
Statistical Integration of ‘Omics Data Increases Biological Knowledge Extracted from Metabolomics Data: Application to Intestinal Exposure to the Mycotoxin Deoxynivalenol

 ,
,
Abstract
:1. Introduction
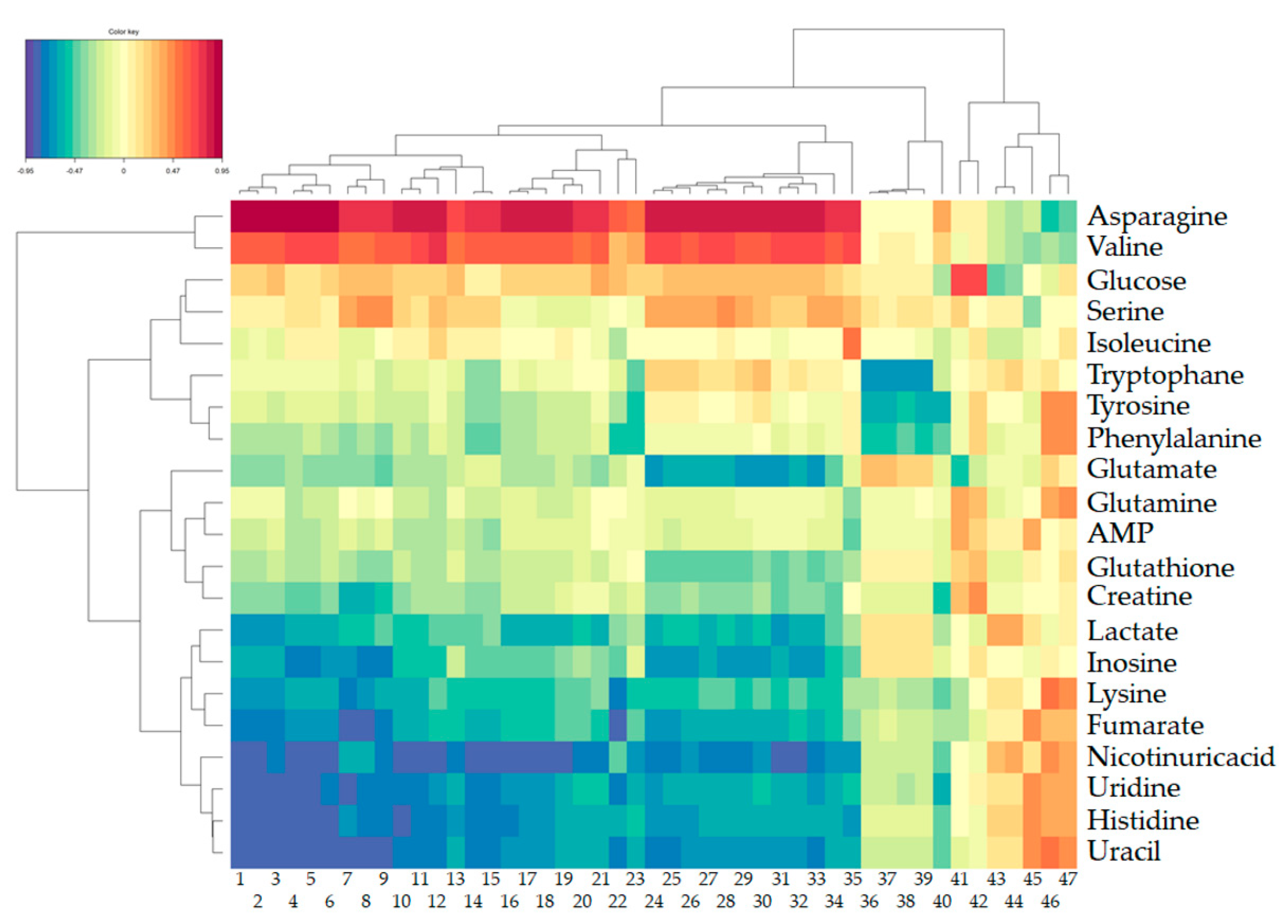
2. Results
2.1. Individual PLS-DA Modeling of ‘Omics Data
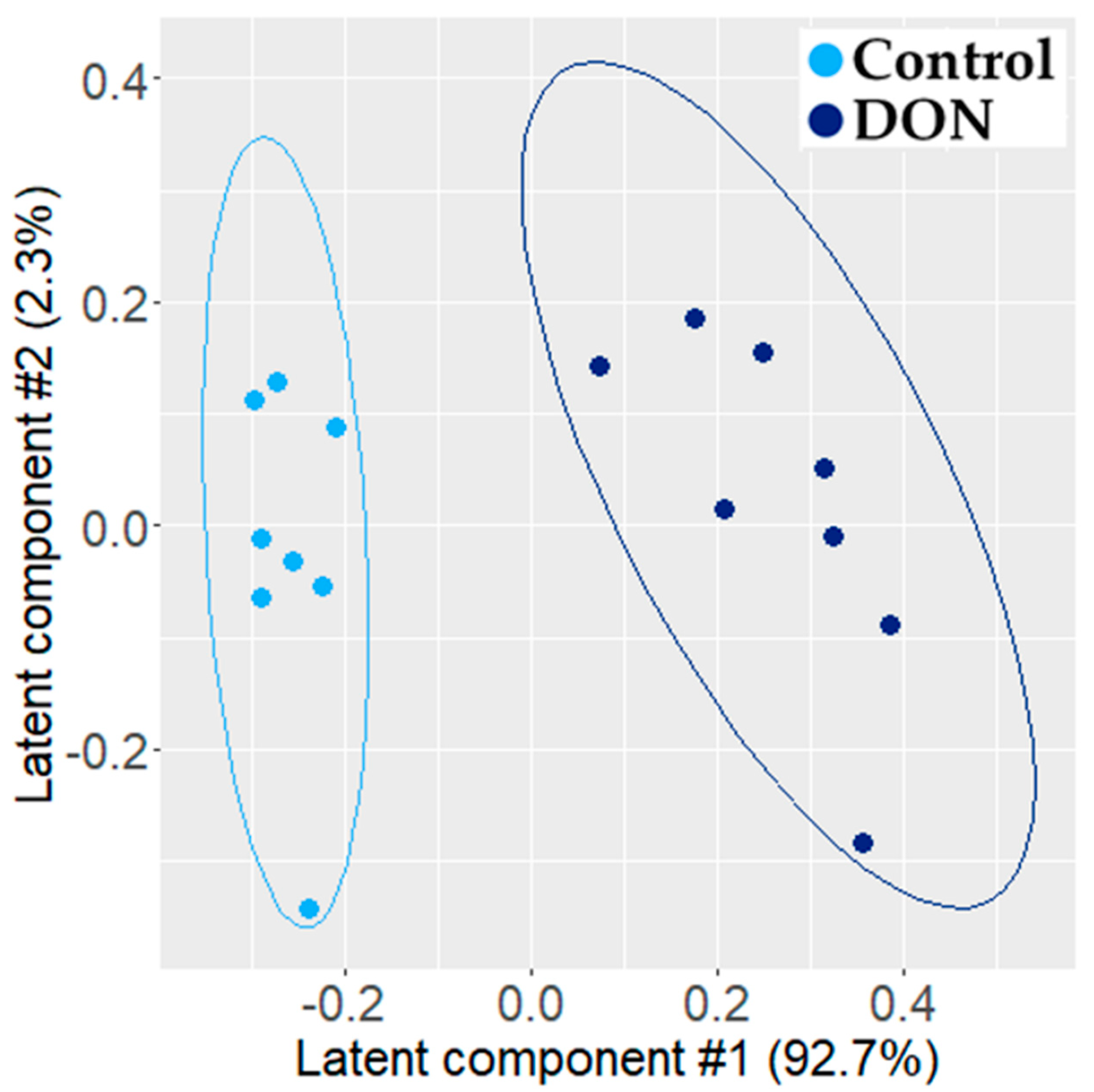
2.1.1. Metabolomic Effect of DON on Intestinal Explants
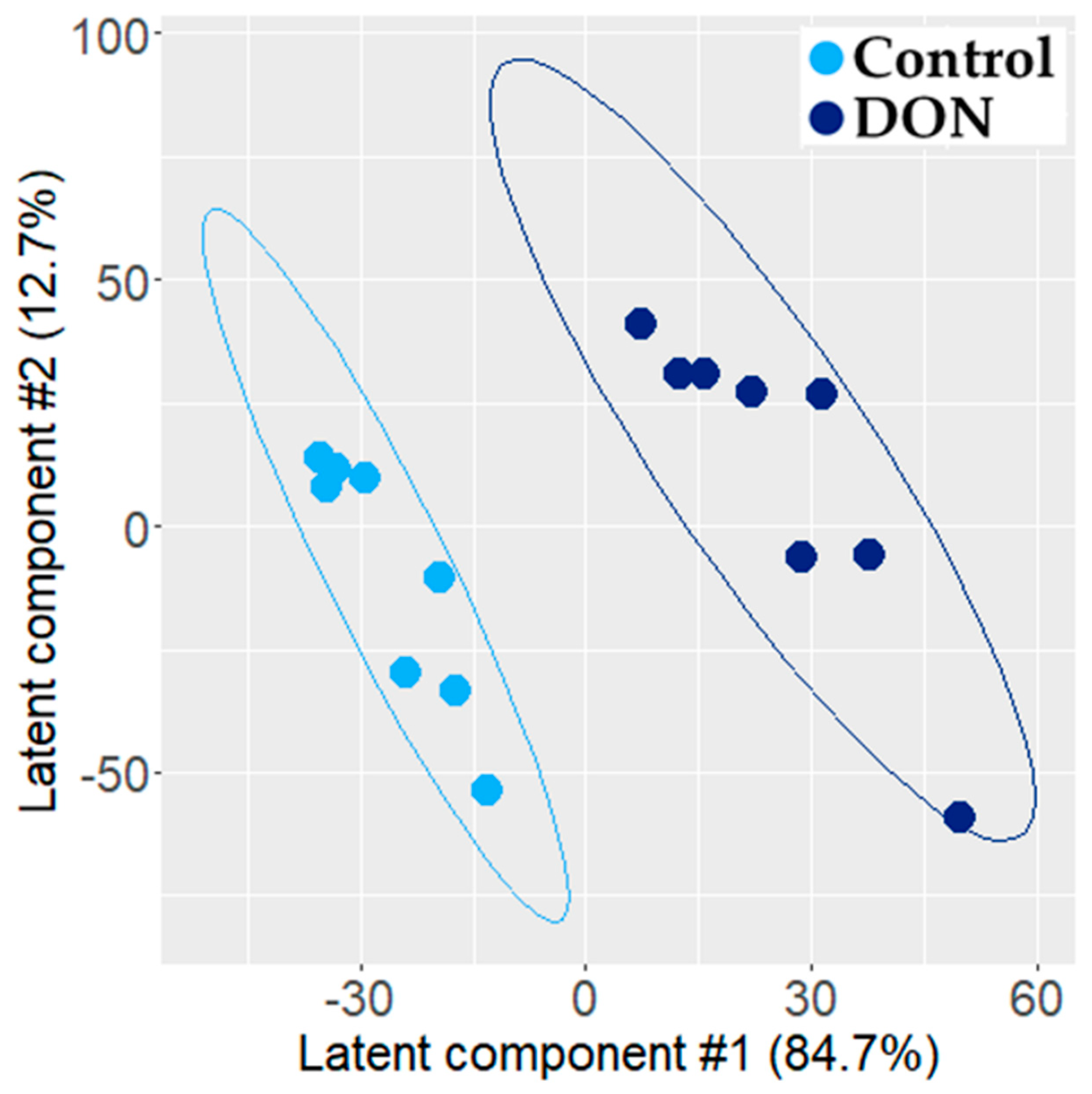
2.1.2. Transcriptomic Effect of DON on Intestinal Explant
2.2. Fusion of Transcriptomic and Metabolomic Data
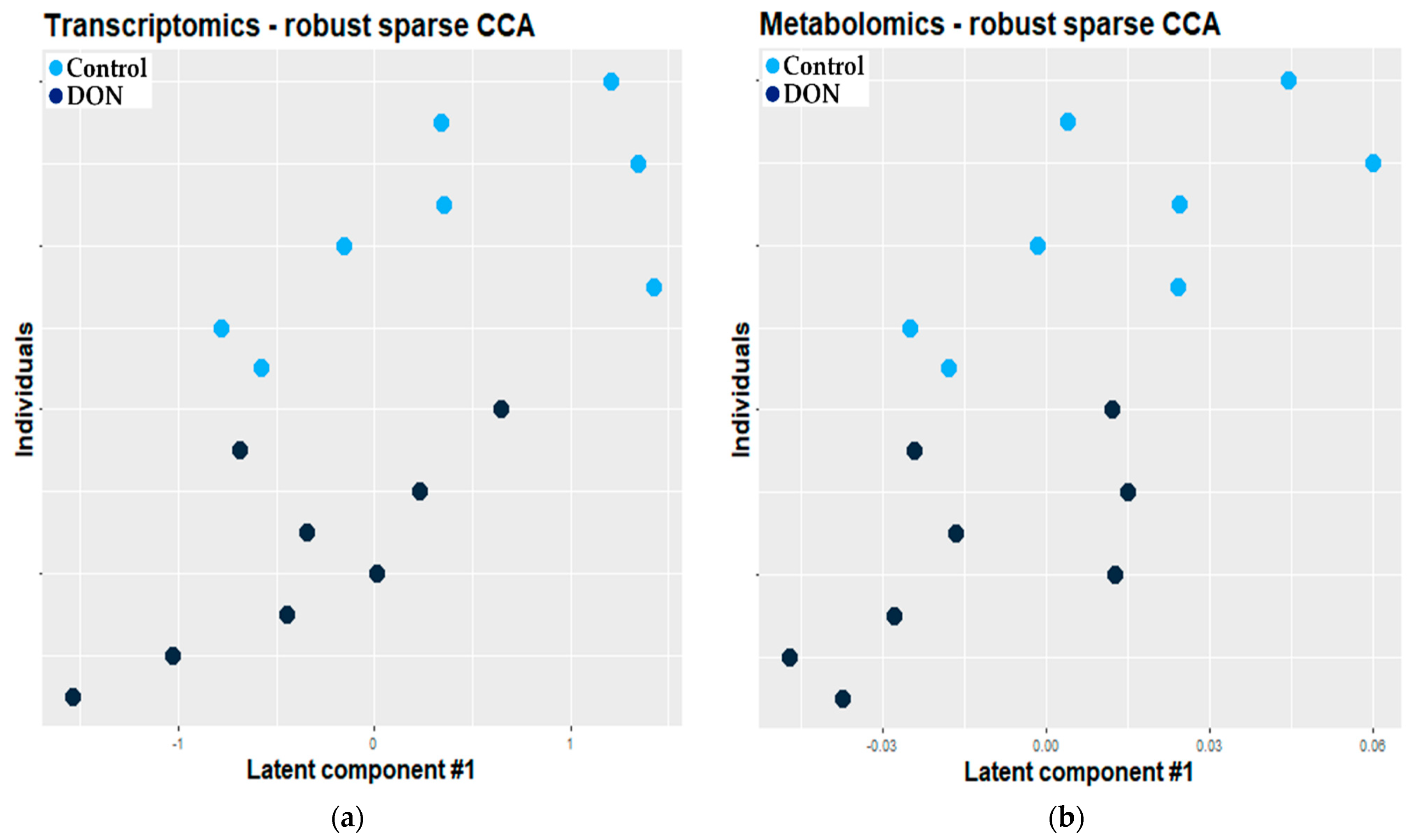
2.2.1. Combination of Robust Sparse CCA and O2-PLS
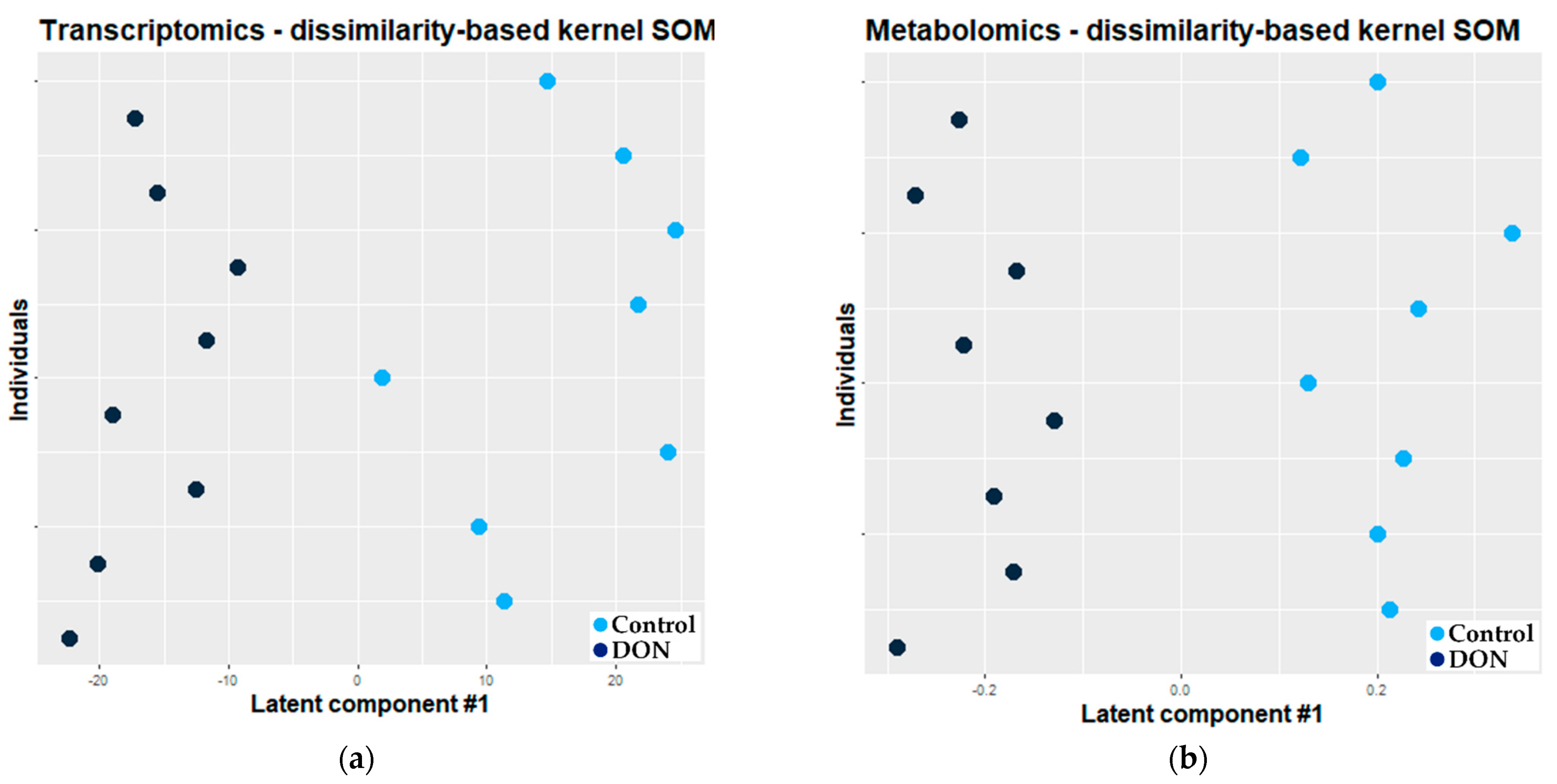
2.2.2. Combination of Self Organizing Map and O2-PLS
3. Discussion
3.1. Individual PLS-DA Modeling of ‘Omics Data
3.2. Fusion of Transcriptomic and Metabolomic Data
3.2.1. Combination of Robust Sparse CCA and O2-PLS
3.2.2. Combination of Kernel Dissimilarity-Based SOM and O2-PLS
4. Materials and Methods
4.1. Experimentation
4.1.1. Chemicals
4.1.2. Animals
4.1.3. Treatment of Jejunum Explants
4.2. ‘Omics Analysis
4.2.1. Transcriptomics
4.2.2. 1H-NMR-Based Metabolomics
4.3. Statistical Analysis
4.3.1. Sparse CCA Analyses
4.3.2. SOM Analyses
4.3.3. PLS-DA Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gruber-Dorninger, C.; Jenkins, T.; Schatzmayr, G. Global Mycotoxin Occurrence in Feed: A Ten-Year Survey. Toxins 2019, 11, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B.; Hogstrand, C.; et al. Risks to Human and Animal Health Related to the Presence of Deoxynivalenol and Its Acetylated and Modified Forms in Food and Feed. EFSA J. 2017, 15, e04718. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Oswald, I.P. Effect of Deoxynivalenol and Other Type B Trichothecenes on the Intestine: A Review. Toxins 2014, 6, 1615–1643. [Google Scholar] [CrossRef] [PubMed]
