar] [CrossRef] [Green Version]
- An, W.G.; Schulte, T.W.; Neckers, L.M. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ.-Publ. Am. Assoc. Cancer Res. 2000, 11, 355–360. [Google Scholar]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. 17-Allylamino-17-demethoxygeldanamycin and Herbimycin A Induce Cell Death by Modulating beta-Catenin and PI3K/AKT Signaling in FRO Anaplastic Thyroid Carcinoma Cells. Anticancer Res. 2015, 35, 5453–5460. [Google Scholar]
- Nakazono, A.; Adachi, N.; Takahashi, H.; Seki, T.; Hamada, D.; Ueyama, T.; Sakai, N.; Saito, N. Pharmacological induction of heat shock proteins ameliorates toxicity of mutant PKCγ in spinocerebellar ataxia type 14. J. Biol. Chem. 2018, 293, 14758–14774. [Google Scholar] [CrossRef] [Green Version]
- Moses, M.A.; Henry, E.C.; Ricke, W.A.; Gasiewicz, T.A. The heat shock protein 90 inhibitor, (-)-epigallocatechin gallate, has anticancer activity in a novel human prostate cancer progression model. Cancer Prev. Res. (Phila) 2015, 8, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.L.; Kim, S.-A.; Choi, H.S.; Yoon, J.-H.; Ahn, S.-G. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer 2010, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Henry, E.C.; Gasiewicz, T.A. (-)-Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry 2009, 48, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, S.; Hirano, H.; Wakimaru, N.; Sarker, K.P.; Kuratsu, J. Inhibitory effect of epigallocatechin-gallate on brain tumor cell lines in vitro. Neuro Oncol. 2001, 3, 22–28. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.X.; Ma, J.W.; Li, H.Y.; Ye, J.C.; Xie, S.M.; Du, B.; Zhong, X.Y. EGCG inhibits properties of glioma stem-like cells and synergizes with temozolomide through downregulation of P-glycoprotein inhibition. J. Neurooncol. 2015, 121, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Udroiu, I.; Marinaccio, J.; Sgura, A. Epigallocatechin-3-gallate induces telomere shortening and clastogenic damage in glioblastoma cells. Environ. Mol. Mutagen. 2019, 60, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Banik, N.L.; Ray, S.K. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer 2010, 116, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.-R.; You, C.-G.; Zhang, N.; Sheng, H.-S.; Zheng, X.-S. Epigallocatechin Gallate Preferentially Inhibits O6-Methylguanine DNA-Methyltransferase Expression in Glioblastoma Cells Rather than in Nontumor Glial Cells. Nutr. Cancer 2018, 70, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B.H. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: Implications for neurodegenerative diseases. J. Neurochem. 2004, 88, 1555–1569. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Ho, C.-T.; Jun, M. Multifaceted neuroprotective effects of (-)-epigallocatechin-3-gallate (EGCG) in Alzheimer’s disease: An overview of pre-clinical studies focused on β-amyloid peptide. Food Sci. Hum. Wellness 2022, 11, 483–493. [Google Scholar] [CrossRef]
- Gonçalves, P.B.; Sodero, A.C.R.; Cordeiro, Y. Green Tea Epigallocatechin-3-gallate (EGCG) Targeting Protein Misfolding in Drug Discovery for Neurodegenerative Diseases. Biomolecules 2021, 11, 767. [Google Scholar] [CrossRef]
- Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules 2022, 12, 371. [Google Scholar] [CrossRef]
- El-Missiry, M.A.; Othman, A.I.; El-Sawy, M.R.; Lebede, M.F. Neuroprotective effect of epigallocatechin-3-gallate (EGCG) on radiation-induced damage and apoptosis in the rat hippocampus. Int. J. Radiat. Biol. 2018, 94, 798–808. [Google Scholar] [CrossRef]
- Wong, S.C.; Kamarudin, M.N.A.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950. [Google Scholar] [CrossRef]
