
Identifying Genetic Variants and Metabolites Associated with Rapid Estimated Glomerular Filtration Rate Decline in Korea Based on Genome–Metabolomic Integrative Analysis
 , , and
, , and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Data Sources and the Study Population
2.2. Exposure Measurements
2.3. GWAS
2.4. Fine-Mapping
2.5. GMIA
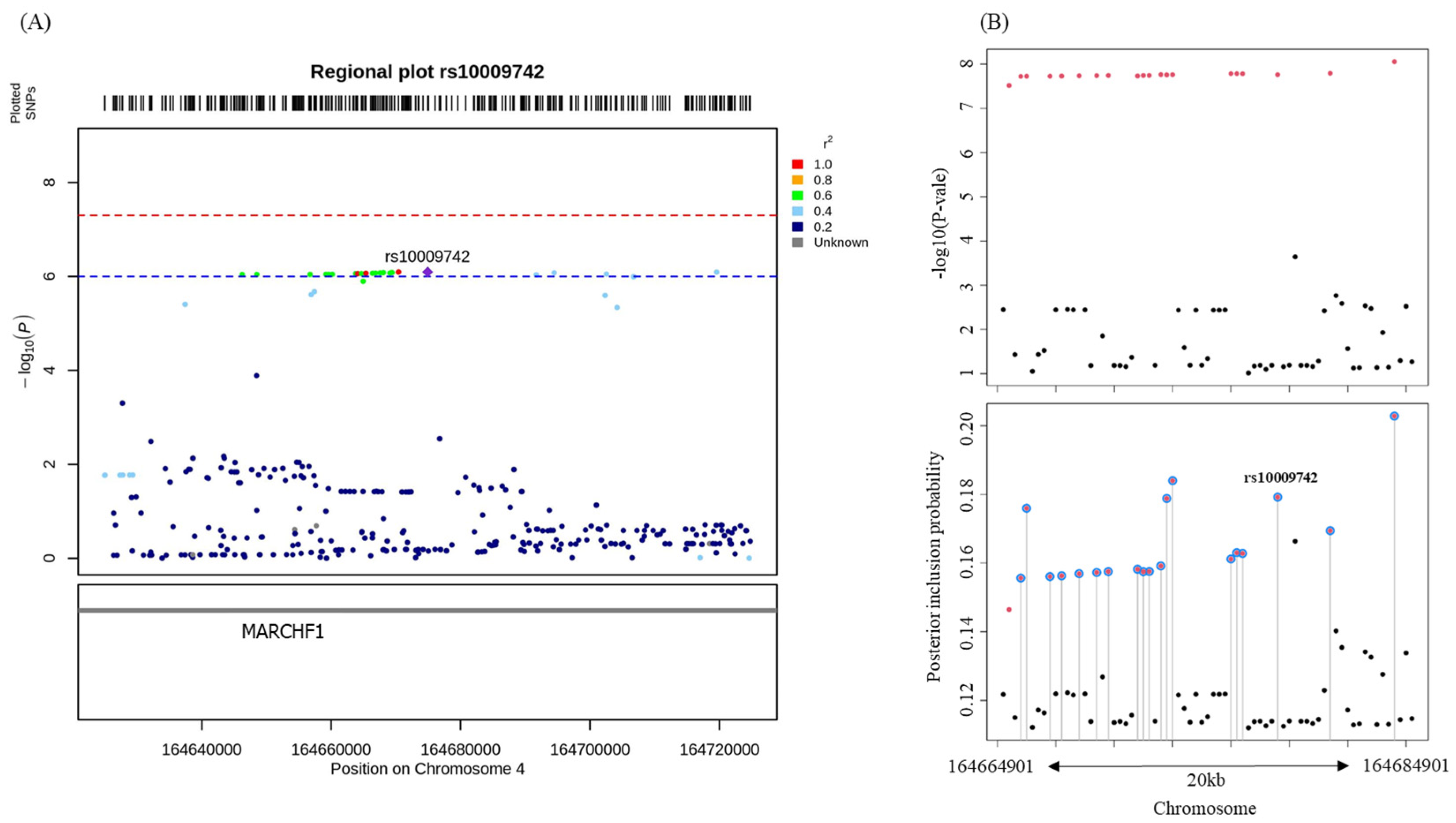
3. Results
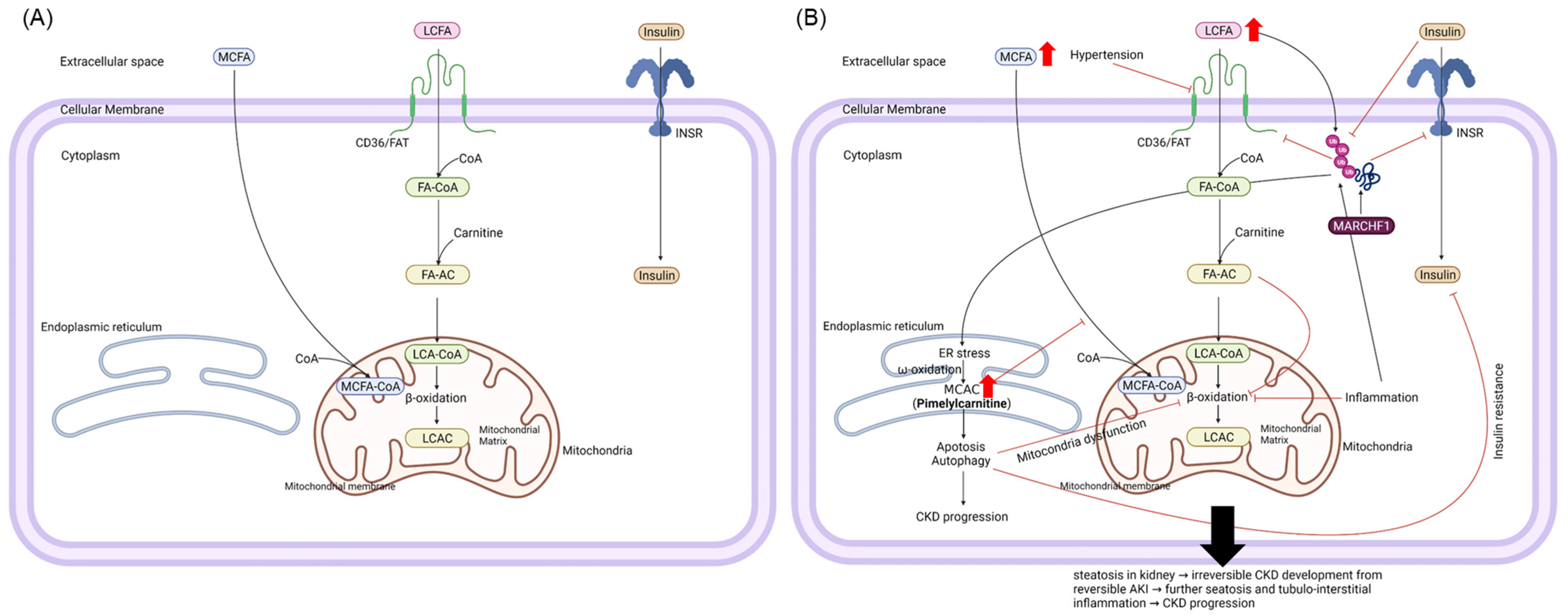
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lv, J.C.; Zhang, L.X. Prevalence and Disease Burden of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Turin, T.C.; Matsushita, K.; Sang, Y.; Ballew, S.H.; Appel, L.J.; Arima, H.; Chadban, S.J.; Cirillo, M.; Djurdjev, O.; et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA 2014, 311, 2518–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Selvin, E.; Bash, L.D.; Franceschini, N.; Astor, B.C.; Coresh, J. Change in estimated GFR associates with coronary heart disease and mortality. J. Am. Soc. Nephrol. 2009, 20, 2617–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.S.; Larson, M.G.; Leip, E.P.; Culleton, B.; Wilson, P.W.; Levy, D. Predictors of new-onset kidney disease in a community-based population. JAMA 2004, 291, 844–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Seaghdha, C.M.; Fox, C.S. Genome-wide association studies of chronic kidney disease: What have we learned? Nat. Rev. Nephrol. 2011, 8, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuttke, M.; Li, Y.; Li, M.; Sieber, K.B.; Feitosa, M.F.; Gorski, M.; Tin, A.; Wang, L.; Chu, A.Y.; Hoppmann, A.; et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 2019, 51, 957–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tin, A.; Köttgen, A. Genome-Wide Association Studies of CKD and Related Traits. Clin. J. Am. Soc. Nephrol. 2020, 15, 1643–1656. [Google Scholar] [CrossRef]
- Köttgen, A.; Glazer, N.L.; Dehghan, A.; Hwang, S.J.; Katz, R.; Li, M.; Yang, Q.; Gudnason, V.; Launer, L.J.; Harris, T.B.; et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 2009, 41, 712–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudu, M.; Schaeffer, C.; Riba, M.; Ikehata, M.; Brambilla, P.; Messa, P.; Martinelli-Boneschi, F.; Rastaldi, M.P.; Rampoldi, L. Early involvement of cellular stress and inflammatory signals in the pathogenesis of tubulointerstitial kidney disease due to UMOD mutations. Sci. Rep. 2017, 7, 7383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, F. Genetic kidney diseases. Lancet 2010, 375, 1287–1295. [Google Scholar] [CrossRef]
