
ABHD5—A Regulator of Lipid Metabolism Essential for Diverse Cellular Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protein Structure of ABHD5
3. Neutral Lipid Storage Disease in Humans—A Disorder with Two Distinct Clinical Phenotypes
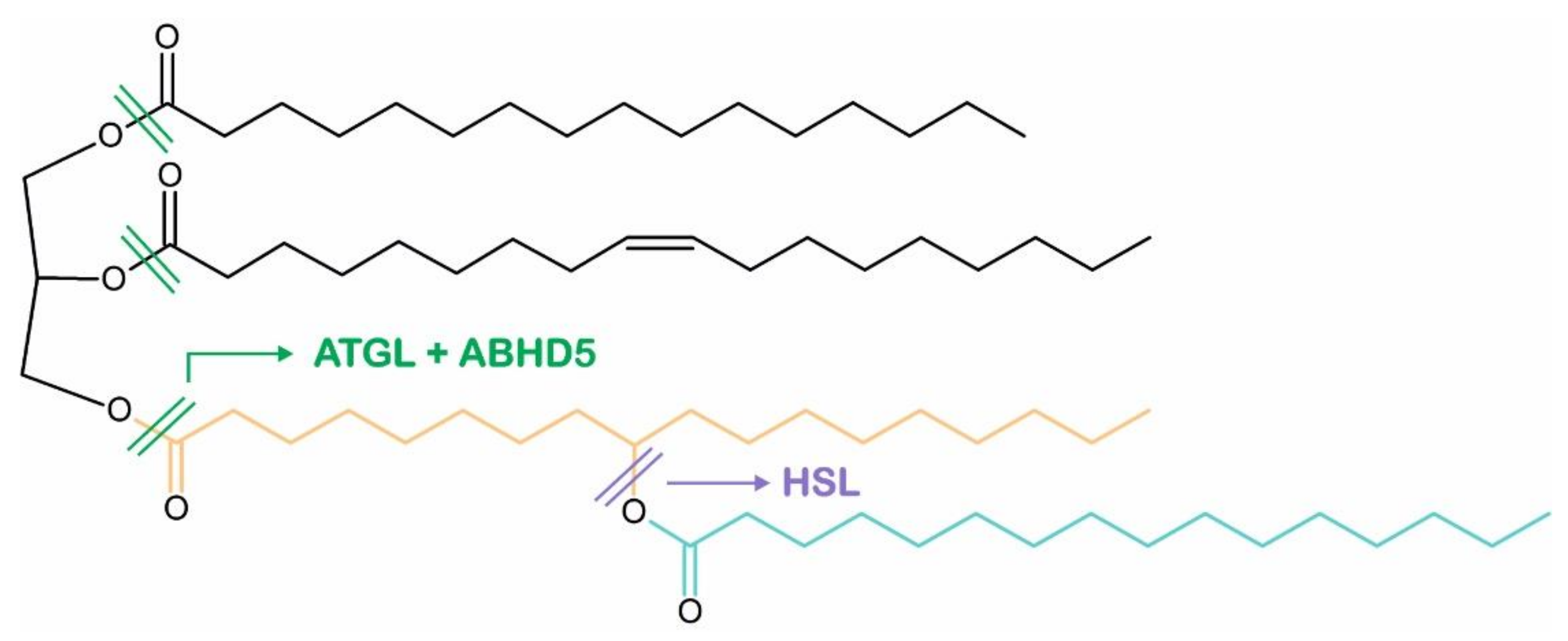
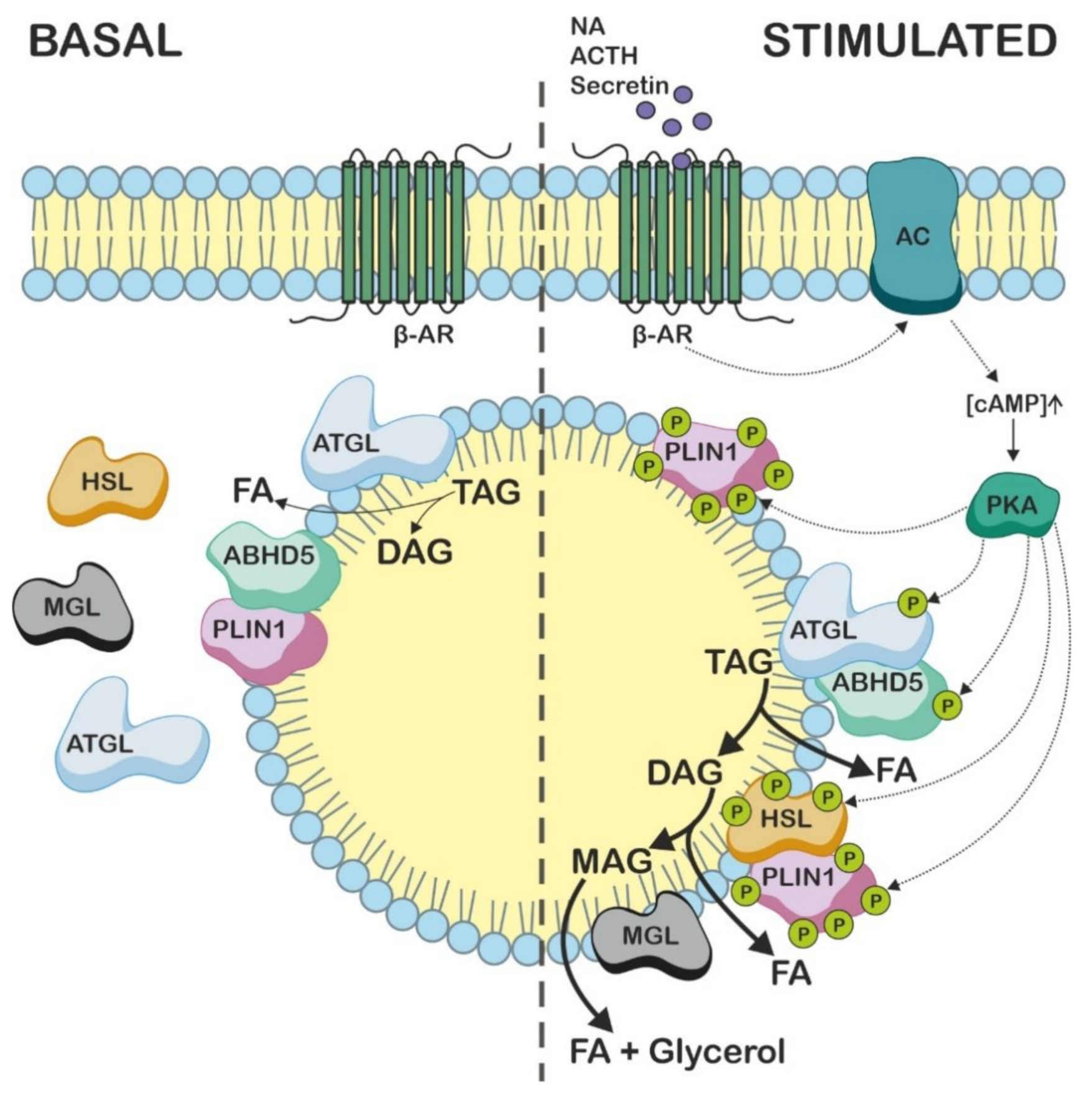
4. ABHD5 Is Involved in the Mobilization of Intracellular TAG Stores
5. Intracellular TAG Hydrolysis Requires the Presence of ABHD5
6. ABHD5 Interacts with LD-Associated Proteins
7. Studies Using Abhd5 Transgenic Mice Show That ABHD5 Plays an Important Role in Adipose and Non-Adipose Tissues
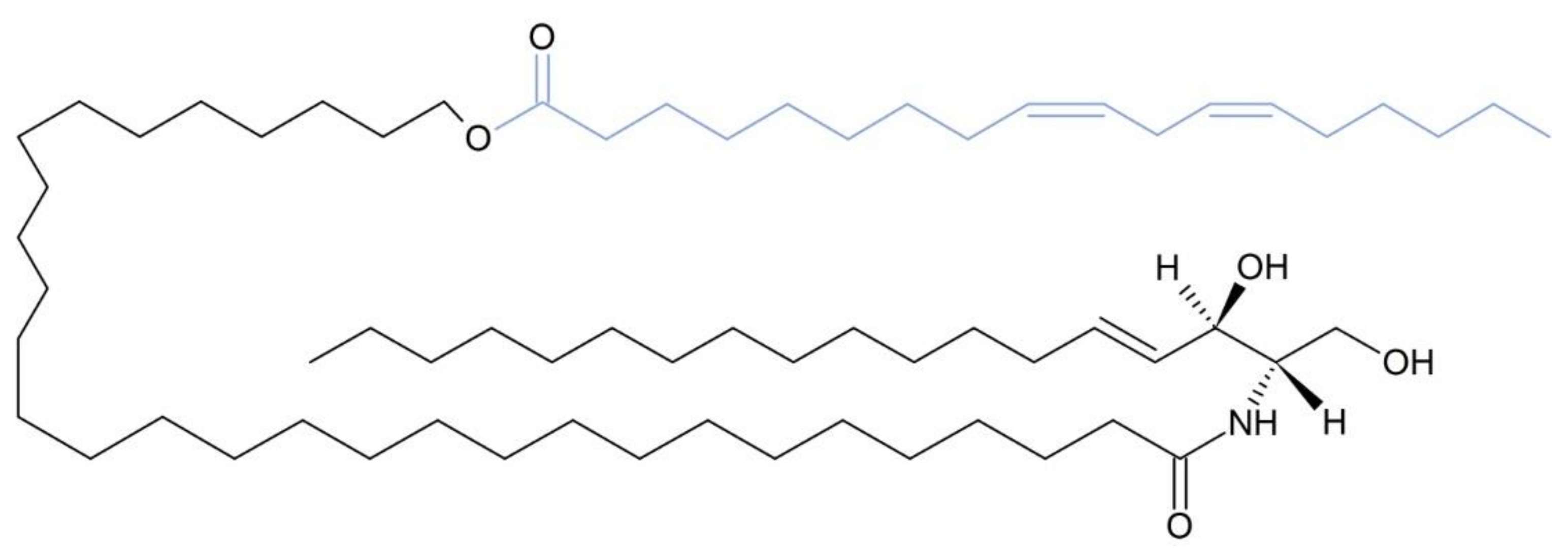
8. ABHD5 Possesses an ATGL-Independent Function in the Skin
9. ABHD5 Co-Activates PNPLA1 Function for Efficient Synthesis of AcylCers
10. ATGL and ABHD5 Exhibit Distinct Functions in Hepatic Lipid Metabolism and Inflammation
11. Protein Interaction of ABHD5 with PNPLA3 Is Critical in the Development of Fatty Liver Disease
12. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W.C. Identification of Novel Human Genes Evolutionarily Conserved in Caenorhabditis Elegans by Comparative Proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose Triglyceride Lipase-Mediated Lipolysis of Cellular Fat Stores is Activated by CGI-58 and Defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, V.; Rotlienberg, A.; Gomez, C.; Cohen, A.W.; Garcia, A.; Bhattacharyya, S.; Shapiro, L.; Dolios, G.; Wang, R.; Lisanti, M.P.; et al. Perilipin A Mediates the Reversible Binding of CGI-58 to Lipid Droplets in 3T3-L1 Adipocytes. J. Biol. Chem. 2004, 279, 42062–42071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Chung, S.; Das, A.; Shelness, G.S.; Rudel, L.L.; Yu, L. CGI-58 Facilitates the Mobilization of Cytoplasmic Triglyceride for Lipoprotein Secretion in Hepatoma Cells. J. Lipid Res. 2007, 48, 2295–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanarin, I.; Patel, A.; Slavin, G.; Wills, E.J.; Andrews, T.M.; Stewart, G. Neutral-Lipid Storage Disease: A New Disorder of Lipid Metabolism. Br. Med. J. 1975, 1, 553–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavin, G.; Wills, E.J.; Richmond, J.E.; Chanarin, I.; Andrews, T.; Stewart, G. Morphological Features in a Neutral Lipid Storage Disease. J. Clin. Pathol. 1975, 28, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, M.L. Ichthyosiform Dermatosis with Systemic Lipidosis. Arch. Dermatol. 1974, 110, 261. [Google Scholar] [CrossRef]
- Kien, B.; Grond, S.; Haemmerle, G.; Lass, A.; Eichmann, T.O.; Radner, F.P.W. ABHD5 Stimulates PNPLA1-Mediated -O-Acylceramide Biosynthesis Essential for a Functional Skin Permeability Barrier. J. Lipid Res. 2018, 59, 2360–2367. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Mottillo, E.P.; Mladenovic-Lucas, L.; Zhou, L.; Granneman, J.G. Dynamic Interactions of ABHD5 with PNPLA3 Regulate Triacylglycerol Metabolism in Brown Adipocytes. Nat. Metab. 2019, 1, 560–569. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Omatsu, N.; Matsushita, S.; Osumi, T. CGI-58 Interacts with Perilipin and is Localized to Lipid Droplets: Possible Involvement of CGI-58 Mislocalization in Chanarin-Dorfman Syndrome. J. Biol. Chem. 2004, 279, 30490–30497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Omatsu, N.; Morimoto, E.; Nakashima, H.; Ueno, K.; Tanaka, T.; Satouchi, K.; Hirose, F.; Osumi, T. CGI-58 Facilitates Lipolysis on Lipid Droplets but is not Involved in the Vesiculation of Lipid Droplets Caused by Hormonal Stimulation. J. Lipid Res. 2007, 48, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granneman, J.G.; Moore, H.P.H.; Granneman, R.L.; Greenberg, A.S.; Obin, M.S.; Zhu, Z. Analysis of Lipolytic Protein Trafficking and Interactions in Adipocytes. J. Biol. Chem. 2007, 282, 5726–5735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Yang, W.; Kozusko, K.; Saudek, V.; Savage, D.B. Perilipins 2 and 3 Lack a Carboxy-Terminal Domain Present in Perilipin 1 Involved in Sequestering ABHD5 and Suppressing Basal Lipolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 9163–9168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granneman, J.G.; Moore, H.-P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with Adipose Triglyceride Lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Bell, M.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; Coleman, R.; et al. Unique Regulation of Adipose Triglyceride Lipase (ATGL) by Perilipin 5, a Lipid Droplet-Associated Protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granneman, J.G.; Moore, H.P.H.; Krishnamoorthy, R.; Rathod, M. Perilipin Controls Lipolysis by Regulating the Interactions of AB-Hydrolase Containing 5 (Abhd5) and Adipose Triglyceride Lipase (Atgl). J. Biol. Chem. 2009, 284, 34538–34544. [Google Scholar] [CrossRef] [Green Version]
- Hofer, P.; Boeszoermenyi, A.; Jaeger, D.; Feiler, U.; Arthanari, H.; Mayer, N.; Zehender, F.; Rechberger, G.; Oberer, M.; Zimmermann, R.; et al. Fatty Acid-Binding Proteins Interact with Comparative Gene Identification-58 Linking Lipolysis with Lipid Ligand Shuttling. J. Biol. Chem. 2015, 290, 18438–18453. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.A.; Madoux, F.; Mladenovic, L.; Zhang, H.; Ye, X.; Angrish, M.; Mottillo, E.P.; Caruso, J.A.; Halvorsen, G.; Roush, W.R.; et al. Endogenous and Synthetic ABHD5 Ligands Regulate ABHD5-Perilipin Interactions and Lipolysis in Fat and Muscle. Cell Metab. 2015, 22, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The α/β Hydrolase Fold. Protein Eng. Des. Sel. 1992, 5, 197–211. [Google Scholar] [CrossRef]
- Lord, C.C.; Thomas, G.; Brown, J.M. Mammalian Alpha Beta Hydrolase Domain (ABHD) Proteins: Lipid Metabolizing Enzymes at the Interface of Cell Signaling and Energy Metabolism. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 792–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.; Cornaciu, I.; Lass, A.; Schweiger, M.; Poeschl, M.; Eder, C.; Kumari, M.; Schoiswohl, G.; Wolinski, H.; Kohlwein, S.D.; et al. The N-Terminal Region of Comparative Gene Identification-58 (CGI-58) is Important for Lipid Droplet Binding and Activation of Adipose Triglyceride Lipase. J. Biol. Chem. 2010, 285, 12289–12298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardini, M.; Dijkstra, B.W. α/β Hydrolase Fold Enzymes: The Family Keeps Growing. Curr. Opin. Struct. Biol. 1999, 9, 732–737. [Google Scholar] [CrossRef]