- Walker, B.C.; Adhikari, S.; Mittal, S. Therapeutic Potential of Curcumin for the Treatment of Malignant Gliomas. In Gliomas; Debinski, W., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Afshari, A.R.; Jalili-Nik, M.; Abbasinezhad-Moud, F.; Javid, H.; Karimi, M.; Mollazadeh, H.; Jamialahmadi, T.; Sathyapalan, T.; Sahebkar, A. Anti-tumor Effects of Curcuminoids in Glioblastoma Multiforme: An Updated Literature Review. Curr. Med. Chem. 2021, 28, 8116–8138. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Jung, C.-W.; Lee, W.S.; Kim, H.-J.; Jeong, H.-J.; Park, M.-J.; Jang, W.I.; Kim, E.H. Interaction of curcumin with glioblastoma cells via high and low linear energy transfer radiation therapy inducing radiosensitization effects. J. Radiat. Res. 2022, 63, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Shen, C.-Y.; Chien, Y.-C.; Chang, W.-S.; Tsai, C.-W.; Lin, Y.-H.; Hwang, J.-J. Validation of Enhancing Effects of Curcumin on Radiotherapy with F98/FGT Glioblastoma-Bearing Rat Model. Int. J. Mol. Sci. 2020, 21, 4385. [Google Scholar] [CrossRef] [PubMed]
- Dhandapani, K.M.; Mahesh, V.B.; Brann, D.W. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFκB transcription factors. J. Neurochem. 2007, 102, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Zoi, V.; Galani, V.; Tsekeris, P.; Kyritsis, A.P.; Alexiou, G.A. Radiosensitization and Radioprotection by Curcumin in Glioblastoma and Other Cancers. Biomedicines 2022, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.K.; Perlow, H.K.; Ritter, A.R.; Upadhyay, R.; Raval, R.R.; Thomas, E.M.; Beyer, S.J.; Pillainayagam, C.; Goranovich, J.; Ong, S.; et al. Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma. Biomedicines 2022, 10, 1763. [Google Scholar] [CrossRef]
- Sminia, P.; van den Berg, J.; van Kootwijk, A.; Hageman, E.; Slotman, B.J.; Verbakel, W.F.A.R. Experimental and clinical studies on radiation and curcumin in human glioma. J. Cancer Res. Clin. Oncol. 2021, 147, 403–409. [Google Scholar] [CrossRef]
- Zoi, V.; Galani, V.; Vartholomatos, E.; Zacharopoulou, N.; Tsoumeleka, E.; Gkizas, G.; Bozios, G.; Tsekeris, P.; Chousidis, I.; Leonardos, I.; et al. Curcumin and Radiotherapy Exert Synergistic Anti-Glioma Effect In Vitro. Biomedicines 2021, 9, 1562. [Google Scholar] [CrossRef]
- Fan, Y.J.; Zhou, Y.X.; Zhang, L.R.; Lin, Q.F.; Gao, P.Z.; Cai, F.; Zhu, L.P.; Liu, B.; Xu, J.H. C1206, a novel curcumin derivative, potently inhibits Hsp90 and human chronic myeloid leukemia cells in vitro. Acta Pharm. Sin. 2018, 39, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Giommarelli, C.; Zuco, V.; Favini, E.; Pisano, C.; Dal Piaz, F.; De Tommasi, N.; Zunino, F. The enhancement of antiproliferative and proapoptotic activity of HDAC inhibitors by curcumin is mediated by Hsp90 inhibition. Cell Mol. Life Sci. 2010, 67, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Chung, I.K. Curcumin inhibits nuclear localization of telomerase by dissociating the Hsp90 co-chaperone p23 from hTERT. Cancer Lett. 2010, 290, 76–86. [Google Scholar] [CrossRef]
- Ye, M.; Huang, W.; Wu, W.-w.; Liu, Y.; Ye, S.-n.; Xu, J.-h. FM807, a curcumin analogue, shows potent antitumor effects in nasopharyngeal carcinoma cells by heat shock protein 90 inhibition. Oncotarget 2017, 8, 15364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Ito, H.; Kamei, K.; Iwamoto, I. Stimulation of the stress-induced expression of stress proteins by curcumin in cultured cells and in rat tissues in vivo. Cell Stress Chaperones 1998, 3, 152–160. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Zhuang, Y.; Ying, Z.; Wu, A.; Gomez-Pinilla, F. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience 2009, 161, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp. Neurol. 2006, 197, 309–317. [Google Scholar] [CrossRef]