- Taylor, K.S.; McLellan, J.; Verbakel, J.Y.; Aronson, J.K.; Lasserson, D.S.; Pidduck, N.; Roberts, N.; Fleming, S.; O’Callaghan, C.A.; Bankhead, C.R.; et al. Effects of antihypertensives, lipid-modifying drugs, glycaemic control drugs and sodium bicarbonate on the progression of stages 3 and 4 chronic kidney disease in adults: A systematic review and meta-analysis. BMJ Open 2019, 9, e030596. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- King, E.A.; Davis, J.W.; Degner, J.F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yang, H.K.; Lee, H.J.; Park, D.J.; Kong, S.H.; Park, S.K. Systematic review of gastric cancer-associated genetic variants, gene-based meta-analysis, and gene-level functional analysis to identify candidate genes for drug development. Front. Genet. 2022, 13, 928783. [Google Scholar] [CrossRef] [PubMed]
- Gorski, M.; Jung, B.; Li, Y.; Matias-Garcia, P.R.; Wuttke, M.; Coassin, S.; Thio, C.H.L.; Kleber, M.E.; Winkler, T.W.; Wanner, V.; et al. Meta-analysis uncovers genome-wide significant variants for rapid kidney function decline. Kidney Int. 2021, 99, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Su, Y.; Paueksakon, P.; Cheng, H.; Chen, X.; Wang, H.; Harris, R.C.; Zent, R.; Pozzi, A. Integrin α1/Akita double-knockout mice on a Balb/c background develop advanced features of human diabetic nephropathy. Kidney Int. 2012, 81, 1086–1097. [Google Scholar] [CrossRef] [Green Version]
- Regner, K.R.; Harmon, A.C.; Williams, J.M.; Stelloh, C.; Johnson, A.C.; Kyle, P.B.; Lerch-Gaggl, A.; White, S.M.; Garrett, M.R. Increased susceptibility to kidney injury by transfer of genomic segment from SHR onto Dahl S genetic background. Physiol. Genom. 2012, 44, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inker, L.A.; Heerspink, H.J.L.; Tighiouart, H.; Levey, A.S.; Coresh, J.; Gansevoort, R.T.; Simon, A.L.; Ying, J.; Beck, G.J.; Wanner, C.; et al. GFR Slope as a Surrogate End Point for Kidney Disease Progression in Clinical Trials: A Meta-Analysis of Treatment Effects of Randomized Controlled Trials. J. Am. Soc. Nephrol. 