- Lefèvre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the Gene Encoding a New Protein of the Esterase/Lipase/Thioesterase Subfamily, in Chanarin-Dorfman Syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Ramakrishnan, G.; Chandramohan, C.; Rajasekharan, R. CGI-58, the Causative Gene for Chanarin-Dorfman Syndrome, Mediates Acylation of Lysophosphatidic Acid. J. Biol. Chem. 2008, 283, 24525–24533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero-Moran, G.; Caviglia, J.M.; McMahon, D.; Rothenberg, A.; Subramanian, V.; Xu, Z.; Lara-Gonzalez, S.; Storch, J.; Carman, G.M.; Brasaemle, D.L. CGI-58/ABHD5 Is a Coenzyme A-Dependent Lysophosphatidic Acid Acyltransferase. J. Lipid Res. 2010, 51, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, D.; Dinh, A.; Kurz, D.; Shah, D.; Han, G.S.; Carman, G.M.; Brasaemle, D.L. Comparative Gene Identification 58/α/β Hydrolase Domain 5 Lacks Lysophosphatidic Acid Acyltransferase Activity. J. Lipid Res. 2014, 55, 1750–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulminskaya, N.; Oberer, M. Protein-Protein Interactions Regulate the Activity of Adipose Triglyceride Lipase in Intracellular Lipolysis. Biochimie 2020, 169, 62–68. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Sanders, M.A.; Zhang, H.; Zhou, L.; Chou, C.-Y.; Granneman, J.G. Structural and Functional Insights into ABHD5, a Ligand-Regulated Lipase Co-Activator. Sci. Rep. 2022, 12, 2565. [Google Scholar] [CrossRef]
- Jordans, G.H. The Familial Occurrence of Fat Containing Vacuoles in the Leukocytes Diagnosed in Two Brothers Suffering from Dystrophia Musculorum Progressiva (ERB.). Acta Med. Scand. 1953, 145, 419–423. [Google Scholar] [CrossRef]
- Rozenszajn, L.; Klajman, A.; Yaffe, D.; Efrati, P. Jordans’ Anomaly in White Blood Cells. Report of Case. Blood 1966, 28, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Vahlquist, A.; Fischer, J.; Törmä, H. Inherited Nonsyndromic Ichthyoses: An Update on Pathophysiology, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2018, 19, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Igal, R.A.; Rhoads, J.M.; Coleman, R.A. Neutral Lipid Storage Disease with Fatty Liver and Cholestasis. J. Pediatr. Gastroenterol. Nutr. 1997, 25, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Bruno, C.; Bertini, E.; Di Rocco, M.; Cassandrini, D.; Ruffa, G.; De Toni, T.; Seri, M.; Spada, M.; Li Volti, G.; D’Amico, A.; et al. Clinical and Genetic Characterization of Chanarin–Dorfman Syndrome. Biochem. Biophys. Res. Commun. 2008, 369, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, C.; Coleman, R.A.; Moro, L.; Dacou-Voutetakis, C.; Elsayed, S.M.; Prati, D.; Colli, A.; Mela, D.; Colombo, R.; Tavian, D. Clinical and Genetic Characterization of Chanarin-Dorfman Syndrome Patients: First Report of Large Deletions in the ABHD5 Gene. Orphanet J. Rare Dis. 2010, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol, R.M.; Gilaberte, M.; Toll, A.; Florensa, L.; Lloreta, J.; Gonzalez-Ensenat, M.A.; Fischer, J.; Azon, A. Erythrokeratoderma Variabilis-like Ichthyosis in Chanarin-Dorfman Syndrome. Br. J. Dermatol. 2005, 153, 838–841. [Google Scholar] [CrossRef]
- Samuelov, L.; Fuchs-Telem, D.; Sarig, O.; Sprecher, E. An Exceptional Mutational Event Leading to Chanarin-Dorfman Syndrome in a Large Consanguineous Family. Br. J. Dermatol. 2011, 164, 1390–1392. [Google Scholar] [CrossRef]
- Cakmak, E.; Alagozlu, H.; Yonem, O.; Ataseven, H.; Citli, S.; Ozer, H. Steatohepatitis and Liver Cirrhosis in Chanarin-Dorfman Syndrome with a New ABDH5 Mutation. Clin. Res. Hepatol. Gastroenterol. 2012, 36, e34–e37. [Google Scholar] [CrossRef]
- Emre, S.; Ünver, N.; Evans, S.E.; Yüzbaşıoğlu, A.; Gürakan, F.; Gümrük, F.; Karaduman, A. Molecular Analysis of Chanarin-Dorfman Syndrome (CDS) Patients: Identification of Novel Mutations in the ABHD5 Gene. Eur. J. Med. Genet. 2010, 53, 141–144. [Google Scholar] [CrossRef]
- Schleinitz, N.; Fischer, J.; Sanchez, A.; Veit, V.; Harle, J.-R.; Pelissier, J.-F. Two New Mutations of the ABHD5 Gene in a New Adult Case of Chanarin Dorfman Syndrome: An Uncommon Lipid Storage Disease. Arch. Dermatol. 2005, 141, 798–800. [Google Scholar] [CrossRef]
- Ben Selma, Z.; Yilmaz, S.; Schischmanoff, P.O.; Blom, A.; Ozogul, C.; Laroche, L.; Caux, F. A Novel S115G Mutation of CGI-58 in a Turkish Patient with Dorfman-Chanarin Syndrome [3]. J. Invest. Dermatol. 2007, 127, 2273–2276. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Hadžić, N.; Fischer, J.; Knisely, A. Steatohepatitis and Unsuspected Micronodular Cirrhosis in Dorfman-Chanarin Syndrome with Documented ABHD5 Mutation. J. Pediatr. 2004, 144, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Sawamura, D.; Shimizu, H.; Nomura, Y.; Sugawara, M. Truncation of CGI-58 Protein Causes Malformation of Lamellar Granules Resulting in Ichthyosis in Dorfman-Chanarin Syndrome. J. Invest. Dermatol. 2003, 121, 1029–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasaraghavan, R.; Krishnamurthy, S.; Chandar, R.; Cassandrini, D.; Mahadevan, S.; Bruno, C.; Santorelli, F.M. Acitretin-Responsive Ichthyosis in Chanarin-Dorfman Syndrome with a Novel Mutation in the ABHD5/CGI-5 8 Gene. Pediatr. Dermatol. 2014, 31, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Tamhankar, P.; Iyer, S.; Sanghavi, S.; Khopkar, U. Chanarin-Dorfman Syndrome: Clinical Report and Novel Mutation in ABHD5 Gene. J. Postgrad. Med. 2014, 60, 332. [Google Scholar] [CrossRef] [PubMed]
