Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease


Abstract
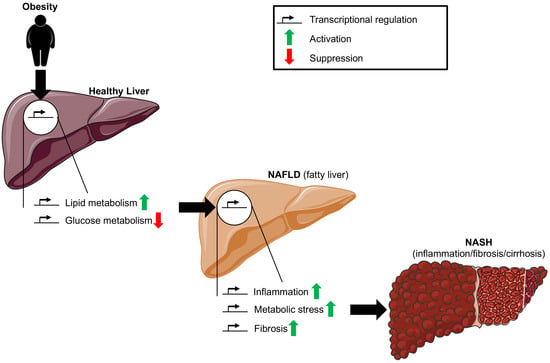
:
1. Introduction
2. Lipid Metabolism
2.1. PPARα
2.2. PPARγ
2.3. PPARδ
2.4. SREBP
2.5. CAR
2.6. LXR
2.7. CREBH
2.8. FXR
2.9. STAT5
2.10. C/EBPα
3. Glucose Metabolism
3.1. ChREBP
3.2. PGC1α
3.3. CREB
3.4. CREBH
3.5. FOXO
3.6. HNF4α
3.7. PPARδ
4. Inflammation
4.1. NF-κB
4.2. IRFs
4.3. STAT
4.4. AP1
4.5. AATF
4.6. SHP
4.7. Runx2
4.8. C/EBPβ
4.9. PPARα
4.10. PPARγ
4.11. CAR
4.12. LXR
5. Metabolic Stress
5.1. Xbp1
5.2. ATF4
5.3. ATF6
5.4. NRF2
5.5. CYP2E1
6. Fibrosis
6.1. TGFβ/SMAD axis
6.2. AEBP1
6.3. YAP
6.4. PPARα
6.5. PPARγ
6.6. RUNX2
6.7. c-Jun
7. Microbiome Dysbiosis
8. Prediction of Transcriptional Regulators by Database Analyses
8.1. Prognostic Biomarkers for Human NAFLD and NASH
8.2. Altered Transcription Factors in Mouse Models of NAFLD and NASH
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Machado, M.V.; Cortez-Pinto, H. Non-alcoholic fatty liver disease: What the clinician needs to know. World J. Gastroenterol. 2014, 20, 12956–12980. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Oseini, A.M.; Sanyal, A.J. Therapies in non-alcoholic steatohepatitis (NASH). Liver Int. 2017, 37, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Kakehashi, A.; Stefanov, V.E.; Ishii, N.; Okuno, T.; Fujii, H.; Kawai, K.; Kawada, N.; Wanibuchi, H. Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas. Int. J. Mol. Sci. 2017, 18, 434. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [Green Version]
- Shiri-Sverdlov, R.; Wouters, K.A.M.; Van Gorp, P.; Gijbels, M.J.; Noël, B.; Buffat, L.; Staels, B.; Maeda, N.; Van Bilsen, M.; Hofker, M.H. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 2006, 44, 732–741. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H.L. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef]
- Nakae, D.; Mizumoto, Y.; Andoh, N.; Tamura, K.; Horiguchi, K.; Endoh, T.; Kobayashi, E.; Tsujiuchi, T.; Denda, A.; Lombardi, B.; et al. Comparative Changes in the Liver of Female Fischer-344 Rats after Short-Term Feeding of a Semipurified or a Semisynthetic L-Amino Acid-Defined Choline-Deficient Diet. Toxicol. Pathol. 1995, 23, 583–590. [Google Scholar] [CrossRef]
- Mamikutty, N.; Thent, Z.; Suhaimi, F. Fructose-Drinking Water Induced Nonalcoholic Fatty Liver Disease and Ultrastructural Alteration of Hepatocyte Mitochondria in Male Wistar Rat. BioMed Res. Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.-I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Sinton, M.; Hay, D.C.; Drake, A.J. Metabolic control of gene transcription in non-alcoholic fatty liver disease: The role of the epigenome. Clin. Epigenet. 2019, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Ponugoti, B.; Kemper, J.K.; Fang, S. Functional Interaction of Hepatic Nuclear Factor-4 and Peroxisome Proliferator-Activated Receptor- Gamma Coactivator 1 alpha in CYP7A1 Regulation Is Inhibited by a Key Lipogenic Activator, Sterol Regulatory Element-Binding Protein-1c. Mol. Endocrinol. 2007, 21, 2698–2712. [Google Scholar] [CrossRef] [PubMed]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.B.; Jang, K.; Jun, D.W.; Lee, B.H.; Shin, K.J. Expression of Liver X Receptor Correlates with Intrahepatic Inflammation and Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2014, 59, 2975–2982. [Google Scholar] [CrossRef] [PubMed]
- Baciu, C.; Pasini, E.; Angeli, M.; Schwenger, K.; Afrin, J.; Humar, A.; Fischer, S.; Patel, K.; Allard, J.; Bhat, M. Systematic integrative analysis of gene expression identifies HNF4A as the central gene in pathogenesis of non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0189223. [Google Scholar] [CrossRef]
- Kaltenecker, D.; Themanns, M.; Mueller, K.M.; Spirk, K.; Suske, T.; Merkel, O.; Kenner, L.; Luís, A.; Kozlov, A.; Haybaeck, J.; et al. Hepatic growth hormone-JAK2-STAT5 signalling: Metabolic function, non-alcoholic fatty liver disease and hepatocellular carcinoma progression. Cytokine 2019, 124, 154569. [Google Scholar] [CrossRef]
