
Benchmarking Non-Targeted Metabolomics Using Yeast-Derived Libraries

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results
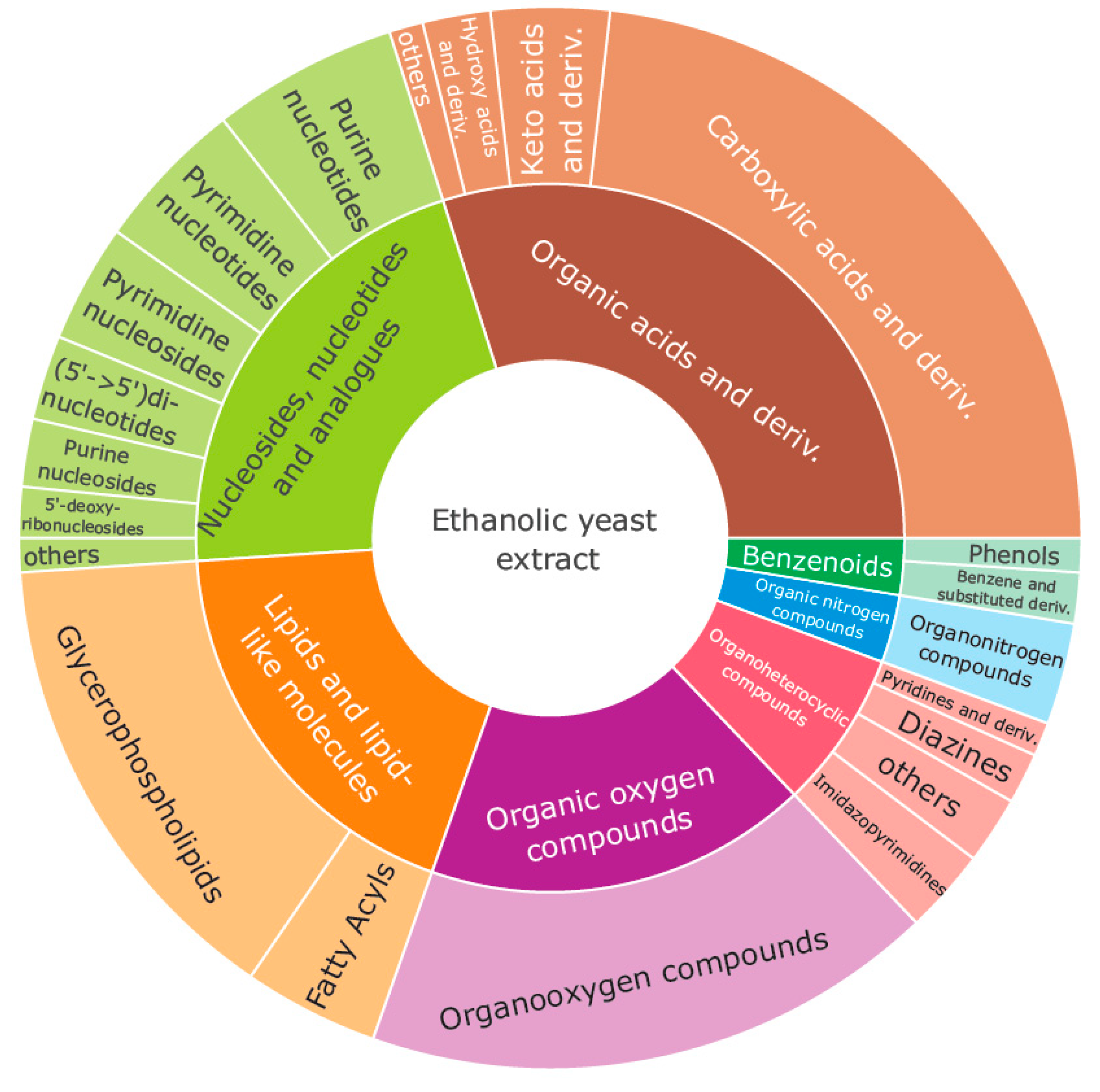
2.1. Metabolite Identification and Quality of the Proposed Benchmark Material
2.1.1. Metabolites and Lipids Present in the Yeast Material
2.1.2. Metabolite Stability and Yeast Fermentation Reproducibility
2.2. Application of the Benchmark Material for Non-Targeted Metabolomics
2.2.1. Yeast Quality Controls for Instrument Performance
2.2.2. Yeast Quality Controls Facilitating Method Development
3. Discussion
4. Materials and Methods
4.1. Standards and Solvents
4.2. Production of Ethanolic Yeast Extracts
4.3. LC–MS Analysis and Data Analysis of Yeast Extracts
Untargeted Metabolomics
4.4. Targeted Metabolomics of Interesting Metabolite Classes
4.4.1. Lipids
4.4.2. Carnitines and Coenzymes
4.4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M + H | RT | Est. Concentration (nM) | ||
|---|---|---|---|---|
| Coenzyme A | C21H36N7O16P3S | 768.1225 | 6.29 | 2792 |
| Acetyl coenzyme A | C23H38N7O17P3S | 810.1330 | 6.60 | 316 |
| Palmitoyl coenzyme A | C37H66N7O17P3S | 1006.3522 | 7.10 | <LOQ |
| Carnitine | C7H15NO3 | 162.1125 | 1.48 | 139 |
| O-acetyl-L-carnitine | C9H17NO4 | 204.1230 | 2.16 | 16 |
| Propionyl-L-carnitine | C10H19NO4 | 218.1387 | 3.79 | 3 |
References
- Baker, M. Metabolomics: From small molecules to big ideas. Nat. Methods 2011, 8, 117–121. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Singer, H.P.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S.; et al. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.R. Metabolomics—The new frontier in food safety and quality research. Food Res. Int. 2015, 72, 80–81. [Google Scholar] [CrossRef]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef] [PubMed]
- González-Riano, C.; Dudzik, D.; Garcia, A.; Gil-de-la-Fuente, A.; Gradillas, A.; Godzien, J.; López-Gonzálvez, Á.; Rey-Stolle, F.; Rojo, D.; Ruperez, F.J.; et al. Recent Developments along the Analytical Process for Metabolomics Workflows. Anal. Chem. 2020, 92, 203–226. [Google Scholar] [CrossRef]
- Metabolomics Society: Standardization in Metabolomics Experiments. Available online: http://metabolomicssociety.org/resources/metabolomics-standards (accessed on 14 July 2020).
- Sansone, S.-A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The metabolomics standards initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Sumner, L.W.; Samuel, T.; Noble, R.; Gmbh, S.D.; Barrett, D.; Beale, M.H.; Hardy, N. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Salek, R.M.; Steinbeck, C.; Viant, M.R.; Goodacre, R.; Dunn, W.B. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience 2013, 2, 1–3. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2019, 48, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.H.; Nellis, M.; Uppal, K.; Ma, C.; Tran, V.; Liang, Y.; Walker, D.I.; Jones, D.P. Reference Standardization for Quantification and Harmonization of Large-Scale Metabolomics. Anal. Chem. 2020, 92, 8836–8844. [Google Scholar] [CrossRef]