- Missaglia, S.; Valadares, E.R.; Moro, L.; Faguntes, E.D.T.; quintão Roque, R.; Giardina, B.; Tavian, D. Early Onset of Chanarin-Dorfman Syndrome with Severe Liver Involvement in a Patient with a Complex Rearrangement of ABHD5 Promoter. BMC Med. Genet. 2014, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Cakmak, E.; Bagci, G. Chanarin-Dorfman Syndrome: A Comprehensive Review. Liver Int. 2021, 41, 905–914. [Google Scholar] [CrossRef]
- Schweiger, M.; Lass, A.; Zimmermann, R.; Eichmann, T.O.; Zechner, R. Neutral Lipid Storage Disease: Genetic Disorders Caused by Mutations in Adipose Triglyceride Lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E289–E296. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Lefèvre, C.; Morava, E.; Mussini, J.M.; Lafoŕt, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The Gene Encoding Adipose Triglyceride Lipase (PNPLA2) is Mutated in Neutral Lipid Storage Disease with Myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef]
- Fiorillo, C.; Brisca, G.; Cassandrini, D.; Scapolan, S.; Astrea, G.; Valle, M.; Scuderi, F.; Trucco, F.; Natali, A.; Magnano, G.; et al. Subclinical Myopathy in a Child with Neutral Lipid Storage Disease and Mutations in the PNPLA2 Gene. Biochem. Biophys. Res. Commun. 2013, 430, 241–244. [Google Scholar] [CrossRef]
- Tavian, D.; Missaglia, S.; Redaelli, C.; Pennisi, E.M.; Invernici, G.; Wessalowski, R.; Maiwald, R.; Arca, M.; Coleman, R.A. Contribution of Novel ATGL Missense Mutations to the Clinical Phenotype of NLSD-M: A Strikingly Low Amount of Lipase Activity May Preserve Cardiac Function. Hum. Mol. Genet. 2012, 21, 5318–5328. [Google Scholar] [CrossRef] [PubMed]
- Samukawa, M.; Nakamura, N.; Hirano, M.; Morikawa, M.; Sakata, H.; Nishino, I.; Izumi, R.; Suzuki, N.; Kuroda, H.; Shiga, K.; et al. Neutral Lipid Storage Disease Associated with the PNPLA2 Gene: Case Report and Literature Review. Eur. Neurol. 2020, 83, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Schweiger, M.; Kloss-Brandstätter, A.; Lamina, C.; Haun, M.; Erhart, G.; Paulweber, B.; Rahman, Y.; Olpin, S.; Wolinski, H.; et al. Investigation and Functional Characterization of Rare Genetic Variants in the Adipose Triglyceride Lipase in a Large Healthy Working Population. PLoS Genet. 2010, 6, e1001239. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.L.; Monger, D.J.; Rutherford, S.L.; Hincenbergs, M.; Rehfeld, S.J.; Grunfeld, C. Neutral Lipid Storage Disease with Ichthyosis: Lipid Content and Metabolism of Fibroblasts. J. Inherit. Metab. Dis. 1988, 11, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; DiMauro, S.; Eastwood, A.; Hays, A.; Johnson, W.G.; Olarte, M.; Whitlock, R.; Mayeux, R.; Rowland, L.P. Lipid Storage Myopathy, Ichthyosis, and Steatorrhea. Muscle Nerve 1979, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Radom, J.; Salvayre, R.; Mussini, J.M.; De Lisle, B.; Negre, A.; Maret, A.; Billaudel, S.; Douste-Blazy, L. Biochemical and Ultrastructural Features of Human Fibroblasts Cultured from a New Variant of Type 3 Lipid Storage Myopathy. Biol. Cell 1988, 62, 39–45. [Google Scholar] [CrossRef]
- Salvayre, R.; Nègre, A.; Radom, J.; Douste-Blazy, L. Independence of Triacylglycerol-Containing Compartments in Cultured Fibroblasts from Wolman Disease and Multisystemic Lipid Storage Myopathy. FEBS Lett. 1989, 250, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Igal, R.A.; Coleman, R.A. Neutral Lipid Storage Disease: A Genetic Disorder with Abnormalities in the Regulation of Phospholipid Metabolism. J. Lipid Res. 1998, 39, 31–43. [Google Scholar] [CrossRef]
- Williams, M.L.; Coleman, R.A.; Placezk, D.; Grunfeld, C. Neutral Lipid Storage Disease: A Possible Functional Defect in Phospholipid-Linked Triacylglycerol Metabolism. Biochim. Biophys. Acta-Mol. Basis Dis. 1991, 1096, 162–169. [Google Scholar] [CrossRef]
- Bergman, R.; Aviram, M.; Bitterman-Deutsch, O.; Oiknine, Y.; Shemer, A.; Srebnik, A.; Brook, J.G.; Friedman-Birnbaum, R. Neutral Lipid Storage Disease with Ichthyosis: Serum Apolipoprotein Levels and Cholesterol Metabolism in Monocyte-derived Macrophages. J. Inherit. Metab. Dis. 1991, 14, 241–246. [Google Scholar] [CrossRef]
- Hilaire, N.; Salvayre, R.; Thiers, J.C.; Bonnafé, M.J.; Nègre-Salvayre, A. The Turnover of Cytoplasmic Triacylglycerols in Human Fibroblasts Involves Two Separate Acyl Chain Length-Dependent Degradation Pathways. J. Biol. Chem. 1995, 270, 27027–27034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igal, R.A.; Coleman, R.A. Acylglycerol Recycling from Triacylglycerol to Phospholipid, Not Lipase Activity, Is Defective in Neutral Lipid Storage Disease Fibroblasts. J. Biol. Chem. 1996, 271, 16644–16651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, D.L.; Beames, B.; Summers, M.D.; Park, W.D. Characterization of the Lipid Acyl Hydrolase Activity of the Major Potato (Solanum Tuberosum) Tuber Protein, Patatin, by Cloning and Abundant Expression in a Baculovirus Vector. Biochem. J. 1988, 252, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydel, T.J.; Williams, J.M.; Krieger, E.; Moshiri, F.; Stallings, W.C.; Brown, S.M.; Pershing, J.C.; Purcell, J.P.; Alibhai, M.F. The Crystal Structure, Mutagenesis, and Activity Studies Reveal that Patatin is a Lipid Acyl Hydrolase with a Ser-Asp Catalytic Dyad. Biochemistry 2003, 42, 6696–6708. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian Patatin Domain Containing Proteins: A Family with Diverse Lipolytic Activities Involved in Multiple Biological Functions. J. Lipid Res. 2009, 50, S63–S68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.A.; Gardner, S.D.; Lambie, N.M.; Commans, S.A.; Crowther, D.J. Characterization of the Human Patatin-like Phospholipase Family. J. Lipid Res. 2006, 47, 1940–1949. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue is Promoted by Adipose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.-H.; Sul, H.S. Desnutrin, an Adipocyte Gene Encoding a Novel Patatin Domain-Containing Protein, is Induced by Fasting and Glucocorticoids. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, Cloning, Expression, and Purification of Three Novel Human Calcium-Independent Phospholipase A2 Family Members Possessing Triacylglycerol Lipase and Acylglycerol Transacylase Activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.C.; Sun, Y.; Li, J.-L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, Regulation, and Triglyceride Hydrolase Activity of Adiponutrin Family Members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular Mechanisms for Lipid Mobilization from Fat Stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, M.; Berger, J.E.; Steinberg, D. Hormone-Sensitive Lipase and Monoglyceride Lipase Activities in Adipose Tissue. J. Biol. Chem. 1964, 239, 401–409. [Google Scholar] [CrossRef]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-Sensitive Lipase Deficiency in Mice Causes Diglyceride Accumulation in Adipose Tissue, Muscle, and Testis. J. Biol. Chem. 2002, 277, 4806–4815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornqvist, H.; Belfrage, P. Purification and Some Properties of a Monoacylglycerol-Hydrolyzing Enzyme of Rat Adipose Tissue. J. Biol. Chem. 1976, 251, 813–819. [Google Scholar] [CrossRef]
- Taschler, U.; Radner, F.P.W.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride Lipase Deficiency in Mice Impairs Lipolysis and Attenuates Diet-Induced Insulin Resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Heckmann, B.L.; Campbell, L.E.; Liu, J. G0S2: A Small Giant Controller of Lipolysis and Adipose-Liver Fatty Acid Flux. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lu, X.; Lombès, M.; Rha, G.B.; Chi, Y.-I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G0/G1 Switch Gene 2 Regulates Adipose Lipolysis through Association with Adipose Triglyceride Lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Padmanabha Das, K.M.; Wechselberger, L.; Liziczai, M.; De la Rosa Rodriguez, M.; Grabner, G.F.; Heier, C.; Viertlmayr, R.; Radler, C.; Lichtenegger, J.; Zimmermann, R.; et al. Hypoxia-Inducible Lipid Droplet-Associated Protein Inhibits Adipose Triglyceride Lipase. J. Lipid Res. 2018, 59, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of Intracellular Lipolysis Promotes Human Cancer Cell Adaptation to Hypoxia. Elife 2017, 6, e31132. [Google Scholar] [CrossRef]
- Riegler-Berket, L.; Wechselberger, L.; Cerk, I.K.; Padmanabha Das, K.M.; Viertlmayr, R.; Kulminskaya, N.; Rodriguez Gamez, C.F.; Schweiger, M.; Zechner, R.; Zimmermann, R.; et al. Residues of the Minimal Sequence of G0S2 Collectively Contribute to ATGL Inhibition while C-and N-Terminal Extensions Promote Binding to ATGL. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2022, 1867, 159105. [Google Scholar] [CrossRef]
- Notari, L.; Baladron, V.; Aroca-Aguilar, J.D.; Balko, N.; Heredia, R.; Meyer, C.; Notario, P.M.; Saravanamuthu, S.; Nueda, M.-L.; Sanchez-Sanchez, F.; et al. Identification of a Lipase-Linked Cell Membrane Receptor for Pigment Epithelium-Derived Factor. J. Biol. Chem. 2006, 281, 38022–38037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
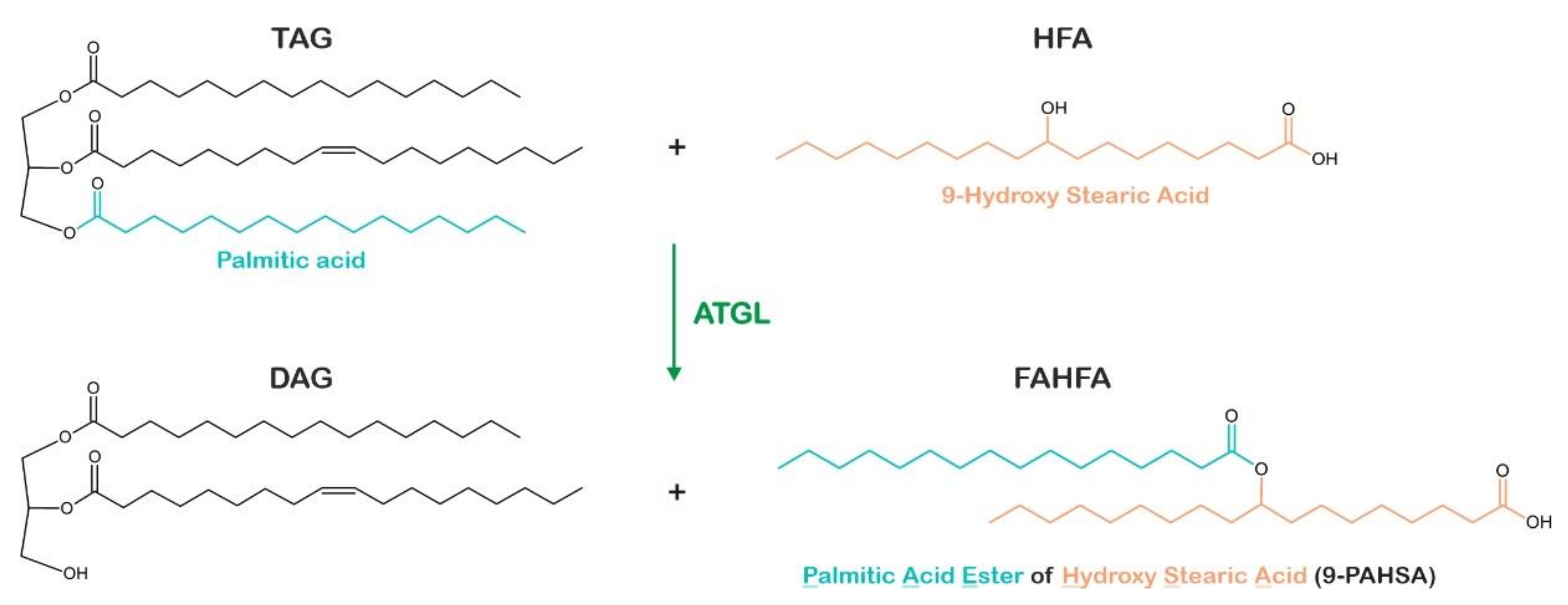
- Patel, R.; Santoro, A.; Hofer, P.; Tan, D.; Oberer, M.; Nelson, A.T.; Konduri, S.; Siegel, D.; Zechner, R.; Saghatelian, A.; et al. ATGL Is a Biosynthetic Enzyme for Fatty Acid Esters of Hydroxy Fatty Acids. Nature 2022, 606, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Brejchova, K.; Balas, L.; Paluchova, V.; Brezinova, M.; Durand, T.; Kuda, O. Understanding FAHFAs: From Structure to Metabolic Regulation. Prog. Lipid Res. 2020, 79, 101053. [Google Scholar] [CrossRef] [PubMed]
- Brejchova, K.; Radner, F.P.W.; Balas, L.; Paluchova, V.; Cajka, T.; Chodounska, H.; Kudova, E.; Schratter, M.; Schreiber, R.; Durand, T.; et al. Distinct Roles of Adipose Triglyceride Lipase and Hormone-Sensitive Lipase in the Catabolism of Triacylglycerol Estolides. Proc. Natl. Acad. Sci. USA 2021, 118, e2020999118. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.A.; Zhang, H.; Mladenovic, L.; Tseng, Y.Y.; Granneman, J.G. Molecular Basis of ABHD5 Lipolysis Activation. Sci. Rep. 2017, 7, 42589. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the Substrate and Stereo/Regioselectivity of Adipose Triglyceride Lipase, Hormone-Sensitive Lipase, and Diacylglycerol-O- Acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457. [Google Scholar] [CrossRef] [Green Version]
- Sztalryd, C.; Brasaemle, D.L. The Perilipin Family of Lipid Droplet Proteins: Gatekeepers of Intracellular Lipolysis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT Proteins, an Ancient Family of Lipid Droplet Proteins that Regulate Cellular Lipid Stores. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2009, 1791, 419–440. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L.; Rubin, B.; Harten, I.A.; Gruia-Gray, J.; Kimmel, A.R.; Londos, C. Perilipin A Increases Triacylglycerol Storage by Decreasing the Rate of Triacylglycerol Hydrolysis. J. Biol. Chem. 2000, 275, 38486–38493. [Google Scholar] [CrossRef] [Green Version]
- Sahu-Osen, A.; Montero-Moran, G.; Schittmayer, M.; Fritz, K.; Dinh, A.; Chang, Y.F.; McMahon, D.; Boeszoermenyi, A.; Cornaciu, I.; Russell, D.; et al. CGI-58/ABHD5 Is Phosphorylated on Ser239 by Protein Kinase A: Control of Subcellular Localization. J. Lipid Res. 2015, 56, 109–121. [Google Scholar] [CrossRef]
- Wang, H.; Hu, L.; Dalen, K.; Dorward, H.; Marcinkiewicz, A.; Russell, D.; Gong, D.; Londos, C.; Yamaguchi, T.; Holm, C.; et al. Activation of Hormone-Sensitive Lipase Requires Two Steps, Protein Phosphorylation and Binding to the PAT-1 Domain of Lipid Droplet Coat Proteins. J. Biol. Chem. 2009, 284, 32116–32125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandotra, S.; Lim, K.; Girousse, A.; Saudek, V.; O’Rahilly, S.; Savage, D.B. Human Frame Shift Mutations Affecting the Carboxyl Terminus of Perilipin Increase Lipolysis by Failing to Sequester the Adipose Triglyceride Lipase (ATGL) Coactivator AB-Hydrolase-Containing 5 (ABHD5). J. Biol. Chem. 2011, 286, 34998–35006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasaemle, D.L.; Barber, T.; Wolins, N.E.; Serrero, G.; Blanchette-Mackie, E.J.; Londos, C. Adipose Differentiation-Related Protein Is an Ubiquitously Expressed Lipid Storage Droplet-Associated Protein. J. Lipid Res. 1997, 38, 2249–2263. [Google Scholar] [CrossRef]