- Waché, Y.J.; Valat, C.; Postollec, G.; Bougeard, S.; Burel, C.; Oswald, I.P.; Fravalo, P. Impact of Deoxynivalenol on the Intestinal Microflora of Pigs. Int. J. Mol. Sci. 2009, 10, 1–17. [Google Scholar] [CrossRef]
- Pierron, A.; Mimoun, S.; Murate, L.S.; Loiseau, N.; Lippi, Y.; Bracarense, A.-P.F.L.; Schatzmayr, G.; He, J.W.; Zhou, T.; Moll, W.-D.; et al. Microbial Biotransformation of DON: Molecular Basis for Reduced Toxicity. Sci. Rep. 2016, 6, 29105. [Google Scholar] [CrossRef] [Green Version]
- Alassane-Kpembi, I.; Canlet, C.; Tremblay-Franco, M.; Jourdan, F.; Chalzaviel, M.; Pinton, P.; Cossalter, A.M.; Achard, C.; Castex, M.; Combes, S. 1H-NMR Metabolomics Response to a Realistic Diet Contamination with the Mycotoxin Deoxynivalenol: Effect of Probiotics Supplementation. Food Chem. Toxicol. 2020, 138, 111222. [Google Scholar] [CrossRef]
- O’Gorman, A.; Brennan, L. The Role of Metabolomics in Determination of New Dietary Biomarkers. Proc. Nutr. Soc. 2017, 76, 295–302. [Google Scholar] [CrossRef]
- Boccard, J.; Rudaz, S. Exploring Omics Data from Designed Experiments Using Analysis of Variance Multiblock Orthogonal Partial Least Squares. Anal. Chim. Acta 2016, 920, 18–28. [Google Scholar] [CrossRef]
- González-Ruiz, V.; Schvartz, D.; Sandström, J.; Pezzatti, J.; Jeanneret, F.; Tonoli, D.; Boccard, J.; Monnet-Tschudi, F.; Sanchez, J.-C.; Rudaz, S. An Integrative Multi-Omics Workflow to Address Multifactorial Toxicology Experiments. Metabolites 2019, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Kalkhof, S.; Dautel, F.; Loguercio, S.; Baumann, S.; Trump, S.; Jungnickel, H.; Otto, W.; Rudzok, S.; Potratz, S.; Luch, A.; et al. Pathway and Time-Resolved Benzo[a]Pyrene Toxicity on Hepa1c1c7 Cells at Toxic and Subtoxic Exposure. J. Proteome Res. 2015, 14, 164–182. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of Integrating Data to Uncover Genotype–Phenotype Interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Lê Cao, K.