- Rahman, S.M.; Janssen, R.C.; Jiang, H.; Qadri, I.; MacLean, K.N.; Friedman, J.E.; Schroeder-Gloeckler, J.M. CCAAT/enhancing binding protein β deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 2007, 45, 1108–1117. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 160–174. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Zappa, M.; Lattuada, E.; Roviaro, G.; Fargion, S. Increased Expression and Activity of the Transcription Factor FOXO1 in Nonalcoholic Steatohepatitis. Diabetes 2008, 57, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Videla, L.; Tapia, G.; Rodrigo, R.; Pettinelli, P.; Haim, D.; Santibáñez, C.; Araya, A.V.; Smok, G.; Csendes, A.; Gutiérrez, L.; et al. Liver NF-κB and AP-1 DNA Binding in Obese Patients. Obesity 2009, 17, 973–979. [Google Scholar] [CrossRef]
- Severa, M.; Islam, S.A.; Waggoner, S.N.; Jiang, Z.; Kim, N.D.; Ryan, G.; Kurt-Jones, E.; Charo, I.; Caffrey, D.R.; Boyartchuk, V.L.; et al. The transcriptional repressor BLIMP1 curbs host defenses by suppressing expression of the chemokine CCL8. J. Immunol. 2014, 192, 2291–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289.e20–1306.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, C.; Engelmann, J.C.; Saugspier, M.; Koch, A.; Hartmann, A.; Müller-Nurasyid, M.; Spang, R.; Bosserhoff, A.K.; Hellerbrand, C. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab. Investig. 2014, 94, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, A.; Magee, N.; Deng, F.; Lehn, S.; Zhong, C.; Zhang, Y. Hepatocyte nuclear receptor SHP suppresses inflammation and fibrosis in a mouse model of nonalcoholic steatohepatitis. J. Biol. Chem. 2018, 293, 8656–8671. [Google Scholar] [CrossRef] [Green Version]
- Shimozono, R.; Asaoka, Y.; Yoshizawa, Y.; Aoki, T.; Noda, H.; Yamada, M.; Kaino, M.; Mochizuki, H. Nrf2 Activators Attenuate the Progression of Nonalcoholic Steatohepatitis–Related Fibrosis in a Dietary Rat Model. Mol. Pharmacol. 2013, 84, 62–70. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, L.; Xue, Q.; Liu, C.; Xu, K.; Shen, W.; Deng, L. Cell-specific elevation of Runx2 promotes hepatic infiltration of macrophages by upregulating MCP-1 in high-fat diet-induced mice NAFLD. J. Cell. Biochem. 2019, 120, 11761–11774. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Rousseau, D.; Patouraux, S.; Bonnafous, S.; Adam, G.; Luciano, F.; Luci, C.; Anty, R.; Iannelli, A.; et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology 2018, 68, 515–532. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of Hepatic Lipogenesis by the Transcription Factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [Green Version]
- Cazanave, S.; Podtelezhnikov, A.; Jensen, K.; Seneshaw, M.; Kumar, D.P.; Min, H.-K.; Santhekadur, P.K.; Banini, B.; Mauro, A.G.; Oseini, A.M.; et al. The Transcriptomic Signature of Disease Development and Progression of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2017, 7, 17193. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; E Miquilena-Colina, M.; Vargas-Castrillón, J.; Iacono, O.L.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, S.; Hwang, S.; Cherrington, N.J.; Ryu, D.-Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 2017, 8, 63370–63381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Liver Physiol. 2014, 308, G298–G312. [Google Scholar] [CrossRef] [PubMed]
- Teratani, T.; Tomita, K.; Suzuki, T.; Furuhashi, H.; Irie, R.; Nishikawa, M.; Yamamoto, J.; Hibi, T.; Miura, S.; Minamino, T.; et al. Aortic carboxypeptidase-like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 128, 1581–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.P.; Santhekadur, P.K.; Seneshaw, M.; Mirshahi, F.; Tuculescu, C.U.; Sanyal, A.J. A Regulatory Role of Apoptosis Antagonizing Transcription Factor in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma. Hepatology 2019, 69, 1520–1534. [Google Scholar] [CrossRef]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2016, 152, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 1012–1019. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2☆. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Greenstein, A.W.; Majumdar, N.; Yang, P.; Subbaiah, P.V.; Kineman, R.D.; Cordoba-Chacon, J. Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2016, 232, 107–121. [Google Scholar] [CrossRef]