2019, 30, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Sang, Y.; Ballew, S.H.; Matsushita, K.; Astor, B.C.; Carrero, J.J.; Chang, A.R.; Inker, L.A.; Kenealy, T.; Kovesdy, C.P.; et al. Evaluating Glomerular Filtration Rate Slope as a Surrogate End Point for ESKD in Clinical Trials: An Individual Participant Meta-Analysis of Observational Data. J. Am. Soc. Nephrol. 2019, 30, 1746–1755. [Google Scholar] [CrossRef]
- Eknoyan, G.; Lameire, N.; Eckardt, K.; Kasiske, B.; Wheeler, D.; Levin, A.; Stevens, P.; Bilous, R.; Lamb, E.; Coresh, J. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2013, 3, 5–14. [Google Scholar]
- Wen, C.P.; Matsushita, K.; Coresh, J.; Iseki, K.; Islam, M.; Katz, R.; McClellan, W.; Peralta, C.A.; Wang, H.; de Zeeuw, D.; et al. Relative risks of chronic kidney disease for mortality and end-stage renal disease across races are similar. Kidney Int. 2014, 86, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Barbour, S.J.; Er, L.; Djurdjev, O.; Karim, M.; Levin, A. Differences in progression of CKD and mortality amongst Caucasian, Oriental Asian and South Asian CKD patients. Nephrol. Dial. Transplant. 2010, 25, 3663–3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, Y.N.; Fuentes, E.F.; Chertow, G.M.; Olson, J.L. Race/ethnicity and disease severity in IgA nephropathy. BMC Nephrol. 2004, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katznelson, S.; Cecka, J.M. The great success of Asian kidney transplant recipients. Transplantation 1997, 64, 1850–1852. [Google Scholar] [CrossRef] [PubMed]
- Hall, Y.N.; Hsu, C.Y.; Iribarren, C.; Darbinian, J.; McCulloch, C.E.; Go, A.S. The conundrum of increased burden of end-stage renal disease in Asians. Kidney Int. 2005, 68, 2310–2316. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Sim, X.; Go, M.J.; Wu, J.Y.; Gu, D.; Takeuchi, F.; Takahashi, A.; Maeda, S.; Tsunoda, T.; Chen, P.; et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 2012, 44, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, Y.J.; Han, S.; Hwang, M.Y.; Shin, D.M.; Park, M.Y.; Lu, Y.; Yoon, K.; Jang, H.M.; Kim, Y.K.; et al. The Korea Biobank Array: Design and Identification of Coding Variants Associated with Blood Biochemical Traits. Sci. Rep. 2019, 9, 1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.H.; Park, S.K.; Park, H.C.; Chin, H.J.; Chae, D.W.; Choi, K.H.; Han, S.H.; Yoo, T.H.; Lee, K.; Kim, Y.S.; et al. KNOW-CKD (KoreaN cohort study for Outcome in patients With Chronic Kidney Disease): Design and methods. BMC Nephrol. 