- Libby, A.E.; Bales, E.; Orlicky, D.J.; McManaman, J.L. Perilipin-2 Deletion Impairs Hepatic Lipid Accumulation by Interfering with Sterol Regulatory Element-Binding Protein (SREBP) Activation and Altering the Hepatic Lipidome. J. Biol. Chem. 2016, 291, 24231–24246. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.H.-J.; Li, L.; Paul, A.; Taniguchi, S.; Nannegari, V.; Heird, W.C.; Chan, L. Protection against Fatty Liver but Normal Adipogenesis in Mice Lacking Adipose Differentiation-Related Protein. Mol. Cell. Biol. 2006, 26, 1063–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManaman, J.L.; Bales, E.S.; Orlicky, D.J.; Jackman, M.; MacLean, P.S.; Cain, S.; Crunk, A.E.; Mansur, A.; Graham, C.E.; Bowman, T.A.; et al. Perilipin-2-Null Mice Are Protected against Diet-Induced Obesity, Adipose Inflammation, and Fatty Liver Disease. J. Lipid Res. 2013, 54, 1346–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, T.D.; Schaack, J.; Orlicky, D.J.; Palmer, C.; Chang, B.H.-J.; Chan, L.; McManaman, J.L. Adipophilin Regulates Maturation of Cytoplasmic Lipid Droplets and Alveolae in Differentiating Mammary Glands. J. Cell Sci. 2011, 124, 3247–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, T.D.; Palmer, C.A.; Orlicky, D.J.; Bales, E.S.; Chang, B.H.-J.; Chan, L.; McManaman, J.L. Mammary Glands of Adipophilin-Null Mice Produce an Amino-Terminally Truncated Form of Adipophilin that Mediates Milk Lipid Droplet Formation and Secretion. J. Lipid Res. 2008, 49, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; McMahan, R.H.; Monks, J.; Rosa, F.G.; McManaman, J.L. Perilipin-2 Promotes Obesity and Progressive Fatty Liver Disease in Mice through Mechanistically Distinct Hepatocyte and Extra-hepatocyte Actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Sohn, J.H.; Han, J.S.; Park, Y.J.; Jeon, Y.G.; Ji, Y.; Dalen, K.T.; Sztalryd, C.; Kimmel, A.R.; Kim, J.B. Perilipin 3 Deficiency Stimulates Thermogenic Beige Adipocytes through PPARα Activation. Diabetes 2018, 67, 791–804. [Google Scholar] [CrossRef]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Schoenfish, M.J.; Tzekov, A.; Bickel, P.E. S3-12, Adipophilin, and TIP47 Package Lipid in Adipocytes. J. Biol. Chem. 2005, 280, 19146–19155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, M.; Wang, H.; Chen, H.; McLenithan, J.C.; Gong, D.W.; Yang, R.Z.; Yu, D.; Fried, S.K.; Quon, M.J.; Londos, C.; et al. Consequences of Lipid Droplet Coat Protein Downregulation in Liver Cells: Abnormal Lipid Droplet Metabolism and Induction of Insulin Resistance. Diabetes 2008, 57, 2037–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, R.M.; Patel, R.T.; Rao, V.; Dhir, R.; Graham, M.J.; Crooke, R.M.; Ahima, R.S. Reduction of TIP47 Improves Hepatic Steatosis and Glucose Homeostasis in Mice. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R996–R1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C.; Nebb, H.I. LSDP5 Is a PAT Protein Specifically Expressed in Fatty Acid Oxidizing Tissues. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2007, 1771, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a Lipid Droplet-Binding Protein, Protects Heart from Oxidative Burden by Sequestering Fatty Acid from Excessive Oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Xie, Z.; Yuan, Y.; Sui, W.; Wang, C.; Gao, X.; Zhao, Y.; Zhang, F.; Gu, Y.; Hu, P.; et al. Plin5 Alleviates Myocardial Ischaemia/Reperfusion Injury by Reducing Oxidative Stress through Inhibiting the Lipolysis of Lipid Droplets. Sci. Rep. 2017, 7, 42574. [Google Scholar] [CrossRef] [Green Version]
- Andersson, L.; Drevinge, C.; Mardani, I.; Dalen, K.T.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Haugen, F.; Adiels, M.; Fogelstrand, P.; et al. Deficiency in Perilipin 5 Reduces Mitochondrial Function and Membrane Depolarization in Mouse Hearts. Int. J. Biochem. Cell Biol. 2017, 91, 9–13. [Google Scholar] [CrossRef]
- Drevinge, C.; Dalen, K.T.; Mannila, M.N.; Täng, M.S.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Mardani, I.; Haugen, F.; Fogelstrand, P.; et al. Perilipin 5 Is Protective in the Ischemic Heart. Int. J. Cardiol. 2016, 219, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sreenivasan, U.; Gong, D.-W.; O’Connell, K.A.; Dabkowski, E.R.; Hecker, P.A.; Ionica, N.; Konig, M.; Mahurkar, A.; Sun, Y.; et al. Cardiomyocyte-Specific Perilipin 5 Overexpression Leads to Myocardial Steatosis and Modest Cardiac Dysfunction. J. Lipid Res. 2013, 54, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective Lipolysis and Altered Energy Metabolism in Mice Lacking Adipose Triglyceride Lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef]
- Pollak, N.M.; Schweiger, M.; Jaeger, D.; Kolb, D.; Kumari, M.; Schreiber, R.; Kolleritsch, S.; Markolin, P.; Grabner, G.F.; Heier, C.; et al. Cardiac-Specific Overexpression of Perilipin 5 Provokes Severe Cardiac Steatosis via the Formation of a Lipolytic Barrier. J. Lipid Res. 2013, 54, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, N.M.; Jaeger, D.; Kolleritsch, S.; Zimmermann, R.; Zechner, R.; Lass, A.; Haemmerle, G. The Interplay of Protein Kinase A and Perilipin 5 Regulates Cardiac Lipolysis*. J. Biol. Chem. 2015, 290, 1295–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a Lipid Droplet-Associated Protein, Provides Physical and Metabolic Linkage to Mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, M.; Kimler, V.A.; Ghazi, F.R.; Rathore, G.K.; Perkins, G.A.; Ellisman, M.H.; Granneman, J.G. Adipocyte Lipolysis Affects Perilipin 5 and Cristae Organization at the Cardiac Lipid Droplet-Mitochondrial Interface. Sci. Rep. 2019, 9, 4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosma, M.; Minnaard, R.; Sparks, L.M.; Schaart, G.; Losen, M.; Baets, M.H.; Duimel, H.; Kersten, S.; Bickel, P.E.; Schrauwen, P.; et al. The Lipid Droplet Coat Protein Perilipin 5 Also Localizes to Muscle Mitochondria. Histochem. Cell Biol. 2012, 137, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.-M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 Fine-Tunes Lipid Oxidation to Metabolic Demand and Protects against Lipotoxicity in Skeletal Muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [Green Version]