- Medina-Gómez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Curtis, R.K.; Jimenez-Liñan, M.; Blount, M.; Yeo, G.S.H.; et al. PPAR gamma 2 Prevents Lipotoxicity by Controlling Adipose Tissue Expandability and Peripheral Lipid Metabolism. PLoS Genet. 2007, 3, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Haluzík, M.; Lambert, G.; Yim, S.-H.; Gavrilova, O.; Ward, J.M.; Brewer, B.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Pettinelli, P.; Videla, L. Up-Regulation of PPAR-γ mRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wang, P.; Luo, J.; Wang, Z.; Song, Y.; Ye, J.; Hou, X. Adipogenic changes of hepatocytes in a high-fat diet-induced fatty liver mice model and non-alcoholic fatty liver disease patients. Endocrine 2014, 48, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2009, 51, 445–453. [Google Scholar] [CrossRef]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann–Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for Nonalcoholic Steatohepatitis: One-Year Results of the Randomized Placebo-Controlled Fatty Liver Improvement With Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus. Ann. Intern. Med. 2016, 165, 305. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Tudor, C.; Engelborghs, Y.; Wahli, W.; Desvergne, B. Fluorescence Imaging Reveals the Nuclear Behavior of Peroxisome Proliferator-activated Receptor/Retinoid X Receptor Heterodimers in the Absence and Presence of Ligand. J. Biol. Chem. 2005, 280, 17880–17890. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [Green Version]
- Bays, H.E.; Schwartz, S.; Littlejohn, T.; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.-J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, A Novel Peroxisome Proliferator Receptor-? Agonist: Lipid and Other Metabolic Effects in Dyslipidemic Overweight Patients Treated with and without Atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of Peroxisome Proliferator-Activated Receptor (PPAR) Promotes Reversal of Multiple Metabolic Abnormalities, Reduces Oxidative Stress, and Increases Fatty Acid Oxidation in Moderately Obese Men. Diabetes 2007, 57, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, H. SREBPs: Physiology and pathophysiology of the SREBP family. FEBS J. 2008, 276, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Fatehi-Hassanabad, Z.; Chan, C.B. Transcriptional regulation of lipid metabolism by fatty acids: A key determinant of pancreatic β-cell function. Nutr. Metab. 2005, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.-Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Claudel, T.; Trauner, M. PNPLA3 expression and its impact on the liver: Current perspectives. Hepatic Med. 2017, 9, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, F.; Fernandez, A.; De Lacy, A.M.; Fernándezcheca, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Hamadeh, H.K.; Webb, H.K.; Yamamoto, Y.; Sueyoshi, T.; Afshari, C.A.; Lehmann, J.M.; Negishi, M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol. Pharmacol. 2002, 61, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, J.; Zhai, Y.; Wada, T.; Xie, W. The Constitutive Androstane Receptor Is an Anti-obesity Nuclear Receptor That Improves Insulin Sensitivity*. J. Biol. Chem. 2009, 284, 25984–25992. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Saha, P.K.; Huang, W.; Chen, W.; Abu-Elheiga, L.; Wakil, S.J.; Stevens, R.D.; Ilkayeva, O.; Newgard, C.B.; Chan, L.; et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. USA 2009, 106, 18831–18836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.B.; Oza, N.A.; Anand, I.S.; Deshpande, S.S.; Patel, C.N. Liver X Receptor: A Novel Therapeutic Target. Indian J. Pharm. Sci. 2008, 70, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-G.; Xu, X.; Cho, S.; Hur, K.Y.; Lee, M.-S.; Kersten, S.; Lee, A.-H. CREBH-FGF21 axis improves hepatic steatosis by suppressing adipose tissue lipolysis. Sci. Rep. 2016, 6, 27938. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Shimano, H. CREBH Regulates Systemic Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 1396. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-W.; Chanda, D.; Yang, J.; Oh, H.; Kim, S.S.; Yoon, Y.-S.; Hong, S.; Park, K.; Lee, I.-K.; Choi, C.S.; et al. Regulation of Hepatic Gluconeogenesis by an ER-Bound Transcription Factor, CREBH. Cell Metab. 2010, 11, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Danno, H.; Ishii, K.-A.; Nakagawa, Y.; Mikami, M.; Yamamoto, T.; Yabe, S.; Furusawa, M.; Kumadaki, S.; Watanabe, K.; Shimizu, H.; et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARα. Biochem. Biophys. Res. Commun. 