-A.; Martin, P.G.; Robert-Granié, C.; Besse, P. Sparse Canonical Methods for Biological Data Integration: Application to a Cross-Platform Study. BMC Bioinform. 2009, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Wilms, I.; Croux, C. Robust Sparse Canonical Correlation Analysis. BMC Syst. Biol. 2016, 10, 72. [Google Scholar] [CrossRef] [Green Version]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. Integration of Transcriptomics and Metabolomics for Understanding of Global Responses to Nutritional Stresses in Arabidopsis Thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H.; et al. Elucidation of Gene-to-Gene and Metabolite-to-Gene Networks in Arabidopsis by Integration of Metabolomics and Transcriptomics. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar] [CrossRef] [Green Version]
- Boccard, J.; Rutledge, D.N. A Consensus Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) Strategy for Multiblock Omics Data Fusion. Anal. Chim. Acta 2013, 769, 30–39. [Google Scholar] [CrossRef]
- El Bouhaddani, S.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.-W. Evaluation of O2PLS in Omics Data Integration. BMC Bioinform. 2016, 17, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandolini-Bunlon, M.; Pétéra, M.; Gaudreau, P.; Comte, B.; Bougeard, S.; Pujos-Guillot, E. Multi-Block PLS Discriminant Analysis for the Joint Analysis of Metabolomic and Epidemiological Data. Metabolomics 2019, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data Integration in Plant Biology: The O2PLS Method for Combined Modeling of Transcript and Metabolite Data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Bylesjö, M.; Nilsson, R.; Srivastava, V.; Grönlund, A.; Johansson, A.I.; Jansson, S.; Karlsson, J.; Moritz, T.; Wingsle, G.; Trygg, J. Integrated Analysis of Transcript, Protein and Metabolite Data To Study Lignin Biosynthesis in Hybrid Aspen. J. Proteome Res. 2009, 8, 199–210. [Google Scholar] [CrossRef]