- Ng, A.P.; Chng, W.J.; Khan, M. Curcumin sensitizes acute promyelocytic leukemia cells to unfolded protein response-induced apoptosis by blocking the loss of misfolded N-CoR protein. Mol. Cancer Res. MCR 2011, 9, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.-z.; Xu, J.-h.; Huang, X.-w.; Wu, L.-x.; Su, Y.; Chen, Y.-z. Curcumin synergistically augments bcr/abl phosphorothioate antisense oligonucleotides to inhibit growth of chronic myelogenous leukemia cells1. Acta Pharm. Sin. 2007, 28, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Fan, Y.; Wu, S.; Cai, X.; Shi, Y. Synergistic effects of curcumin and bortezomib on multiple myeloma cells. Int. J. Clin. Exp. 2016, 9, 21787–21793. [Google Scholar]
- Lu, P.Z.; Lai, C.Y.; Chan, W.H. Caffeine induces cell death via activation of apoptotic signal and inactivation of survival signal in human osteoblasts. Int. J. Mol. Sci. 2008, 9, 698–718. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.S.; Ferris, R.K.; Downs, R.M.; Kinsey, S.T.; Baumgarner, B.L. Caffeine promotes autophagy in skeletal muscle cells by increasing the calcium-dependent activation of AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2014, 453, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Sc, Y.; Muralidhara. Beneficial Role of Coffee and Caffeine in Neurodegenerative Diseases: A Minireview. AIMS Public Health 2016, 3, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-C.; Ding, Y.-M.; Hueng, D.-Y.; Chen, J.-Y.; Chen, Y. Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. J. Nutr. Biochem. 2016, 33, 63–72. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-Mediated Inhibition of Calcium Release Channel Inositol 1,4,5-Trisphosphate Receptor Subtype 3 Blocks Glioblastoma Invasion and Extends Survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Saccone, S.; Federico, C.; Magro, G.; Cavallaro, S.; D’Agata, V. Caffeine Effect on HIFs/VEGF Pathway in Human Glioblastoma Cells Exposed to Hypoxia. Anticancer Agents Med. Chem. 2018, 18, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, P.; Kiang, K.M.Y.; Cheng, Y.S.; Leung, G.K.K. Caffeine Sensitizes U87-MG Human Glioblastoma Cells to Temozolomide through Mitotic Catastrophe by Impeding G2 Arrest. Biomed. Res. Int. 2018, 2018, 5364973. [Google Scholar] [CrossRef] [PubMed]
- Beckner, M.E.; Agostino, N.R.; Pollack, I.F. Migration of Glioblastoma Cells Indicates Invasion Is Mediated by a Network of Proteins Stimulated by HGF/Met and Suppressed by Radicicol. FASEB J. 2007, 21, A26–A27. [Google Scholar] [CrossRef]
- Sohn, M.-J.; Noh, H.-J.; Yoo, I.-D.; Kim, W.-G. Protective effect of radicicol against LPS/IFN-γ-induced neuronal cell death in rat cortical neuron–glia cultures. Life Sci. 2007, 80, 1706–1712. [Google Scholar] [CrossRef]
- Brandt, G.E.; Schmidt, M.D.; Prisinzano, T.E.; Blagg, B.S. Gedunin, a novel hsp90 inhibitor: Semisynthesis of derivatives and preliminary structure-activity relationships. J. Med. Chem. 2008, 51, 6495–6502. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.; Lin, S.; Stouffer, M.; Wandling, E.; Noland, L.; Huanyun, D.; Jean-Loius, D.; Darkwah, B. Gedunin, A Novel HSP90 Inhibitor, Decreases Cellular Growth and Induces Apoptosis In Glioblastoma Cell Lines. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Yang, W.; Xie, J.; Qiang, Q.; Li, L.; Lin, X.; Ren, Y.; Ren, W.; Liu, Q.; Zhou, G.; Wei, W.; et al. Gedunin Degrades Aggregates of Mutant Huntingtin Protein and Intranuclear Inclusions via the Proteasomal Pathway in Neurons and Fibroblasts from Patients with Huntington’s Disease. Neurosci. Bull. 2019, 35, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Tom, S.; Rane, A.; Katewa, A.S.; Chamoli, M.; Matsumoto, R.R.; Andersen, J.K.; Chinta, S.J. Gedunin Inhibits Oligomeric Aβ1–42-Induced Microglia Activation Via Modulation of Nrf2-NF-κB Signaling. Mol. Neurobiol. 2019, 56, 7851–7862. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [Green Version]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.; Kawahara, T.L.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, D.; Ganey, N.J.; Laporte, V.; Patel, N.S.; Beaulieu-Abdelahad, D.; Bachmeier, C.; March, A.; Ait-Ghezala, G.; Mullan, M.J. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2010, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Sreeramulu, S.; Gande, S.L.; Göbel, M.; Schwalbe, H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. Engl. 2009, 48, 5853–5855. [Google Scholar] [CrossRef]
- Lin, M.-W.; Lin, C.C.; Chen, Y.-H.; Yang, H.-B.; Hung, S.-Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy. Antioxidants 2020, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, M.; Wang, B.; Su, Z.; Guo, B.; Qin, L.; Zhang, W.; Zheng, R. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 2021, 47, 102134. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, S.; Morgese, M.G.; Tucci, P.; Trabace, L. The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches. Molecules 2021, 26, 4700. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.; Zhao, P.; Zhao, H.; Gao, W.; Wang, L. Celastrol Suppresses Glioma Vasculogenic Mimicry Formation and Angiogenesis by Blocking the PI3K/Akt/mTOR Signaling Pathway. Front. Pharm. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhao, P.; Wang, X.; Wang, L.; Zhu, Y.; Song, Y.; Gao, W. Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J. Exp. Clin. Cancer Res. 2019, 38, 184. [Google Scholar] [CrossRef] [PubMed]
- Boridy, S.; Le, P.U.; Petrecca, K.; Maysinger, D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014, 5, e1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, Z.; Cheng, J.; Xiang, H.; Qin, J.; He, Y.; Peng, Z.; Jia, J.; Yu, H. Celastrol enhances TRAIL-induced apoptosis in human glioblastoma via the death receptor pathway. Cancer Chemother. Pharm. 2019, 84, 719–728. [Google Scholar] [CrossRef] [PubMed]
- He, Z. Celastrol enhances ER stress regulated apoptosis in human glioblastoma via PERK/Qrich1 pathway. Brain Tumor Res. Treat. 2022, 10, S280. [Google Scholar] [CrossRef]
- Kao, T.C.; Shyu, M.H.; Yen, G.C. Glycyrrhizic acid and 18beta-glycyrrhetinic acid inhibit inflammation via PI3K/Akt/GSK3beta signaling and glucocorticoid receptor activation. J. Agric. Food Chem. 2010, 58, 8623–8629. [Google Scholar] [CrossRef]
- Yan, D.; Saito, K.; Ohmi, Y.; Fujie, N.; Ohtsuka, K. Paeoniflorin, a novel heat shock protein-inducing compound. Cell Stress Chaperones 2004, 9, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoh, T.; Nakashima, T.; Sumida, Y.; Kakisaka, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T. Glycyrrhizin effects rat hepatocytes via the reduction of heat shock protein 90 expression. Gastroenterology 2001, 120, A357. [Google Scholar] [CrossRef]
- Yuan-yuan, H.O.U.; Yang, Y.; Yang, Y.A.O.; Gang, B.A.I. Neuroprotection of Glycyrrhizin against Ischemic Vascular Dementia in vivo and Glutamate-induced Damage in vitro. Chin. Herb. Med. (CHM) 2010, 2, 125–131. [Google Scholar]