2010, 391, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Park, J.-G.; So, J.-S.; Hur, K.Y.; Lee, A.-H. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J. Lipid Res. 2014, 55, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Xu, X.; Simon, T.; Boudyguina, E.; Deng, Z.; Verhague, M.; Lee, A.-H.; Shelness, G.S.; Weinberg, R.B.; Parks, J.S. Very Low Density Lipoprotein Assembly Is Required for cAMP-responsive Element-binding Protein H Processing and Hepatic Apolipoprotein A-IV Expression. J. Biol. Chem. 2016, 291, 23793–23803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Giannikopoulos, P.; Duncan, S.A.; Wang, J.; Johansen, C.T.; Brown, J.D.; Plutzky, J.; Hegele, R.A.; Glimcher, L.H.; Lee, A.-H. The transcription factor cyclic AMP–responsive element–binding protein H regulates triglyceride metabolism. Nat. Med. 2011, 17, 812–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Han, S.-I.; Araki, M.; Nakagawa, Y.; Ohno, H.; Mizunoe, Y.; Kumagai, K.; Murayama, Y.; Osaki, Y.; Iwasaki, H.; et al. CREBH Improves Diet-Induced Obesity, Insulin Resistance, and Metabolic Disturbances by FGF21-Dependent and FGF21-Independent Mechanisms. iScience 2020, 23, 100930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kast-Woelbern, H.R.; Edwards, P.A. Natural Structural Variants of the Nuclear Receptor Farnesoid X Receptor Affect Transcriptional Activation. J. Biol. Chem. 2002, 278, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Li, H. Role of farnesoid X receptor in hepatic steatosis in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2020, 121, 109609. [Google Scholar] [CrossRef]
- Kalaany, N.Y.; Mangelsdorf, D.J. LXRS AND FXR: The Yin and Yang of Cholesterol and Fat Metabolism. Annu. Rev. Physiol. 2006, 68, 159–191. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X.-Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2014, 36, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, J.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.E.; Waters, M.J. GH-Dependent STAT5 Signaling Plays an Important Role in Hepatic Lipid Metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.; Nam, Y.S.; Piao, M.; Kang, H.J.; Park, S.J.; Lee, J.-H. Liver-specific deletion of the signal transducer and activator of transcription 5 gene aggravates fatty liver in response to a high-fat diet in mice. J. Nutr. Biochem. 2016, 29, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-Y.; Chang, W.-C.; Wang, J.-M. Biological roles of CCAAT/Enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015, 22, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Gavrilova, O.; Lambert, G.; Brewer, H.B.; Ward, J.M.; Inoue, Y.; Leroith, D.; Gonzalez, F.J. Hepatic CCAAT/Enhancer Binding Protein α Mediates Induction of Lipogenesis and Regulation of Glucose Homeostasis in Leptin-Deficient Mice. Mol. Endocrinol. 2004, 18, 2751–2764. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.M.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1951–1956. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Krawczyk, S.A.; Doridot, L.; Fowler, A.J.; Wang, J.X.; Trauger, S.A.; Noh, H.-L.; Kang, H.J.; Meissen, J.K.; Blatnik, M.; et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Investig. 2016, 126, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; So, J.-S.; Park, J.-G.; Lee, A.-H. Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; St-Pierre, J. PGC1 and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erion, D.M.; Ignatova, I.D.; Yonemitsu, S.; Nagai, Y.; Chatterjee, P.; Weismann, D.; Hsiao, J.J.; Zhang, D.; Iwasaki, T.; Stark, R.; et al. Prevention of Hepatic Steatosis and Hepatic Insulin Resistance by Knockdown of cAMP Response Element-Binding Protein. Cell Metab. 2009, 10, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.C. FOXO transcription factors in non-alcoholic fatty liver disease☆. Liver Res. 2017, 1, 168–173. [Google Scholar] [CrossRef]
- Sparks, J.; Dong, H.H. FoxO1 and hepatic lipid metabolism. Curr. Opin. Lipidol. 2009, 20, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Lu, H. Crosstalk of HNF4α with extracellular and intracellular signaling pathways in the regulation of hepatic metabolism of drugs and lipids. Acta Pharm. Sin. B 2016, 6, 393–408. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lou, G.; Li, C.; Wang, X.; Cederbaum, A.I.; Gan, L.; Xie, B. HBx Inhibits CYP2E1 Gene Expression via Downregulating HNF4α in Human Hepatoma Cells. PLoS ONE 2014, 9, e107913. [Google Scholar] [CrossRef]