2014, 15, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.J.; Kwak, S.Y.; Jo, G.; Song, T.J.; Shin, M.J. Serum metabolite profile associated with incident type 2 diabetes in Koreans: Findings from the Korean Genome and Epidemiology Study. Sci. Rep. 2018, 8, 8207. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Elsworth, B.L.; Lyon, M.S.; Alexander, T.; Liu, Y.; Matthews, P.; Hallett, J.; Bates, P.; Palmer, T.; Haberland, V.; Smith, G.D. The MRC IEU OpenGWAS data infrastructure. bioRxiv 2020. [Google Scholar]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Wang, G.; Sarkar, A.; Carbonetto, P.; Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2020, 82, 1273–1300. [Google Scholar] [CrossRef]
- Major, T.; Takei, R. LocusZoom-Like Plots for Human GWAS Results (v2.1); Zenodo: Zurich, Switzerland, 2021. [Google Scholar] [CrossRef]
- Jee, S.H.; Sull, J.W.; Lee, J.E.; Shin, C.; Park, J.; Kimm, H.; Cho, E.Y.; Shin, E.S.; Yun, J.E.; Park, J.W.; et al. Adiponectin concentrations: A genome-wide association study. Am. J. Hum. Genet. 2010, 87, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Stein, T.I.; Levitt, J.; Gershoni, M.; Morrey, C.P.; Safran, M.; Lancet, D. MalaCards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef] [Green Version]
- Asleh, R.; Snipelisky, D.; Hathcock, M.; Kremers, W.; Liu, D.; Batzler, A.; Jenkins, G.; Kushwaha, S.; Pereira, N.L. Genomewide association study reveals novel genetic loci associated with change in renal function in heart transplant recipients. Clin. Transplant. 2018, 32, e13395. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, H.; Lee, I.H.; Du, J.; Mitch, W.E. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J. Clin. Investig. 2009, 119, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Medina, R.; Grieber, S.; May, R.C.; England, B.K.; Price, S.R.; Bailey, J.L.; Goldberg, A.L. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J. Clin. Investig. 1994, 93, 2127–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecker, S.H.; Mitch, W.E. Proteolysis by the ubiquitin-proteasome system and kidney disease. J. Am. Soc. Nephrol. 