- Kien, B.; Kolleritsch, S.; Kunowska, N.; Heier, C.; Chalhoub, G.; Tilp, A.; Wolinski, H.; Stelzl, U.; Haemmerle, G. Lipid Droplet-Mitochondria Coupling via Perilipin 5 Augments Respiratory Capacity but is Dispensable for FA Oxidation. J. Lipid Res. 2022, 63, 100172. [Google Scholar] [CrossRef]
- Radner, F.P.W.; Streith, I.E.; Schoiswohl, G.; Schweiger, M.; Kumari, M.; Eichmann, T.O.; Rechberger, G.; Koefeler, H.C.; Eder, S.; Schauer, S.; et al. Growth Retardation, Impaired Triacylglycerol Catabolism, Hepatic Steatosis, and Lethal Skin Barrier Defect in Mice Lacking Comparative Gene Identification-58 (CGI-58). J. Biol. Chem. 2010, 285, 7300–7311. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Betters, J.L.; Lord, C.; Ma, Y.; Han, X.; Yang, K.; Alger, H.M.; Melchior, J.; Sawyer, J.; Shah, R.; et al. CGI-58 Knockdown in Mice Causes Hepatic Steatosis but Prevents Diet-Induced Obesity and Glucose Intolerance. J. Lipid Res. 2010, 51, 3306–3315. [Google Scholar] [CrossRef] [Green Version]
- Cantley, J.L.; Yoshimura, T.; Camporez, J.P.G.; Zhang, D.; Jornayvaz, F.R.; Kumashiro, N.; Guebre-Egziabher, F.; Jurczak, M.J.; Kahn, M.; Guigni, B.A.; et al. CGI-58 Knockdown Sequesters Diacylglycerols in Lipid Droplets/ER-Preventing Diacylglycerol-Mediated Hepatic Insulin Resistance. Proc. Natl. Acad. Sci. USA. 2013, 110, 1869–1874. [Google Scholar] [CrossRef]
- Lord, C.C.; Betters, J.L.; Ivanova, P.T.; Milne, S.B.; Myers, D.S.; Madenspacher, J.; Thomas, G.; Chung, S.; Liu, M.; Davis, M.A.; et al. CGI-58/ABHD5-Derived Signaling Lipids Regulate Systemic Inflammation and Insulin Action. Diabetes 2012, 61, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviglia, J.M.; Sparks, J.D.; Toraskar, N.; Brinker, A.M.; Yin, T.C.; Dixon, J.L.; Brasaemle, D.L. ABHD5/CGI-58 Facilitates the Assembly and Secretion of Apolipoprotein B Lipoproteins by McA RH7777 Rat Hepatoma Cells. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2009, 1791, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Ma, Y.; Kadegowda, A.K.G.; Betters, J.L.; Xie, P.; Liu, G.; Liu, X.; Miao, H.; Ou, J.; Su, X.; et al. Deficiency of Liver Comparative Gene Identification-58 Causes Steatohepatitis and Fibrosis in Mice. J. Lipid Res. 2013, 54, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, D.; Schoiswohl, G.; Hofer, P.; Schreiber, R.; Schweiger, M.; Eichmann, T.O.; Pollak, N.M.; Poecher, N.; Grabner, G.F.; Zierler, K.A.; et al. Fasting-Induced G0/G1 Switch Gene 2 and FGF21 Expression in the Liver Are under Regulation of Adipose Tissue Derived Fatty Acids. J. Hepatol. 2015, 63, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Zierler, K.A.; Jaeger, D.; Pollak, N.M.; Eder, S.; Rechberger, G.N.; Radner, F.P.W.; Woelkart, G.; Kolb, D.; Schmidt, A.; Kumari, M.; et al. Functional Cardiac Lipolysis in Mice Critically Depends on Comparative Gene Identification-58. J. Biol. Chem. 2013, 288, 9892–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P.; Kadegowda, A.K.G.; Ma, Y.; Guo, F.; Han, X.; Wang, M.; Groban, L.; Xue, B.; Shi, H.; Li, H.; et al. Muscle-Specific Deletion of Comparative Gene Identification-58 (CGI-58) Causes Muscle Steatosis but Improves Insulin Sensitivity in Male Mice. Endocrinol 2015, 156, 1648–1658. [Google Scholar] [CrossRef]
- Badin, P.M.; Loubière, C.; Coonen, M.; Louche, K.; Tavernier, G.; Bourlier, V.; Mairal, A.; Rustan, A.C.; Smith, S.R.; Langin, D.; et al. Regulation of Skeletal Muscle Lipolysis and Oxidative Metabolism by the Co-Lipase CGI-58. J. Lipid Res. 2012, 53, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Jebessa, Z.H.; Shanmukha, K.D.; Dewenter, M.; Lehmann, L.H.; Xu, C.; Schreiter, F.; Siede, D.; Gong, X.M.; Worst, B.C.; Federico, G.; et al. The Lipid-Droplet-Associated Protein ABHD5 Protects the Heart through Proteolysis of HDAC4. Nat. Metab. 2019, 1, 1157–1167. [Google Scholar] [CrossRef]
- Xie, P.; Guo, F.; Ma, Y.; Zhu, H.; Wang, F.; Xue, B.; Shi, H.; Yang, J.; Yu, L. Intestinal Cgi-58 Deficiency Reduces Postprandial Lipid Absorption. PLoS ONE 2014, 9, e91652. [Google Scholar] [CrossRef] [Green Version]
- Korbelius, M.; Vujic, N.; Sachdev, V.; Obrowsky, S.; Rainer, S.; Gottschalk, B.; Graier, W.F.; Kratky, D. ATGL/CGI-58-Dependent Hydrolysis of a Lipid Storage Pool in Murine Enterocytes. Cell Rep. 2019, 28, 1923–1934.e4. [Google Scholar] [CrossRef]
- Goeritzer, M.; Schlager, S.; Radovic, B.; Madreiter, C.T.; Rainer, S.; Thomas, G.; Lord, C.C.; Sacks, J.; Brown, A.L.; Vujic, N.; et al. Deletion of CGI-58 or Adipose Triglyceride Lipase Differently Affects Macrophage Function and Atherosclerosis. J. Lipid Res. 2014, 55, 2562–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Radović, B.; Chandak, P.G.; Kolb, D.; Eisenberg, T.; Ring, J.; Fertschai, I.; Uellen, A.; Wolinski, H.; Kohlwein, S.-D.; et al. Triacylglycerol Accumulation Activates the Mitochondrial Apoptosis Pathway in Macrophages. J. Biol. Chem. 2011, 286, 7418–7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandak, P.G.; Radović, B.; Aflaki, E.; Kolb, D.; Buchebner, M.; Fröhlich, E.; Magnes, C.; Sinner, F.; Haemmerle, G.; Zechner, R.; et al. Efficient Phagocytosis Requires Triacylglycerol Hydrolysis by Adipose Triglyceride Lipase. J. Biol. Chem. 2010, 285, 20192–20201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Doddapattar, P.; Radović, B.; Povoden, S.; Kolb, D.; Vujić, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 Ceramide Is Crucial for Triacylglycerol-Induced Apoptosis in Macrophages. Cell Death Dis. 2012, 3, e280. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Balenga, N.A.B.; Luschnig-Schratl, P.; Wolinski, H.; Povoden, S.; Chandak, P.G.; Bogner-Strauss, J.G.; Eder, S.; Konya, V.; Kohlwein, S.