- Yu, D.; Chen, G.; Pan, M.; Zhang, J.; He, W.; Liu, Y.; Nian, X.; Sheng, L.; Xu, B. High fat diet-induced oxidative stress blocks hepatocyte nuclear factor 4α and leads to hepatic steatosis in mice. J. Cell. Physiol. 2018, 233, 4770–4782. [Google Scholar] [CrossRef]
- Ravnskjaer, K.; Frigerio, F.; Boergesen, M.; Nielsen, T.; Maechler, P.; Mandrup, S. PPARδ is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J. Lipid Res. 2009, 51, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, J.; Barg, S.; Vallois, D.; Lahiri, S.; Roger, C.; Yessoufou, A.; Pradevand, S.; McDonald, A.; Bonal, C.; Reimann, F.; et al. PPARβ/δ affects pancreatic β cell mass and insulin secretion in mice. J. Clin. Investig. 2012, 122, 4105–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.-W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smale, S.T.; Natoli, G. Transcriptional Control of Inflammatory Responses. Cold Spring Harb. Perspect. Biol. 2014, 6, a016261. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Pena, A.D.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-κB Activation, Rather Than TNF, Mediates Hepatic Inflammation in a Murine Dietary Model of Steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Cortez-Pinto, H.; Solá, S.; Castro, R.E.; Ramalho, R.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M.P.; Baptista, A. Hepatocyte Apoptosis, Expression of Death Receptors, and Activation of NF-κB in the Liver of Nonalcoholic and Alcoholic Steatohepatitis Patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef]
- Wang, X.-A.; Zhang, R.; Zhang, S.; Deng, S.; Jiang, D.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S.; et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am. J. Physiol. Metab. 2013, 305, E485–E495. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Upadhyay, P.K.; Majumdar, S.S.; Nagarajan, P. Animal Models Correlating Immune Cells for the Development of NAFLD/NASH. J. Clin. Exp. Hepatol. 2015, 5, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.M.S.; Castro, R.E.; Machado, M.V.; Evangelista, T.; Silvestre, A.; Costa, A.; Coutinho, J.; Carepa, F.; Cortez-Pinto, H.; Rodrigues, C.M.P. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetology 2011, 54, 1788–1798. [Google Scholar] [CrossRef]
- Schreck, I.; Al-Rawi, M.; Mingot, J.-M.; Scholl, C.; Diefenbacher, M.E.; O’Donnell, P.; Bohmann, D.; Weiss, C. c-Jun localizes to the nucleus independent of its phosphorylation by and interaction with JNK and vice versa promotes nuclear accumulation of JNK. Biochem. Biophys. Res. Commun. 2011, 407, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Bruno, T.; Iezzi, S.; De Nicola, F.; Di Padova, M.; DeSantis, A.; Scarsella, M.; Di Certo, M.G.; Leonetti, C.; Floridi, A.; Passananti, C.; et al. Che-1 activates XIAP expression in response to DNA damage. Cell Death Differ. 2007, 15, 515–520. [Google Scholar] [CrossRef]
- DeSantis, A.; Bruno, T.; Catena, V.; De Nicola, F.; Goeman, F.; Iezzi, S.; Sorino, C.; Ponzoni, M.; Bossi, G.; Federico, V.; et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J. 2015, 34, 1214–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, A.; Bruno, T.; Catena, V.; De Nicola, F.; Goeman, F.; Iezzi, S.; Sorino, C.; Gentileschi, M.P.; Germoni, S.; Monteleone, V.; et al. Che-1 modulates the decision between cell cycle arrest and apoptosis by its binding to p53. Cell Death Dis. 2015, 6, e1764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuk, J.-M.; Shin, N.-M.; Lee, H.-M.; Kim, J.-J.; Kim, S.-W.; Jin, H.S.; Yang, C.-S.; Park, K.A.; Chanda, D.; Kim, N.-K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef]
- Yang, C.-S.; Kim, J.-J.; Kim, T.S.; Jo, E.-K.; Kim, S.Y.; Lee, H.-M.; Shin, N.-M.; Nguyen, L.T.; Lee, M.-S.; Jin, H.S.; et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, V. The Role of C/EBP Isoforms in the Control of Inflammatory and Native Immunity Functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [Green Version]