- Hassani, S.; Martens, H.; Qannari, E.M.; Hanafi, M.; Kohler, A. Model Validation and Error Estimation in Multi-Block Partial Least Squares Regression. Chemom. Intell. Lab. Syst. 2012, 117, 42–53. [Google Scholar] [CrossRef]
- Mehl, F.; Marti, G.; Merle, P.; Delort, E.; Baroux, L.; Sommer, H.; Wolfender, J.-L.; Rudaz, S.; Boccard, J. Integrating Metabolomic Data from Multiple Analytical Platforms for a Comprehensive Characterisation of Lemon Essential Oils: Lemon Oil Characterisation by Multiblock Metabolomic Analysis. Flavour Fragr. J. 2015, 30, 131–138. [Google Scholar] [CrossRef]
- Moyon, T.; Le Marec, F.; Qannari, E.M.; Vigneau, E.; Le Plain, A.; Courant, F.; Antignac, J.-P.; Parnet, P.; Alexandre-Gouabau, M.-C. Statistical Strategies for Relating Metabolomics and Proteomics Data: A Real Case Study in Nutrition Research Area. Metabolomics 2012, 8, 1090–1101. [Google Scholar] [CrossRef]
- Alassane-Kpembi, I.; Gerez, J.R.; Cossalter, A.-M.; Neves, M.; Laffitte, J.; Naylies, C.; Lippi, Y.; Kolf-Clauw, M.; Bracarense, A.P.L.; Pinton, P.; et al. Intestinal Toxicity of the Type B Trichothecene Mycotoxin Fusarenon-X: Whole Transcriptome Profiling Reveals New Signaling Pathways. Sci. Rep. 2017, 7, 7530. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Dall’Asta, C. Forthcoming Challenges in Mycotoxins Toxicology Research for Safer Food—A Need for Multi-Omics Approach. Toxins 2017, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Pierron, A.; Mimoun, S.; Murate, L.S.; Loiseau, N.; Lippi, Y.; Bracarense, A.-P.F.L.; Liaubet, L.; Schatzmayr, G.; Berthiller, F.; Moll, W.-D.; et al. Intestinal Toxicity of the Masked Mycotoxin Deoxynivalenol-3-β-d-Glucoside. Arch. Toxicol. 2016, 90, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Xiao, H.; Ren, W.; Yin, J.; Hu, J.; Duan, J.; Liu, G.; Tan, B.; Xiong, X.; Oso, A.O.; et al. An NMR-Based Metabolomic Approach to Investigate the Effects of Supplementation with Glutamic Acid in Piglets Challenged with Deoxynivalenol. PLoS ONE 2014, 9, e113687. [Google Scholar] [CrossRef]