- Su, X.-Q.; Wang, X.-Y.; Gong, F.-T.; Feng, M.; Bai, J.-J.; Zhang, R.-R.; Dang, X.-Q. Oral treatment with glycyrrhizin inhibits NLRP3 inflammasome activation and promotes microglial M2 polarization after traumatic spinal cord injury. Brain Res. Bull. 2020, 158, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, Y.; Weng, Z.; Zhou, T.; Feng, T.; Lin, Y. Glycyrrhizin protects brain against ischemia–reperfusion injury in mice through HMGB1-TLR4-IL-17A signaling pathway. Brain Res. 2014, 1582, 176–186. [Google Scholar] [CrossRef]
- Kim, S.-W.; Jin, Y.; Shin, J.-H.; Kim, I.-D.; Lee, H.-K.; Park, S.; Han, P.-L.; Lee, J.-K. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti-inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol. Dis. 2012, 46, 147–156. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Semple, B.; Piperi, C.; Othman, I.; Shaikh, M.F. Potential Neuroprotective Effect of the HMGB1 Inhibitor Glycyrrhizin in Neurological Disorders. ACS Chem. Neurosci. 2020, 11, 485–500. [Google Scholar] [CrossRef]
- Gao, T.; Chen, Z.; Chen, H.; Yuan, H.; Wang, Y.; Peng, X.; Wei, C.; Yang, J.; Xu, C. Inhibition of HMGB1 mediates neuroprotection of traumatic brain injury by modulating the microglia/macrophage polarization. Biochem. Biophys. Res. Commun. 2018, 497, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Davenport, J.; Manjarrez, J.R.; Peterson, L.; Krumm, B.; Blagg, B.S.; Matts, R.L. Gambogic acid, a natural product inhibitor of Hsp90. J. Nat. Prod. 2011, 74, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thida, M.; Kim, D.W.; Tran, T.T.T.; Pham, M.Q.; Lee, H.; Kim, I.; Lee, J.W. Gambogic acid induces apoptotic cell death in T98G glioma cells. Bioorganic Med. Chem. Lett. 2016, 26, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Yang, Y.; You, Q.-D.; Ma, Y.-J.; Yang, L.; Nie, F.-F.; Gu, H.-Y.; Zhao, L.; Lu, N.; Qi, Q.; et al. Inhibition of glioblastoma growth and angiogenesis by gambogic acid: An in vitro and in vivo study. Biochem. Pharm. 2008, 75, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Hao, J.; Liu, T.; Zhang, D.; Lv, H.; Song, E.; Zhu, C. Hesperetin Suppresses Inflammatory Responses in Lipopolysaccharide-Induced RAW 264.7 Cells via the Inhibition of NF-κB and Activation of Nrf2/HO-1 Pathways. Inflammation 2016, 39, 964–973. [Google Scholar] [CrossRef]
- Ren, J.; Li, L.; Wang, Y.; Zhai, J.; Chen, G.; Hu, K. Gambogic acid induces heme oxygenase-1 through Nrf2 signaling pathway and inhibits NF-κB and MAPK activation to reduce inflammation in LPS-activated RAW264.7 cells. Biomed. Pharm. 2019, 109, 555–562. [Google Scholar] [CrossRef]
- Sharma, V.; Kaur, A.; Singh, T.G. Counteracting role of nuclear factor erythroid 2-related factor 2 pathway in Alzheimer’s disease. Biomed. Pharm. 2020, 129, 110373. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Deb, C.R.; Ahmed, S.U.; Saratchandra, Y.; Konwar, B.K. Molecular docking simulation analysis of the interaction of dietary flavonols with heat shock protein 90. J. Biomed. Res. 2015, 30, 67. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Asthana, P.; Kumar, A.; Ambasta, R.K.; Kumar, P. Quercetin mediated reduction of angiogenic markers and chaperones in DLA-induced solid tumours. Asian Pac. J. Cancer Prev. 2011, 12, 2829–2835. [Google Scholar]