2011, 22, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, J.; Dannenhoffer, J.; Rutter, A. Acute Kidney Injury. Prim. Care 2020, 47, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Hocher, B.; Adamski, J. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 269–284. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; L, I.J.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired β-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Marcovina, S.M.; Sirtori, C.; Peracino, A.; Gheorghiade, M.; Borum, P.; Remuzzi, G.; Ardehali, H. Translating the basic knowledge of mitochondrial functions to metabolic therapy: Role of L-carnitine. Transl. Res. 2013, 161, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, B.; Merlen, C.; Vaillant, F.; McDuff, J.; Bouchard, B.; Beguin, P.C.; Dolinsky, V.W.; Foisy, S.; Villeneuve, L.R.; Labarthe, F.; et al. Post-translational modifications, a key process in CD36 function: Lessons from the spontaneously hypertensive rat heart. J. Mol. Cell Cardiol. 2011, 51, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.R. Inherited disorders of mitochondrial fatty acid oxidation: A new responsibility for the neonatologist. Semin. Neonatol. 2002, 7, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Petersen, M.C.; Nasiri, A.R.; Butrico, G.; Fung, A.; Ruan, H.B.; Kursawe, R.; Caprio, S.; Thibodeau, J.; Bourgeois-Daigneault, M.C.; et al. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat. Commun. 2016, 7, 12639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.S.; Liu, M.H.; Cheng, M.L.; Wang, C.H. Decreases in Circulating Concentrations of Short-Chain Acylcarnitines are Associated with Systolic Function Improvement After Decompensated Heart Failure. Int. Heart J. 2020, 61, 1014–1021. [Google Scholar] [CrossRef]
- Liu, J.J.; Ghosh, S.; Kovalik, J.P.; Ching, J.; Choi, H.W.; Tavintharan, S.; Ong, C.N.; Sum, C.F.; Summers, S.A.; Tai, E.S.; et al. Profiling of Plasma Metabolites Suggests Altered Mitochondrial Fuel Usage and Remodeling of Sphingolipid Metabolism in Individuals With Type 2 Diabetes and Kidney Disease. Kidney Int. Rep. 2017, 2, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Gansevoort, R.T.; Coresh, J.; Inker, L.A.; Heerspink, H.L.; Grams, M.E.; Greene, T.; Tighiouart, H.; Matsushita, K.; Ballew, S.H.; et al. Change in Albuminuria and GFR as End Points for Clinical Trials in Early Stages of CKD: A Scientific Workshop Sponsored by the National Kidney Foundation in Collaboration With the US Food and Drug Administration and European Medicines Agency. Am. J. Kidney Dis. 2020, 75, 84–104. [Google Scholar] [CrossRef] [Green Version]