- Berghe, W.V.; Vermeulen, L.; Delerive, P.; De Bosscher, K.; Staels, B.; Haegeman, G. A Paradigm for Gene Regulation: Inflammation, NF-κB and PPAR. Adv. Exp. Med. Biol. 2003, 544, 181–196. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Muller, M. Peroxisome Proliferator-Activated Receptor α Protects against Obesity-Induced Hepatic Inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Morán-Salvador, E.; Titos, E.; Rius, B.; González-Périz, A.; García-Alonso, V.; López-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Claria, J. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskin-Bey, E.S.; Anan, A.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Constitutive androstane receptor agonist, TCPOBOP, attenuates steatohepatitis in the methionine choline-deficient diet-fed mouse. World J. Gastroenterol. 2007, 13, 5635–5641. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kakizaki, S.; Horiguchi, N.; Sohara, N.; Sato, K.; Takagi, H.; Mori, M.; Negishi, M. The role of the nuclear receptor constitutive androstane receptor in the pathogenesis of non-alcoholic steatohepatitis. Gut 2006, 56, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venteclef, N.; Jakobsson, T.; Ehrlund, A.; Damdimopoulos, A.; Mikkonen, L.; Ellis, E.; Nilsson, L.-M.; Parini, P.; Jänne, O.A.; Gustafsson, J.-Å.; et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRβ in the hepatic acute phase response. Genes Dev. 2010, 24, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Griffett, K.; Solt, L.A.; Elgendy, B.; Kamenecka, T.M.; Burris, T.P. A Liver-Selective LXR Inverse Agonist That Suppresses Hepatic Steatosis. ACS Chem. Biol. 2012, 8, 559–567. [Google Scholar] [CrossRef]
- Griffett, K.; Welch, R.D.; Flaveny, C.A.; Kolar, G.R.; Neuschwander-Tetri, B.A.; Burris, T.P. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol. Metab. 2015, 4, 353–357. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.-S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gökalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrema, H.; Zhou, Y.; Zhang, D.; Lee, J.; Hernandez, M.A.S.; Shulman, G.I.; Ozcan, U. XBP1s Is an Anti-lipogenic Protein. J. Biol. Chem. 2016, 291, 17394–17404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Henkel, A.S.; Lecuyer, B.E.; Schipma, M.J.; Anderson, K.A.; Green, R.M. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am. J. Physiol. Liver Physiol. 2015, 309, G965–G974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.; Zhang, T.; Yu, S.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Ringquist, S.; Dong, H.H. ATF4 Protein Deficiency Protects against High Fructose-induced Hypertriglyceridemia in Mice*. J. Biol. Chem. 2013, 288, 25350–25361. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice through Peroxisome Proliferator-Activated Receptor Alpha. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y.-J. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Jiang, Y.-F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef]
- Gupte, A.A.; Lyon, C.J.; Hsueh, W.A. Nuclear Factor (Erythroid-Derived 2)-Like-2 Factor (Nrf2), a Key Regulator of the Antioxidant Response to Protect Against Atherosclerosis and Nonalcoholic Steatohepatitis. Curr. Diabetes Rep. 2013, 13, 362–371. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 367–398. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.; Ashford, M.L.; Hayes, J. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Liu, Z.; Dou, W.; Ni, Z.; Wen, Q.; Zhang, R.; Qin, M.; Wang, X.; Tang, H.; Cao, Y.; Wang, J.; et al. Deletion of Nrf2 leads to hepatic insulin resistance via the activation of NF-κB in mice fed a high-fat diet. Mol. Med. Rep. 2016, 14, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodcroft, K.J.; Hafner, M.S.; Novak, R.F. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology 2002, 35, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.-M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelicen, P.; Tindberg, N. Lipopolysaccharide Induces CYP2E1 in Astrocytes through MAP Kinase Kinase-3 and C/EBPbeta and -Delta. J. Biol. Chem. 2003, 279, 15734–15742. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.K. Relevance of CYP2E1 to Non-alcoholic Fatty Liver Disease. Membr. Biog. 2013, 67, 165–175. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Gonzalez, F.J. Role of the Liver-Enriched Transcription Factor HNF-1α in Expression of theCYP2E1Gene. DNA Cell Biol. 1995, 14, 285–293. [Google Scholar] [CrossRef]