- Xiao, H.; Wu, M.M.; Shao, F.Y.; Tan, B.E.; Li, T.J.; Ren, W.K.; Yin, J.; Wang, J.; He, Q.H.; Yin, Y.L.; et al. Metabolic Profiles in the Response to Supplementation with Composite Antimicrobial Peptides in Piglets Challenged with Deoxynivalenol1. J. Anim. Sci. 2015, 93, 1114–1123. [Google Scholar] [CrossRef]
- Adeva-Andany, M.; López-Ojén, M.; Funcasta-Calderón, R.; Ameneiros-Rodríguez, E.; Donapetry-García, C.; Vila-Altesor, M.; Rodríguez-Seijas, J. Comprehensive Review on Lactate Metabolism in Human Health. Mitochondrion 2014, 17, 76–100. [Google Scholar] [CrossRef]
- Huang, N.-J.; Lin, Y.-C.; Lin, C.-Y.; Pishesha, N.; Lewis, C.A.; Freinkman, E.; Farquharson, C.; Millán, J.L.; Lodish, H. Enhanced Phosphocholine Metabolism Is Essential for Terminal Erythropoiesis. Blood 2018, 131, 2955–2966. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, E.A.; Carrié, A.L. Quantitative Aspects of Glucose and Glutamine Metabolism by Intestinal Cells. Gut 1994, 35, S13–S17. [Google Scholar] [CrossRef] [Green Version]
- Adesso, S.; Autore, G.; Quaroni, A.; Popolo, A.; Severino, L.; Marzocco, S. The Food Contaminants Nivalenol and Deoxynivalenol Induce Inflammation in Intestinal Epithelial Cells by Regulating Reactive Oxygen Species Release. Nutrients 2017, 9, 1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alassane-Kpembi, I.; Pinton, P.; Hupé, J.-F.; Neves, M.; Lippi, Y.; Combes, S.; Castex, M.; Oswald, I. Saccharomyces Cerevisiae Boulardii Reduces the Deoxynivalenol-Induced Alteration of the Intestinal Transcriptome. Toxins 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelaert, J.; Bendhaiba, L.; Olteanu, M.; Villa-Vialaneix, N. SOMbrero: An R Package for Numeric and Non-numeric Self-Organizing Maps. In Advances in Self-Organizing Maps and Learning Vector Quantization; Villmann, T., Schleif, F.