-D.; et al. Impaired Rho GTPase Activation Abrogates Cell Polarization and Migration in Macrophages with Defective Lipolysis. Cell. Mol. Life Sci. 2011, 68, 3933–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, B.; Chandak, P.G.; Aflaki, E.; Van Puijvelde, G.H.M.; Radovic, B.; Hildebrand, R.B.; Meurs, I.; Out, R.; Kuiper, J.; Van Berkel, T.J.C.; et al. Macrophage Adipose Triglyceride Lipase Deficiency Attenuates Atherosclerotic Lesion Development in Low-Density Lipoprotein Receptor Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Ou, J.; Ma, Y.; Guo, F.; Yang, Z.; Wiggins, M.; Liu, C.; Song, W.; Han, X.; Wang, M.; et al. Macrophage CGI-58 Deficiency Activates Ros-Inflammasome Pathway to Promote Insulin Resistance in Mice. Cell Rep. 2014, 7, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Xie, P.; Zeng, X.; Xiao, J.; Sun, B.; Yang, D. Transgenic CGI-58 Expression in Macrophages Alleviates the Atherosclerotic Lesion Development in ApoE Knockout Mice. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 1683–1690. [Google Scholar] [CrossRef]
- Elias, P.M.; Williams, M.L.; Holleran, W.M.; Jiang, Y.J.; Schmuth, M. Thematic Review Series: Skin Lipids. Pathogenesis of Permeability Barrier Abnormalities in the Ichthyoses: Inherited Disorders of Lipid Metabolism. J. Lipid Res. 2008, 49, 697–714. [Google Scholar] [CrossRef] [Green Version]
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised Nomenclature and Classification of Inherited Ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef]
- Fischer, J.; Bourrat, E. Genetics of Inherited Ichthyoses and Related Diseases. Acta Derm. Venereol. 2020, 100, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Traupe, H.; Fischer, J.; Oji, V. Nonsyndromic Types of Ichthyoses-an Update. JDDG J. Der Dtsch. Dermatol. Ges. 2014, 12, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmuth, M.; Martinz, V.; Janecke, A.R.; Fauth, C.; Schossig, A.; Zschocke, J.; Gruber, R. Inherited Ichthyoses/Generalized Mendelian Disorders of Cornification. Eur. J. Hum. Genet. 2013, 21, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grond, S.; Radner, F.P.W.; Eichmann, T.O.; Kolb, D.; Grabner, G.F.; Wolinski, H.; Gruber, R.; Hofer, P.; Heier, C.; Schauer, S.; et al. Skin Barrier Development Depends on CGI-58 Protein Expression during Late-Stage Keratinocyte Differentiation. J. Investig. Dermatol. 2017, 137, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Epp, N.; Fürstenberger, G.; Müller, K.; de Juanes, S.; Leitges, M.; Hausser, I.; Thieme, F.; Liebisch, G.; Schmitz, G.; Krieg, P. 12R-Lipoxygenase Deficiency Disrupts Epidermal Barrier Function. J. Cell Biol. 2007, 177, 173–182. [Google Scholar] [CrossRef]
- Yanagi, T.; Akiyama, M.; Nishihara, H.; Sakai, K.; Nishie, W.; Tanaka, S.; Shimizu, H. Harlequin Ichthyosis Model Mouse Reveals Alveolar Collapse and Severe Fetal Skin Barrier Defects. Hum. Mol. Genet. 2008, 17, 3075–3083. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, N.; Yamamoto, M.; Imai, Y.; Sakaguchi, Y.; Takizawa, T.; Ohta, N.; Yagi, N.; Hatta, I.; Hitomi, K.; Takizawa, T.; et al. Knocking-in the R142C Mutation in Transglutaminase 1 Disrupts the Stratum Corneum Barrier and Postnatal Survival of Mice. J. Dermatol. Sci. 2012, 65, 196–206. [Google Scholar] [CrossRef]
- Jennemann, R.; Rabionet, M.; Gorgas, K.; Epstein, S.; Dalpke, A.; Rothermel, U.; Bayerle, A.; van der Hoeven, F.; Imgrund, S.; Kirsch, J.; et al. Loss of Ceramide Synthase 3 Causes Lethal Skin Barrier Disruption. Hum. Mol. Genet. 2012, 21, 586–608. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Ito, M.; Sanaki, T.; Okuda, T.; Tsuchiya, N.; Yoshimoto, R.; Yukioka, H. Acsl1 Is Essential for Skin Barrier Function through the Activation of Linoleic Acid and Biosynthesis of ω-O-Acylceramide in Mice. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2022, 1867, 159085. [Google Scholar] [CrossRef]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V. Lipopenia and Skin Barrier Abnormalities in DGAT2-Deficient Mice. J. Biol. Chem. 2004, 279, 11767–11776. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-Based Tight Junctions are Crucial for the Mammalian Epidermal Barrier. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sandhoff, R.; Kono, M.; Zerfas, P.; Hoffmann, V.; Ding, B.C.-H.; Proia, R.L.; Deng, C.-X. Depletion of Ceramides with Very Long Chain Fatty Acids Causes Defective Skin Permeability Barrier Function, and Neonatal Lethality in ELOVL4 Deficient Mice. Int. J. Biol. Sci. 2007, 3, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, Y.; Cho, Y.; Moradian, S.; Kim, J.; Nakajima, K.; Crumrine, D.; Park, K.; Ujihara, M.; Akiyama, M.; Shimizu, H.; et al. Neutral Lipid Storage Leads to Acylceramide Deficiency, Likely Contributing to the Pathogenesis of Dorfman–Chanarin Syndrome. J. Invest. Dermatol. 2010, 130, 2497–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, P.M.; Gruber, R.; Crumrine, D.; Menon, G.; Williams, M.L.; Wakefield, J.S.; Holleran, W.M.; Uchida, Y. Formation and Functions of the Corneocyte Lipid Envelope (CLE). Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Demerjian, M.; Crumrine, D.A.; Milstone, L.M.; Williams, M.L.; Elias, P.M. Barrier Dysfunction and Pathogenesis of Neutral Lipid Storage Disease with Ichthyosis (Chanarin–Dorfman Syndrome). J. Invest. Dermatol. 2006, 126, 2032–2038. [Google Scholar] [CrossRef] [Green Version]
- Grall, A.; Guaguère, E.; Planchais, S.; Grond, S.; Bourrat, E.; Hausser, I.; Hitte, C.; Le Gallo, M.; Derbois, C.; Kim, G.J.; et al. PNPLA1 Mutations Cause Autosomal Recessive Congenital Ichthyosis in Golden Retriever Dogs and Humans. Nat. Genet. 2012, 44, 140–147. [Google Scholar] [CrossRef]
- Zeng, F.; Qin, W.; Huang, F.; Chang, P. PNPLA1-Mediated Acylceramide Biosynthesis and Autosomal Recessive Congenital Ichthyosis. Metabolites 2022, 12, 685. [Google Scholar] [CrossRef]