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S. Regulation of cytochrome P450 2e1 expression by ethanol: Role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death Dis. 2013, 4, e554. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Koyama, Y.; Xu, J.; Liu, X.; Brenner, D.A. New Developments on the Treatment of Liver Fibrosis. Dig. Dis. 2016, 34, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.-Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Wobser, H.; Dorn, C.; Weiss, T.S.; Amann, T.; Bollheimer, C.; Büttner, R.; Schölmerich, J.; Hellerbrand, C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009, 19, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012, 586, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2006, 26, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.-Y.; Oh, M.-A.; Kim, W.H.; Sohn, H.-Y.; Park, S.I. AMP-activated protein kinase inhibits TGF-?-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. J. Cell. Physiol. 2011, 227, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Liu, Z.; Zhou, Y.; Chen, M.; Wang, L.; Li, J. MicroRNA-142-3p inhibits trophoblast cell migration and invasion by disrupting the TGF-β1/Smad3 signaling pathway. Mol. Med. Rep. 2019, 19, 3775–3782. [Google Scholar] [CrossRef]
- Gerhard, G.S.; Hanson, A.; Wilhelmsen, D.; Piras, I.S.; Still, C.D.; Chu, X.; Petrick, A.T.; Distefano, J.K. AEBP1 expression increases with severity of fibrosis in NASH and is regulated by glucose, palmitate, and miR-372-3p. PLoS ONE 2019, 14, e0219764. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-W.; Muise, A.M.; Lyons, P.J.; Ro, H.-S. Regulation of Adipogenesis by a Transcriptional Repressor That Modulates MAPK Activation. J. Biol. Chem. 2001, 276, 10199–10206. [Google Scholar] [CrossRef] [Green Version]
- Jager, M.; Lee, M.-J.; Li, C.; Farmer, S.R.; Fried, S.K.; Layne, M.D. Aortic carboxypeptidase-like protein enhances adipose tissue stromal progenitor differentiation into myofibroblasts and is upregulated in fibrotic white adipose tissue. PLoS ONE 2018, 13, e0197777. [Google Scholar] [CrossRef]
- Majdalawieh, A.; Zhang, L.; Fuki, I.V.; Rader, D.J.; Ro, H.-S. Adipocyte enhancer-binding protein 1 is a potential novel atherogenic factor involved in macrophage cholesterol homeostasis and inflammation. Proc. Natl. Acad. Sci. USA 2006, 103, 2346–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.; Chen, Y.-D.; Sun, F.-R.; Shi, J.-P.; Song, Y.; Yang, J. Potential Regulators Driving the Transition in Nonalcoholic Fatty Liver Disease: A Stage-Based View. Cell. Physiol. Biochem. 2017, 41, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, N.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of YAP and TAZ in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2019. [Google Scholar] [CrossRef]
- Song, K.; Kwon, H.; Han, C.; Chen, W.; Zhang, J.; Ma, W.; Dash, S.; Gandhi, C.R.; Wu, T. YAP in Kupffer cells enhances the production of pro-inflammatory cytokines and promotes the development of non-alcoholic steatohepatitis. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Sakaida, I.; Tsuchiya, M.; Omori, K.; Takami, T.; Okita, K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient l-amino acid-defined diet. Biochem. Biophys. Res. Commun. 2004, 315, 187–195. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Sempoux, C.; Stärkel, P.; Horsmans, Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut 2006, 55, 1020–1029. [Google Scholar] [CrossRef]
- Colca, J.R.; McDonald, W.G.; Cavey, G.S.; Cole, S.L.; Holewa, D.D.; Brightwell-Conrad, A.S.; Wolfe, C.L.; Wheeler, J.S.; Coulter, K.R.; Kilkuskie, P.M.; et al. Identification of a Mitochondrial Target of Thiazolidinedione Insulin Sensitizers (mTOT)—Relationship to Newly Identified Mitochondrial Pyruvate Carrier Proteins. PLoS ONE 2013, 8, e61551. [Google Scholar] [CrossRef]