- Beekman, M.; Nederstigt, C.; Suchiman, H.E.D.; Kremer, D.; van der Breggen, R.; Lakenberg, N.; Alemayehu, W.G.; de Craen, A.J.; Westendorp, R.G.; Boomsma, D.I. Genome-wide association study (GWAS)-identified disease risk alleles do not compromise human longevity. Proc. Natl. Acad. Sci. USA 2010, 107, 18046–18049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, G.M.; Waikar, S.S. Biomarkers in nephrology: Core Curriculum 2013. Am. J. Kidney Dis. 2013, 62, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Haneuse, S. Distinguishing Selection Bias and Confounding Bias in Comparative Effectiveness Research. Med. Care 2016, 54, e23–e29. [Google Scholar] [CrossRef] [Green Version]
- Song, J.W.; Chung, K.C. Observational studies: Cohort and case-control studies. Plast Reconstr. Surg. 2010, 126, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Gaunt, T.; McArdle, W.; Ho, K.; Duggirala, A.; Shihab, H.; Woodward, G.; Lyttleton, O.; Evans, D.M.; Reik, W.; et al. Data Resource Profile: Accessible Resource for Integrated Epigenomic Studies (ARIES). Int. J. Epidemiol. 2015, 44, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, S.; Smith, G.D. Commentary: Should we always deliberately be non-representative? Int. J. Epidemiol. 2013, 42, 1022–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.D. The Wright stuff: Genes in the interrogation of correlation and causation. Eur. J. Pers. 2012, 26, 395–397. [Google Scholar]
- Smith, G.D.; Lawlor, D.A.; Harbord, R.; Timpson, N.; Day, I.; Ebrahim, S. Clustered environments and randomized genes: A fundamental distinction between conventional and genetic epidemiology. PLoS Med. 2007, 4, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinilnikova, O.M.; Dondon, M.G.; Eon-Marchais, S.; Damiola, F.; Barjhoux, L.; Marcou, M.; Verny-Pierre, C.; Sornin, V.; Toulemonde, L.; Beauvallet, J.; et al. GENESIS: A French national resource to study the missing heritability of breast cancer. BMC Cancer 