- Moses, M.A.; Zuehlke, A.D.; Neckers, L. Molecular Chaperone Inhibitors. In Heat Shock Proteins in the Immune System; Springer: Berlin/Heidelberg, Germany, 2018; pp. 21–40. [Google Scholar]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Béliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 318, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.-F.; Wang, G.; Wu, C.-P.; Zhou, X.-M.; Wang, J.; Chen, Z.-P.; To, S.-S.T.; Li, W.-P. A Multi-targeted Natural Flavonoid Myricetin Suppresses Lamellipodia and Focal Adhesions Formation and Impedes Glioblastoma Cell Invasiveness and Abnormal Motility. CNS Neurol. Disord.-Drug Targets-CNS Neurol. Disord. 2018, 17, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Gaiser, T.; Habel, A.; Siegelin, Y. Myricetin sensitizes malignant glioma cells to TRAIL-mediated apoptosis by down-regulation of the short isoform of FLIP and bcl-2. Cancer Lett. 2009, 283, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Ray, S.K. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: Overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis 2016, 21, 312–328. [Google Scholar] [CrossRef]
- Yi, C.; Li, G.; Ivanov, D.N.; Wang, Z.; Velasco, M.X.; Hernández, G.; Kaundal, S.; Villarreal, J.; Gupta, Y.K.; Qiao, M.; et al. Luteolin inhibits Musashi1 binding to RNA and disrupts cancer phenotypes in glioblastoma cells. RNA Biol. 2018, 15, 1420–1432. [Google Scholar] [CrossRef]
- Cheng, W.-Y.; Chiao, M.-T.; Liang, Y.-J.; Yang, Y.-C.; Shen, C.-C.; Yang, C.-Y. Luteolin inhibits migration of human glioblastoma U-87 MG and T98G cells through downregulation of Cdc42 expression and PI3K/AKT activity. Mol. Biol. Rep. 2013, 40, 5315–5326. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, H.; Jia, Y.; Ding, H.; Zhang, L.; Pan, H. Luteolin reduces migration of human glioblastoma cell lines via inhibition of the p-IGF-1R/PI3K/AKT/mTOR signaling pathway. Oncol. Lett. 2017, 14, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Anson, D.M.; Wilcox, R.M.; Huseman, E.D.; Stump, T.A.; Paris, R.L.; Darkwah, B.O.; Lin, S.; Adegoke, A.O.; Gryka, R.J.; Jean-Louis, D.S.; et al. Luteolin Decreases Epidermal Growth Factor Receptor-Mediated Cell Proliferation and Induces Apoptosis in Glioblastoma Cell Lines. Basic Clin. Pharmacol. Toxicol. 2018, 123, 678–686. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Ray, S.K. Synergistic anti-tumor actions of luteolin and silibinin prevented cell migration and invasion and induced apoptosis in glioblastoma SNB19 cells and glioblastoma stem cells. Brain Res. 2015, 1629, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cerretti, G.; Cecchin, D.; Denaro, L.; Caccese, M.; Padovan, M.; Zagonel, V.; Lombardi, G. Impressive response to dabrafenib and trametinib plus silybin in a heavily pretreated IDH wild-type glioblastoma patient with BRAFV600E-mutant and SOX2 amplification. Anticancer Drugs 2022, 10, 1097. [Google Scholar] [CrossRef] [PubMed]
- Dizaji, M.Z.; Malehmir, M.; Ghavamzadeh, A.; Alimoghaddam, K.; Ghaffari, S.H. Synergistic Effects of Arsenic Trioxide and Silibinin on Apoptosis and Invasion in Human Glioblastoma U87MG Cell Line. Neurochem. Res. 2012, 37, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Gülden, M.; Appel, D.; Syska, M.; Uecker, S.; Wages, F.; Seibert, H. Chrysin and silibinin sensitize human glioblastoma cells for arsenic trioxide. Food Chem. Toxicol. 2017, 105, 486–497. [Google Scholar] [CrossRef]
- Bai, Z.-L.; Tay, V.; Guo, S.-Z.; Ren, J.; Shu, M.-G. Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. BioMed Res. Int. 2018, 2018, 6165192. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.-J.; Ding, Y.; Zhang, H.-N.; Sun, T.; Zhang, K.; Yang, L.; Guo, Y.-Y.; Liu, S.-B.; Zhao, M.-G.; et al. Silibinin Prevents Autophagic Cell Death upon Oxidative Stress in Cortical Neurons and Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2016, 53, 932–943. [Google Scholar] [CrossRef]