- Altelaar, A.F.M.F.M.; Frese, C.K.; Preisinger, C.; Hennrich, M.L.; Schram, A.W.; Timmers, H.T.M.T.M.; Heck, A.J.R.; Mohammed, S. Benchmarking stable isotope labeling based quantitative proteomics. J. Proteom. 2013, 88, 14–26. [Google Scholar] [CrossRef]
- Ahn, N.G.; Shabb, J.B.; Old, W.M.; Resing, K.A. Achieving in-depth proteomics profiling by mass spectrometry. ACS Chem. Biol. 2007, 2, 39–52. [Google Scholar] [CrossRef]
- Navarro, P.; Kuharev, J.; Gillet, L.C.; Bernhardt, O.M.; MacLean, B.; Röst, H.L.; Tate, S.A.; Tsou, C.C.; Reiter, L.; Distler, U.; et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016, 34, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelstrup, C.D.; Bekker-Jensen, D.B.; Arrey, T.N.; Hogrebe, A.; Harder, A.; Olsen, J.V. Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J. Proteome Res. 2018, 17, 727–738. [Google Scholar] [CrossRef]
- Köcher, T.; Pichler, P.; Swart, R.; Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanolc-ms/ms using ultralong gradients. Nat. Protoc. 2012, 7, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Ridgeway, M.E.; Bleiholder, C.; Mann, M.; Park, M.A. Trends in trapped ion mobility—Mass spectrometry instrumentation. TrAC Trends Anal. Chem. 2019, 116, 324–331. [Google Scholar] [CrossRef]
- Nielsen, J. Systems biology of lipid metabolism: From yeast to human. FEBS Lett. 2009, 583, 3905–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaiger, M.; Rampler, E.; Hermann, G.; Miklos, W.; Berger, W.; Koellensperger, G. Anion-Exchange Chromatography Coupled to High-Resolution Mass Spectrometry: A Powerful Tool for Merging Targeted and Non-targeted Metabolomics. Anal. Chem. 2017, 89, 7667–7674. [Google Scholar] [CrossRef]
- Rampler, E.; Coman, C.; Hermann, G.; Sickmann, A.; Ahrends, R.; Koellensperger, G. LILY-lipidome isotope labeling of yeast: In vivo synthesis of 13 C labeled reference lipids for quantification by mass spectrometry. Analyst 2017, 142, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, S.; Haberhauer-Troyer, C.; Klavins, K.; Russmayer, H.; Steiger, M.G.; Gasser, B.; Sauer, M.; Mattanovich, D.; Hann, S.; Koellensperger, G. U13C cell extract of Pichia pastoris—A powerful tool for evaluation of sample preparation in metabolomics. J. Sep. Sci. 2012, 35, 3091–3105. [Google Scholar] [CrossRef]
- Hermann, G.; Schwaiger, M.; Volejnik, P.; Koellensperger, G. 13C-labelled yeast as internal standard for LC–MS/MS and LC high resolution MS based amino acid quantification in human plasma. J. Pharm. Biomed. Anal. 2018, 155, 329–334. [Google Scholar] [CrossRef]
- Schwaiger, M.; Schoeny, H.; El Abiead, Y.; Hermann, G.; Rampler, E.; Koellensperger, G. Merging metabolomics and lipidomics into one analytical run. Analyst 2019, 144, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Rampler, E.; Criscuolo, A.; Zeller, M.; El Abiead, Y.; Schoeny, H.; Hermann, G.; Sokol, E.; Cook, K.; Peake, D.A.; Delanghe, B.; et al. A Novel Lipidomics Workflow for Improved Human Plasma Identification and Quantification Using RPLC-MSn Methods and Isotope Dilution Strategies. Anal. Chem. 2018, 90, 6494–6501. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Si-Hung, L.; Troyer, C.; Causon, T.; Hann, S. Sensitive quantitative analysis of phosphorylated primary metabolites using selective metal oxide enrichment and GC- and IC- MS/MS. Talanta 2019, 205, 120147. [Google Scholar] [CrossRef]