2016, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Heizhati, M.; Wang, L.; Gan, L.; Li, M.; Yang, W.; Yao, L.; Wang, Z.; Yang, Z.; Abudoyreyimu, R. Prevalence and associated factors of kidney dysfunction in patients with hypertension and/or diabetes mellitus from a primary care population in Northwest China. Int. J. Gen. Med. 2021, 14, 7567. [Google Scholar] [CrossRef]
- Cheung, B.M.; Li, C. Diabetes and hypertension: Is there a common metabolic pathway? Curr. Atheroscler. Rep. 2012, 14, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Jang, H.B.; Yoo, M.G.; Park, S.I.; Lee, H.J. Amino Acid Metabolites Associated with Chronic Kidney Disease: An Eight-Year Follow-Up Korean Epidemiology Study. Biomedicines 2020, 8, 222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Discovery a (N = 115) | Validation b (N = 69) | p-Value |
|---|---|---|---|
| Mean (SD) | Mean (SD) | ||
| Age at baseline | 57.2 (8.5) | 52.9 (11.9) | 0.01 |
| Systolic BP (mmHg) | 121.9 (10.6) | 134.0 (21.4) | <0.01 |
| Diastolic BP (mmHg) | 73.6 (7.4) | 78.9 (12.1) | <0.01 |
| Body mass index (kg/m2) | 24.2 (2.7) | 25.5 (3.3) | <0.01 |
| Hemoglobin (g/dL) | 13.8 (1.7) | 11.7 (1.9) | <0.01 |
| Serum albumin | 4.5 (0.3) | 3.7 (0.7) | <0.01 |
| eGFR (mL/min/1.73 m2) | 89.2 (14.5) | 53.6 (24.4) | <0.01 |
| eGFR slope (mL/min/1.73 m2/year) | −7.1 (2.1) | −6.4 (1.9) | 0.05 |
| Median [IQR] | Median [IQR] | ||
| Follow-up (years) | 3.9 [3.6, 4.2] | 1.8 [1.4, 2.4] | <0.01 |
| N (%) | N (%) | ||
| Sex (male) | 64 (55.7) | 49 (71.0) | 0.06 |
| K-CHIP Consortium 1 | KNOW-CKD 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Position | SNP | Function | Gene | Allele | MAF | Beta (SE) | p-Value | Beta (SE) | p-Value |
| 4q32.3 | 164664101 | rs5012631 | intronic | MARCHF1 | C/G | 0.03 | −4.113 (0.790) | 8.69 × 10−7 | −2.303 (1.118) | 0.04 |
| 4q32.3 | 164665383 | rs6852270 | intronic | MARCHF1 | C/T | 0.03 | −4.116 (0.790) | 8.55 × 10−7 | −2.315 (1.118) | 0.04 |
| 4q32.3 | 164670375 | rs71600637 | intronic | MARCHF1 | C/CAT | 0.03 | −4.127 (0.790) | 8.08 × 10−7 | −2.377 (1.117) | 0.04 |
| 4q32.3 | 164670444 | rs1390835129 | intronic | MARCHF1 | CT/C | 0.03 | −4.129 (0.790) | 8.01 × 10−7 | −2.365 (1.118) | 0.04 |
| 4q32.3 | 164674901 | rs10009742 | intronic | MARCHF1 | C/T | 0.03 | −4.128 (0.790) | 8.01 × 10−7 | −2.361 (1.118) | 0.04 |