- Liu, X.; Liu, W.; Wang, C.; Chen, Y.; Liu, P.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. Silibinin attenuates motor dysfunction in a mouse model of Parkinson’s disease by suppression of oxidative stress and neuroinflammation along with promotion of mitophagy. Physiol. Behav. 2021, 239, 113510. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Kempuraj, D.D.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Govindarajan, R.; Chandrasekaran, P.N.; et al. Neuroprotective effects of flavone luteolin in neuroinflammation and neurotrauma. BioFactors 2021, 47, 190–197. [Google Scholar] [CrossRef]
- Ahmad, S.; Jo, M.H.; Ikram, M.; Khan, A.; Kim, M.O. Deciphering the Potential Neuroprotective Effects of Luteolin against Aβ1–42-Induced Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9583. [Google Scholar] [CrossRef]
- Chen, H.-Q.; Jin, Z.-Y.; Wang, X.-J.; Xu, X.-M.; Deng, L.; Zhao, J.-W. Luteolin protects dopaminergic neurons from inflammation-induced injury through inhibition of microglial activation. Neurosci. Lett. 2008, 448, 175–179. [Google Scholar] [CrossRef]
- Jang, S.; Dilger, R.N.; Johnson, R.W. Luteolin Inhibits Microglia and Alters Hippocampal-Dependent Spatial Working Memory in Aged Mice. J. Nutr. 2010, 140, 1892–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Li, L.; Sun, B.; Liu, F.; Yang, P.; Chen, T.; Li, T.; Liu, X. Luteolin Inhibits Behavioral Sensitization by Blocking Methamphetamine-Induced MAPK Pathway Activation in the Caudate Putamen in Mice. PLoS ONE 2014, 9, e98981. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Rahul; Jyoti, S.; Naz, F.; Ashafaq, M.; Shahid, M.; Siddique, Y.H. Therapeutic potential of luteolin in transgenic Drosophila model of Alzheimer’s disease. Neurosci. Lett. 2019, 692, 90–99. [Google Scholar] [CrossRef]
- Zhao, H.; Brandt, G.E.; Galam, L.; Matts, R.L.; Blagg, B.S.J. Identification and initial SAR of silybin: An Hsp90 inhibitor. Bioorganic Med. Chem. Lett. 2011, 21, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Chen, D.; Zhao, B.; Zhao, Z.; Zhou, J.; Xu, Y.; Xin, Y.; Liu, C.; Luo, L.; Yin, Z. Luteolin Induces Carcinoma Cell Apoptosis through Binding Hsp90 to Suppress Constitutive Activation of STAT3. PLoS ONE 2012, 7, e49194. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Ning, C.; Wang, Y.; Ma, T.; Huang, H.; Ge, Y.; Liu, J.; Jiang, Y. Natural plant flavonoid apigenin directly disrupts Hsp90/Cdc37 complex and inhibits pancreatic cancer cell growth and migration. J. Funct. Foods 2015, 18, 10–21. [Google Scholar] [CrossRef]
- Shendge, A.K.; Chaudhuri, D.; Mandal, N. The natural flavones, acacetin and apigenin, induce Cdk-Cyclin mediated G2/M phase arrest and trigger ROS-mediated apoptosis in glioblastoma cells. Mol. Biol. Rep. 2021, 48, 539–549. [Google Scholar] [CrossRef]
- Kim, B.; Jung, N.; Lee, S.; Sohng, J.K.; Jung, H.J. Apigenin Inhibits Cancer Stem Cell-Like Phenotypes in Human Glioblastoma Cells via Suppression of c-Met Signaling. Phytother. Res. 2016, 30, 1833–1840. [Google Scholar] [CrossRef]
- Stump, T.A.; Santee, B.N.; Williams, L.P.; Kunze, R.A.; Heinze, C.E.; Huseman, E.D.; Gryka, R.J.; Simpson, D.S.; Amos, S. The antiproliferative and apoptotic effects of apigenin on glioblastoma cells. J. Pharm. Pharm. 2017, 69, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, Z.; Dai, X.; Zhang, L.; Li, M. Apigenin and Temozolomide Synergistically Inhibit Glioma Growth Through the PI3K/AKT Pathway. Cancer Biother. Radiopharm. 2021. [Google Scholar] [CrossRef]
- Losi, G.; Puia, G.; Garzon, G.; de Vuono, M.C.; Baraldi, M. Apigenin modulates GABAergic and glutamatergic transmission in cultured cortical neurons. Eur. J. Pharm. 2004, 502, 41–46. [Google Scholar] [CrossRef]