-M., Kaden, M., Lange, M., Eds.; Advances in Intelligent Systems and Computing; Springer International Publishing: Cham, Switzerland, 2014; Volume 295, pp. 219–228. ISBN 978-3-319-07694-2. [Google Scholar]
- Mariette, J.; Villa-Vialaneix, N. Unsupervised Multiple Kernel Learning for Heterogeneous Data Integration. Bioinformatics 2017, 34, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payros, D.; Alassane-Kpembi, I.; Pierron, A.; Loiseau, N.; Pinton, P.; Oswald, I.P. Toxicology of Deoxynivalenol and Its Acetylated and Modified Forms. Arch. Toxicol. 2016, 90, 2931–2957. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. Encinitas Calif 2014, 13, 8–12. [Google Scholar]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wu, C.; Li, P.; Li, N.; Zhang, D.; Zhu, Q.; Ren, W.; Peng, Y. Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. BioMed Res. Int. 2018, 2018, 9171905. [Google Scholar] [CrossRef]
- Ravindran, R.; Loebbermann, J.; Nakaya, H.I.; Khan, N.; Ma, H.; Gama, L.; Machiah, D.K.; Lawson, B.; Hakimpour, P.; Wang, Y.-C.; et al. The Amino Acid Sensor GCN2 Controls Gut Inflammation by Inhibiting Inflammasome Activation. Nature 2016, 531, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Cano, P.M.; Seeboth, J.; Meurens, F.; Cognie, J.; Abrami, R.; Oswald, I.P.; Guzylack-Piriou, L. Deoxynivalenol as a New Factor in the Persistence of Intestinal Inflammatory Diseases: An Emerging Hypothesis through Possible Modulation of Th17-Mediated Response. PLoS ONE 2013, 8, e53647. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic Profiling, Metabolomic and Metabonomic Procedures for NMR Spectroscopy of Urine, Plasma, Serum and Tissue Extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Usal, M.; Veyrenc, S.; Darracq-Ghitalla-Ciock, M.; Regnault, C.; Sroda, S.; Fini, J.-B.; Canlet, C.; Tremblay-Franco, M.; Raveton, M.; Reynaud, S. Transgenerational Metabolic Disorders and Reproduction Defects Induced by Benzo[a]Pyrene in Xenopus Tropicalis. Environ. Pollut. 2021, 269, 116109. [Google Scholar] [CrossRef] [PubMed]