| SNP | Function | Gene | Alleles | MAF | Beta (SE) 1 | p-Value 1 | Metabolites | Beta (SE) 2 | p-Value 2 | FDR2 |
|---|---|---|---|---|---|---|---|---|---|---|
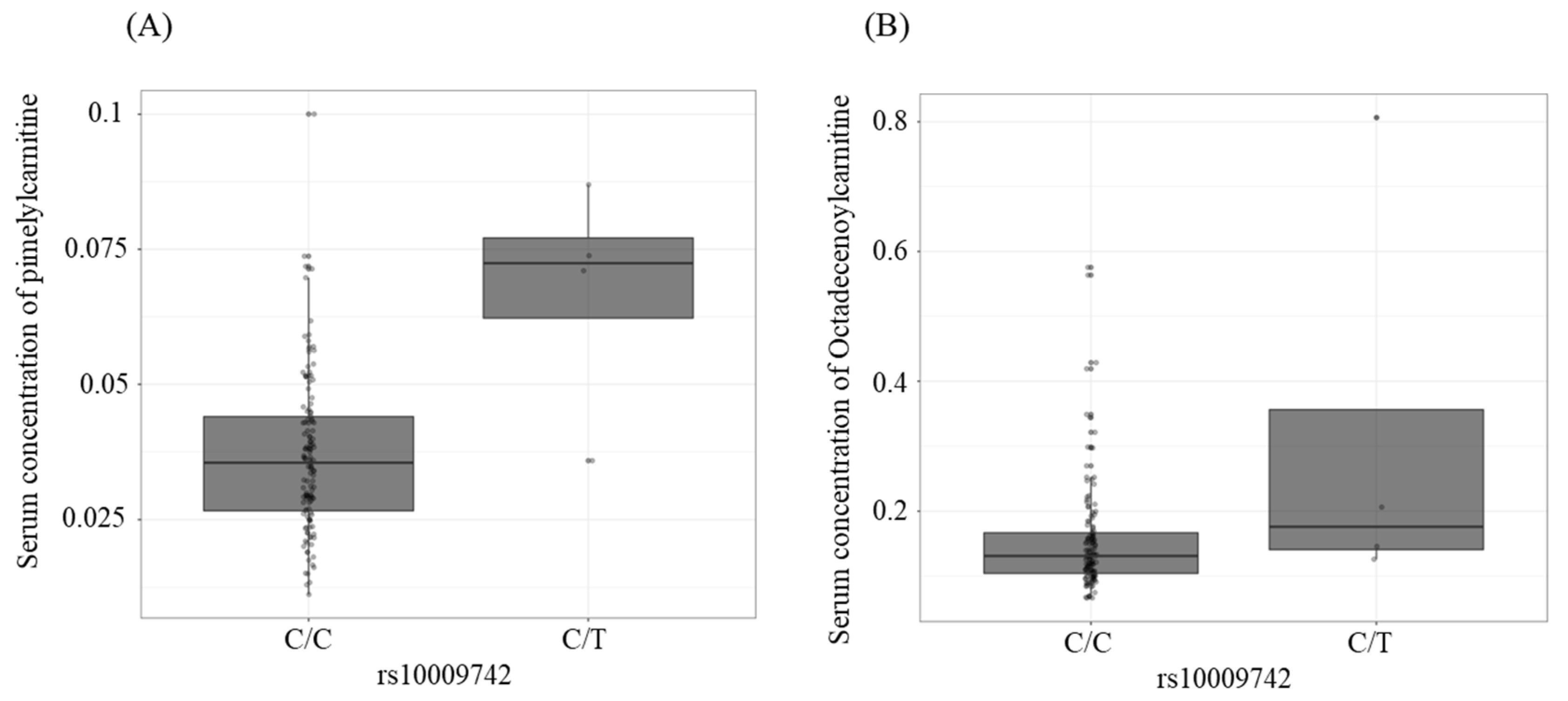
| rs10009742 | intronic | MARCHF1 | C/T | 0.027 | −4.128 (0.790) | 8.01 × 10−7 | C7-DC | 0.030 (0.007) | 7.10 × 10−5 | 1.44 × 10−2 |
| rs10009742 | intronic | MARCHF1 | C/T | 0.027 | −4.128 (0.790) | 8.01 × 10−7 | C18:1 | 0.167 (0.049) | 8.11 × 10−4 | 8.21 × 10−2 |
| rs1390835129 | intronic | MARCHF1 | CT/C | 0.027 | −4.129 (0.790) | 8.01 × 10−7 | C7-DC | 0.030 (0.007) | 7.10 × 10−5 | 1.44 × 10−2 |
| rs1390835129 | intronic | MARCHF1 | CT/C | 0.027 | −4.129 (0.790) | 8.01 × 10−7 | C18:1 | 0.167 (0.049) | 8.11 × 10−4 | 8.21 × 10−2 |
| rs71600637 | intronic | MARCHF1 | C/CAT | 0.027 | −4.127 (0.790) | 8.08 × 10−7 | C7-DC | 0.030 (0.007) | 7.10 × 10−5 | 1.44 × 10−2 |
| rs71600637 | intronic | MARCHF1 | C/CAT | 0.027 | −4.127 (0.790) | 8.08 × 10−7 | C18:1 | 0.167 (0.049) | 8.11 × 10−4 | 8.21 × 10−2 |
| rs6852270 | intronic | MARCHF1 | C/T | 0.027 | −4.116 (0.790) | 8.55 × 10−7 | C7-DC | 0.030 (0.007) | 7.10 × 10−5 | 1.44 × 10−2 |
| rs6852270 | intronic | MARCHF1 | C/T | 0.027 | −4.116 (0.790) | 8.55 × 10−7 | C18:1 | 0.167 (0.049) | 8.11 × 10−4 | 8.21 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Han, M.; Moon, S.; Kim, K.; An, W.J.; Ryu, H.; Oh, K.-H.; Park, S.K. Identifying Genetic Variants and Metabolites Associated with Rapid Estimated Glomerular Filtration Rate Decline in Korea Based on Genome–Metabolomic Integrative Analysis. Metabolites 2022, 12, 1139. https://doi.org/10.3390/metabo12111139
Lee S, Han M, Moon S, Kim K, An WJ, Ryu H, Oh K-H, Park SK. Identifying Genetic Variants and Metabolites Associated with Rapid Estimated Glomerular Filtration Rate Decline in Korea Based on Genome–Metabolomic Integrative Analysis. Metabolites. 2022; 12(11):1139. https://doi.org/10.3390/metabo12111139
Chicago/Turabian StyleLee, Sangjun, Miyeun Han, Sungji Moon, Kyungsik Kim, Woo Ju An, Hyunjin Ryu, Kook-Hwan Oh, and Sue K. Park. 2022. "Identifying Genetic Variants and Metabolites Associated with Rapid Estimated Glomerular Filtration Rate Decline in Korea Based on Genome–Metabolomic Integrative Analysis" Metabolites 12, no. 11: 1139. https://doi.org/10.3390/metabo12111139