- Liu, S.-Q.; Su, F.; Fang, L.-M.; Xia, Q.; Zhang, X. Protective effect of apigenin on neurons against oxygen-glucose deprivation/reperfusion induced injury. FASEB J. 2010, 24, 604–615. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, R.; XU, J.; Xie, Y. Effect of apigenin on hippocampus neuron injury induced by A?-amyloid 25-35. Chin. Pharm. Bull. 2005, 21, 996–998. [Google Scholar]
- Ding, Y.M.; Lin, J.T.; Fang, L.M.; Lou, Z.Q.; Liang, G.N.; Zhang, X.Y.; Li, A.Q.; Zhang, X. The neuroprotective effect of apigenin against OGD/R injury in rat hippocampal neurons. Pak. J. Pharm. Sci. 2020, 33, 1527–1533. [Google Scholar] [PubMed]
- Dourado, N.S.; Souza, C.d.S.; de Almeida, M.M.A.; Bispo da Silva, A.; dos Santos, B.L.; Silva, V.D.A.; De Assis, A.M.; da Silva, J.S.; Souza, D.O.; Costa, M.d.F.D.; et al. Neuroimmunomodulatory and Neuroprotective Effects of the Flavonoid Apigenin in in vitro Models of Neuroinflammation Associated With Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Yarim, G.F.; Kazak, F.; Yarim, M.; Sozmen, M.; Genc, B.; Ertekin, A.; Gokceoglu, A. Apigenin alleviates neuroinflammation in a mouse model of Parkinson’s disease. Int. J. Neurosci. 2022, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Wang, X.; Du, Q.; Gong, H.; Yang, Y.; Wang, T.; Wu, N.; Liu, X.; Li, W.; Zhao, C.; et al. Chetomin, a Hsp90/HIF1α pathway inhibitor, effectively targets lung cancer stem cells and non-stem cells. Cancer Biol. Ther. 2020, 21, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Hagemann, C.; Carta, F.; Katzer, A.; Polat, B.; Staab, A.; Scozzafava, A.; Anacker, J.; Vince, G.H.; Flentje, M.; et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including Acetazolamide in human Glioblastoma. Bioorg. Med. Chem. 2013, 21, 3949–3957. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1α inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10, 605. [Google Scholar] [CrossRef] [Green Version]
- Chua, L.L.; Ho, P.; Toh, J.; Tan, E.K. Chetomin rescues pathogenic phenotype of LRRK2 mutation in drosophila. Aging 2020, 12, 18561–18570. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Harun Ai Rashid, M.; Mridul, M.; Mohanty, C.; Swayamsiddha, S. Application of Artificial Intelligence in COVID-19 drug repurposing. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Cea, M.; Borghi, R.; Odetti, P.; Nencioni, A. Do Cancer Drugs Counteract Neurodegeneration? Repurposing for Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, J.; North, B.J.; Wei, W. Functional analyses of major cancer-related signaling pathways in Alzheimer’s disease etiology. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2017, 1868, 341–358. [Google Scholar] [CrossRef]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The Therapeutic Target Hsp90 and Cancer Hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitra, S.; Dash, R.; Munni, Y.A.; Selsi, N.J.; Akter, N.; Uddin, M.N.; Mazumder, K.; Moon, I.S. Natural Products Targeting Hsp90 for a Concurrent Strategy in Glioblastoma and Neurodegeneration. Metabolites 2022, 12, 1153. https://doi.org/10.3390/metabo12111153
Mitra S, Dash R, Munni YA, Selsi NJ, Akter N, Uddin MN, Mazumder K, Moon IS. Natural Products Targeting Hsp90 for a Concurrent Strategy in Glioblastoma and Neurodegeneration. Metabolites. 2022; 12(11):1153. https://doi.org/10.3390/metabo12111153
Chicago/Turabian StyleMitra, Sarmistha, Raju Dash, Yeasmin Akter Munni, Nusrat Jahan Selsi, Nasrin Akter, Md Nazim Uddin, Kishor Mazumder, and Il Soo Moon. 2022. "Natural Products Targeting Hsp90 for a Concurrent Strategy in Glioblastoma and Neurodegeneration" Metabolites 12, no. 11: 1153. https://doi.org/10.3390/metabo12111153