- Cabaton, N.J.; Poupin, N.; Canlet, C.; Tremblay-Franco, M.; Audebert, M.; Cravedi, J.-P.; Riu, A.; Jourdan, F.; Zalko, D. An Untargeted Metabolomics Approach to Investigate the Metabolic Modulations of HepG2 Cells Exposed to Low Doses of Bisphenol A and 17β-Estradiol. Front. Endocrinol. 2018, 9, 571. [Google Scholar] [CrossRef] [PubMed]
- Cottret, L.; Frainay, C.; Chazalviel, M.; Cabanettes, F.; Gloaguen, Y.; Camenen, E.; Merlet, B.; Heux, S.; Portais, J.-C.; Poupin, N.; et al. MetExplore: Collaborative Edition and Exploration of Metabolic Networks. Nucleic Acids Res. 2018, 46, W495–W502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotelling, H. Relations between Two Sets of Variates. Biometrika 1936, 28, 321–377. [Google Scholar] [CrossRef]
- Kohonen, T. Self-Organized Formation of Topologically Correct Feature Maps. Biol. Cybern. 1982, 43, 59–69. [Google Scholar] [CrossRef]
- Olteanu, M.; Villa-Vialaneix, N. Using SOMbrero for Clustering and Visualizing Graphs. J. Soc. Fr. Stat. 2015, 156, 25. [Google Scholar]
- Trygg, J. O2-PLS for Qualitative and Quantitative Analysis in Multivariate Calibration. J. Chemom. 2002, 16, 283–293. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Bylesjö, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS Discriminant Analysis: Combining the Strengths of PLS-DA and SIMCA Classification. J. Chemom. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- El Bouhaddani, S.; Uh, H.-W.; Jongbloed, G.; Hayward, C.; Klarić, L.; Kiełbasa, S.M.; Houwing-Duistermaat, J. Integrating Omics Datasets with the OmicsPLS Package. BMC Bioinform. 2018, 19, 371. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Name | Number of Mapped Metabolites | Coverage (%) | BH-Corrected p-Value |