- Mairinger, T.; Sanderson, J.; Hann, S. GC–QTOFMS with a low-energy electron ionization source for advancing isotopologue analysis in 13C-based metabolic flux analysis. Anal. Bioanal. Chem. 2019, 411, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Demarest, T.G.; Truong, G.T.D.; Lovett, J.; Mohanty, J.G.; Mattison, J.A.; Mattson, M.P.; Ferrucci, L.; Bohr, V.A.; Moaddel, R. Assessment of NAD + metabolism in human cell cultures, erythrocytes, cerebrospinal fluid and primate skeletal muscle. Anal. Biochem. 2019, 572, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Si-Hung, L.; Causon, T.J.; Hann, S. Comparison of fully wettable RPLC stationary phases for LC-MS-based cellular metabolomics. Electrophoresis 2017, 38, 2287–2295. [Google Scholar] [CrossRef]
- Galvez, L.; Rusz, M.; Schwaiger-Haber, M.; El Abiead, Y.; Hermann, G.; Jungwirth, U.; Berger, W.; Keppler, B.K.; Jakupec, M.A.; Koellensperger, G. Preclinical studies on metal based anticancer drugs as enabled by integrated metallomics and metabolomics. Metallomics 2019, 11, 1716–1728. [Google Scholar] [CrossRef] [Green Version]
- Mairinger, T.; Weiner, M.; Hann, S.; Troyer, C. Selective and Accurate Quantification of N -Acetylglucosamine in Biotechnological Cell Samples via GC–MS/MS and GC–TOFMS. Anal. Chem. 2020, 92, 4875–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puleston, D.J.; Buck, M.D.; Klein Geltink, R.I.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab. 2019, 30, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Swain, A.; Bambouskova, M.; Kim, H.; Andhey, P.S.; Duncan, D.; Auclair, K.; Chubukov, V.; Simons, D.M.; Roddy, T.P.; Stewart, K.M.; et al. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat. Metab. 2020, 2, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.B.; Troyer, C.; Mairinger, T.; Ortmayr, K.; Neubauer, S.; Koellensperger, G.; Hann, S. Isotopologue analysis of sugar phosphates in yeast cell extracts by gas chromatography chemical ionization time-of-flight mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Mairinger, T.; Hann, S. Implementation of data-dependent isotopologue fragmentation in 13C-based metabolic flux analysis. Anal. Bioanal. Chem. 2017, 409, 3713–3718. [Google Scholar] [CrossRef] [Green Version]
- Schwaiger-Haber, M.; Hermann, G.; El Abiead, Y.; Rampler, E.; Wernisch, S.; Sas, K.; Pennathur, S.; Koellensperger, G. Proposing a validation scheme for 13C metabolite tracer studies in high-resolution mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 3103–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtzman, C.P. Biotechnological strains of Komagataella (Pichia) pastoris are Komagataella phaffii as determined from multigene sequence analysis. J. Ind. Microbiol. Biotechnol. 2009, 36, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Chu, D.B.; Marx, H.; Sauer, M.; Hann, S.; Koellensperger, G. LC-MS/MS-based analysis of coenzyme A and short-chain acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 2015, 407, 6681–6688. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent Topics in Mass Spectrometry-Based Metabolomics and Lipidomics—Standardization, Coverage, and Throughput. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Liebisch, G.; Vizcaíno, J.A.; Köfeler, H.; Trötzmüller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Feunang, Y.D.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Summers, S.A. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat. Rev. Endocrinol. 2017, 13, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H.H. A simple method for isolation and purification of total lipides from animal tissue. J. Biol. Chem 1952, 226, 497–509. [Google Scholar] [CrossRef]
- Grillitsch, K.; Tarazona, P.; Klug, L.; Wriessnegger, T.; Zellnig, G.; Leitner, E.; Feussner, I.; Daum, G. Isolation and characterization of the plasma membrane from the yeast Pichia pastoris. Biochim. Biophys. Acta 2014, 1838, 1889–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klug, L.; Tarazona, P.; Gruber, C.; Grillitsch, K.; Gasser, B.; Trötzmüller, M.; Köfeler, H.; Leitner, E.; Feussner, I.; Mattanovich, D.; et al. The lipidome and proteome of microsomes from the methylotrophic yeast Pichia pastoris. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.J.; Pratt, B.; Bose, N.; Dubois, L.G., St.; John-Williams, L.; Perrott, K.M.; Ky, K.; Kapahi, P.; Sharma, V.; Maccoss, M.J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19, 1447–1458. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Schoeny, H.; Mitic, B.M.; El Abiead, Y.; Schwaiger, M.; Koellensperger, G. Simultaneous non-polar and polar lipid analysis by on-line combination of HILIC, RP and high resolution MS. Analyst 2018, 143, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampler, E.; Schoeny, H.; Schwaiger-Haber, M.; Koellensperger, G. Novel LC-MS Workflows for Improved Lipid Identification and Quantification. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–11. ISBN 9780081005965. [Google Scholar]
- Wernisch, S.; Pennathur, S. Evaluation of coverage, retention patterns, and selectivity of seven liquid chromatographic methods for metabolomics. Anal. Bioanal. Chem. 2016, 408, 6079–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rampler, E.; Hermann, G.; Grabmann, G.; El Abiead, Y.; Schoeny, H.; Baumgartinger, C.; Köcher, T.; Koellensperger, G. Benchmarking Non-Targeted Metabolomics Using Yeast-Derived Libraries. Metabolites 2021, 11, 160. https://doi.org/10.3390/metabo11030160
Rampler E, Hermann G, Grabmann G, El Abiead Y, Schoeny H, Baumgartinger C, Köcher T, Koellensperger G. Benchmarking Non-Targeted Metabolomics Using Yeast-Derived Libraries. Metabolites. 2021; 11(3):160. https://doi.org/10.3390/metabo11030160
Chicago/Turabian StyleRampler, Evelyn, Gerrit Hermann, Gerlinde Grabmann, Yasin El Abiead, Harald Schoeny, Christoph Baumgartinger, Thomas Köcher, and Gunda Koellensperger. 2021. "Benchmarking Non-Targeted Metabolomics Using Yeast-Derived Libraries" Metabolites 11, no. 3: 160. https://doi.org/10.3390/metabo11030160