- Chang, P.-A.; Sun, Y.-J.; Huang, F.-F.; Qin, W.-Z.; Chen, Y.-Y.; Zeng, X.; Wu, Y.-J. Identification of Human Patatin-like Phospholipase Domain-Containing Protein 1 and a Mutant in Human Cervical Cancer HeLa Cells. Mol. Biol. Rep. 2013, 40, 5597–5605. [Google Scholar] [CrossRef]
- Grond, S.; Eichmann, T.O.; Dubrac, S.; Kolb, D.; Schmuth, M.; Fischer, J.; Crumrine, D.; Elias, P.M.; Haemmerle, G.; Zechner, R.; et al. PNPLA1 Deficiency in Mice and Humans Leads to a Defect in the Synthesis of Omega-O-Acylceramides. J. Invest. Dermatol. 2017, 137, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.-A.; Han, L.-P.; Sun, L.-X.; Huang, F.-F. Identification Mouse Patatin-like Phospholipase Domain Containing Protein 1 as a Skin-Specific and Membrane-Associated Protein. Gene 2016, 591, 344–350. [Google Scholar] [CrossRef]
- Zimmer, A.D.; Kim, G.-J.; Hotz, A.; Bourrat, E.; Hausser, I.; Has, C.; Oji, V.; Stieler, K.; Vahlquist, A.; Kunde, V.; et al. Sixteen Novel Mutations in PNPLA1 in Patients with Autosomal Recessive Congenital Ichthyosis Reveal the Importance of an Extended Patatin Domain in PNPLA1 that is Essential for Proper Human Skin Barrier Function. Br. J. Dermatol. 2017, 177, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, T.; Anjo, T.; Kaneko, A.; Senoo, Y.; Shibata, A.; Takama, H.; Yokoyama, K.; Nishito, Y.; Ono, T.; Taya, C.; et al. PNPLA1 Has a Crucial Role in Skin Barrier Function by Directing Acylceramide Biosynthesis. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichery, M.; Huchenq, A.; Sandhoff, R.; Severino-Freire, M.; Zaafouri, S.; Opálka, L.; Levade, T.; Soldan, V.; Bertrand-Michel, J.; Lhuillier, E.; et al. PNPLA1 Defects in Patients with Autosomal Recessive Congenital Ichthyosis and KO Mice Sustain PNPLA1 Irreplaceable Function in Epidermal Omega-Oacylceramide Synthesis and Skin Permeability Barrier. Hum. Mol. Genet. 2017, 26, 1787–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Kamiyama, N.; Nakamichi, S.; Kihara, A. PNPLA1 Is a Transacylase Essential for the Generation of the Skin Barrier Lipid ω-O-Acylceramide. Nat. Commun. 2017, 8, 14610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, C.C.; Yang, H.; Soni, K.G.; Wang, S.P.; Mitchell, G.A.; Wu, J.W. An Epistatic Interaction between Pnpla2 and Lipe Reveals New Pathways of Adipose Tissue Lipolysis. Cells 2019, 8, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Nara, A.; Nakamichi, S.; Kihara, A. Molecular Mechanism of the Ichthyosis Pathology of Chanarin–Dorfman Syndrome: Stimulation of PNPLA1-Catalyzed ω-O-Acylceramide Production by ABHD5. J. Dermatol. Sci. 2018, 92, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerges, S.H.; Wahdan, S.A.; Elsherbiny, D.A.; El-Demerdash, E. Non-Alcoholic Fatty Liver Disease: An Overview of Risk Factors, Pathophysiological Mechanisms, Diagnostic Procedures, and Therapeutic Interventions. Life Sci. 2021, 271, 119220. [Google Scholar] [CrossRef]
- Lord, C.C.; Ferguson, D.; Thomas, G.; Brown, A.L.; Schugar, R.C.; Burrows, A.; Gromovsky, A.D.; Betters, J.; Neumann, C.; Sacks, J.; et al. Regulation of Hepatic Triacylglycerol Metabolism by CGI-58 does not Require ATGL Co-Activation. Cell Rep. 2016, 16, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin Deficiency in Mice does not Contribute to Fatty Liver Disease or Metabolic Syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 Has Retinyl-Palmitate Lipase Activity in Human Hepatic Stellate Cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and Characterization of a PNPLA3 Protein Isoform (I148M) Associated with Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin Functions as a Nutritionally Regulated Lysophosphatidic Acid Acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like Phospholipase Domain-Containing 3/Adiponutrin Deficiency in Mice is not Associated with Fatty Liver Disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Nischalke, H.D.; Berger, C.; Luda, C.; Berg, T.; Müller, T.; Grünhage, F.; Lammert, F.; Coenen, M.; Krämer, B.; Körner, C.; et al. The PNPLA3 Rs738409 148M/M Genotype is a Risk Factor for Liver Cancer in Alcoholic Cirrhosis but Shows no or Weak Association in Hepatitis C Cirrhosis. PLoS ONE 2011, 6, e27087. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speliotes, E.K.; Butler, J.L.; Palmer, C.D.; Voight, B.F.; Hirschhorn, J.N. PNPLA3 Variants Specifically Confer Increased Risk for Histologic Nonalcoholic Fatty Liver Disease but Not Metabolic Disease. Hepatology 2010, 52, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 Is Associated with Alcoholic Liver Disease. Nat. Genet. 2010, 42, 21–23. [Google Scholar] [CrossRef]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic Overexpression of PNPLA3I148M in Mouse Liver Causes Hepatic Steatosis. J. Clin. Invest. 2012, 122, 4130–4144. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A Sequence Variation (I148M) in PNPLA3 Associated with Nonalcoholic Fatty Liver Disease Disrupts Triglyceride Hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 Variant Associated with Fatty Liver Disease (I148M) Accumulates on Lipid Droplets by Evading Ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.H.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid Obesity Exposes the Association between PNPLA3 I148M (Rs738409) and Indices of Hepatic Injury in Individuals of European Descent. Int. J. Obes. 2010, 34, 190–194. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schratter, M.; Lass, A.; Radner, F.P.W. ABHD5—A Regulator of Lipid Metabolism Essential for Diverse Cellular Functions. Metabolites 2022, 12, 1015. https://doi.org/10.3390/metabo12111015
Schratter M, Lass A, Radner FPW. ABHD5—A Regulator of Lipid Metabolism Essential for Diverse Cellular Functions. Metabolites. 2022; 12(11):1015. https://doi.org/10.3390/metabo12111015
Chicago/Turabian StyleSchratter, Margarita, Achim Lass, and Franz P. W. Radner. 2022. "ABHD5—A Regulator of Lipid Metabolism Essential for Diverse Cellular Functions" Metabolites 12, no. 11: 1015. https://doi.org/10.3390/metabo12111015