|---|---|---|---|
| Aminoacyl-tRNA biosynthesis | 10 | 21.3 | 1.73 × 10−9 |
| Biosynthesis of amino acids | 8 | 12.7 | 9.48 × 10−6 |
| Alanine, aspartate and glutamate metabolism | 5 | 15.2 | 5.79 × 10−4 |
| Arginine biosynthesis | 3 | 21.4 | 5.18 × 10−3 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 2 | 50.0 | 6.48 × 10−3 |
| D-Glutamine and D-glutamate metabolism | 2 | 40.0 | 9.18 × 10−3 |
| Nitrogen metabolism | 2 | 33.3 | 0.01 |
| 2-Oxocarboxylic acid metabolism | 3 | 14.3 | 0.01 |
| Valine, leucine and isoleucine biosynthesis | 2 | 25.0 | 0.018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremblay-Franco, M.; Canlet, C.; Pinton, P.; Lippi, Y.; Gautier, R.; Naylies, C.; Neves, M.; Oswald, I.P.; Debrauwer, L.; Alassane-Kpembi, I. Statistical Integration of ‘Omics Data Increases Biological Knowledge Extracted from Metabolomics Data: Application to Intestinal Exposure to the Mycotoxin Deoxynivalenol. Metabolites 2021, 11, 407. https://doi.org/10.3390/metabo11060407
Tremblay-Franco M, Canlet C, Pinton P, Lippi Y, Gautier R, Naylies C, Neves M, Oswald IP, Debrauwer L, Alassane-Kpembi I. Statistical Integration of ‘Omics Data Increases Biological Knowledge Extracted from Metabolomics Data: Application to Intestinal Exposure to the Mycotoxin Deoxynivalenol. Metabolites. 2021; 11(6):407. https://doi.org/10.3390/metabo11060407
Chicago/Turabian StyleTremblay-Franco, Marie, Cécile Canlet, Philippe Pinton, Yannick Lippi, Roselyne Gautier, Claire Naylies, Manon Neves, Isabelle P. Oswald, Laurent Debrauwer, and Imourana Alassane-Kpembi. 2021. "Statistical Integration of ‘Omics Data Increases Biological Knowledge Extracted from Metabolomics Data: Application to Intestinal Exposure to the Mycotoxin Deoxynivalenol" Metabolites 11, no. 6: 407. https://doi.org/10.3390/metabo11060407