- McCommis, K.S.; Hodges, W.T.; Brunt, E.M.; Nalbantoglu, I.; McDonald, W.G.; Holley, C.; Fujiwara, H.; Schaffer, J.E.; Colca, J.; Finck, B.N. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017, 65, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Skat-Rørdam, J.; Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Hattori, S.; Dhar, D.K.; Hara, N.; Tonomoto, Y.; Onoda, T.; Ono, T.; Yamanoi, A.; Tachibana, M.; Tsuchiya, M.; Nagasue, N. FR-167653, a selective p38 MAPK inhibitor, exerts salutary effect on liver cirrhosis through downregulation of Runx2. Lab. Investig. 2007, 87, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Sun, B.; Zhao, X.; Zhang, Y.; Gu, Q.; Liang, X.; Dong, X.; Zhao, N. The Expression and Functional Significance of Runx2 in Hepatocellular Carcinoma: Its Role in Vasculogenic Mimicry and Epithelial–Mesenchymal Transition. Int. J. Mol. Sci. 2017, 18, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, A.; Malladi, P.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am. J. Physiol. Liver Physiol. 2004, 287, G264–G273. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2018, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Liu, J.; Jackson, M.I.; Zhao, F.-Q.; Yan, L.; Combs, G.F. Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high-fat diet. J. Nutr. 2013, 143, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Li, H.; Yang, X.; Xue, X.; Deng, L.; Shen, J.; Zhang, M.; Zhao, L.; Zhang, C. Genetically Obese Human Gut Microbiota Induces Liver Steatosis in Germ-Free Mice Fed on Normal Diet. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Ching, Y.-H.; Li, Y.-P.; Liu, J.-Y.; Huang, Y.-T.; Huang, Y.-W.; Yang, S.-S.; Huang, W.-C.; Chuang, H.-L. Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis. Nutrients 2017, 9, 1220. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Parnell, J.A.; Eksteen, B.; Raman, M.; Bomhof, M.R.; Rioux, K.P.; Madsen, K.L.; Reimer, R.A. Gut microbiota manipulation with prebiotics in patients with non-alcoholic fatty liver disease: A randomized controlled trial protocol. BMC Gastroenterol. 2015, 15, 169. [Google Scholar] [CrossRef] [Green Version]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Therapeutic Implications of the Gut Microbiome and Probiotics in Patients with NAFLD. Diseases 2019, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-W.; Phelps, E.; Ganapini, V.; Khan, N.; Ouyang, F.; Xu, H.; Khanna, S.; Tariq, R.; Friedman-Moraco, R.J.; Woodworth, M.H.; et al. Fecal microbiota transplantation for the treatment of recurrent and severe Clostridium difficileinfection in solid organ transplant recipients: A multicenter experience. Arab. Archaeol. Epigr. 2018, 19, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clément, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2016, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Ramón, V.; Chinchilla-López, P.; Ramírez-Pérez, O.; Méndez-Sánchez, N. Bile Acids in Nonalcoholic Fatty Liver Disease: New Concepts and Therapeutic Advances. Ann. Hepatol. 2017, 16, S58–S67. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Wang, L.; Wan, Y.-J.Y. The role of gut microbiota in liver disease development and treatment. Liver Res. 2019, 3, 3–18. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor–Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2016, 66, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2014, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besten, G.D.; Van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Gallo, D.J.; Green, A.M.; Williams, D.L.; Gong, X.; Shapiro, R.A.; Gambotto, A.A.; Humphris-Narayanan, E.; Vodovotz, Y.; Billiar, T.R. Role of Toll-Like Receptors in Changes in Gene Expression and NF-κB Activation in Mouse Hepatocytes Stimulated with Lipopolysaccharide. Infect. Immun. 2002, 70, 3433–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, S.A.; Oseini, A.; Feaver, R.E.; Cole, B.K.; Asgharpour, A.; Vincent, R.; Siddiqui, M.S.; Lawson, M.J.; Day, N.; Taylor, J.; et al. Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 12541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.; McGilvray, I.D.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, J.A.; Furuya, S.; Konganti, K.; Luo, Y.-S.; McDonald, T.J.; Iwata, Y.; Chiu, W.A.; Threadgill, D.W.; Pogribny, I.P.; Rusyn, I. Impact of Nonalcoholic Fatty Liver Disease on Toxicokinetics of Tetrachloroethylene in Mice. J. Pharmacol. Exp. Ther. 2017, 361, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Maeso-Diaz, R.; Boyer-Diaz, Z.; Lozano, J.J.; Ortega-Ribera, M.; Peralta, C.; Bosch, J.; Gracia-Sancho, J. New rat model of advanced nash mimicking pathophysiological features and transcriptomic signature of the human disease. Cells 2019, 8, 1062. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.; Wang, G.Z.; Guo, D.D.; Bai, R.-X.; Wang, M.; Du, S.Y. Deletion of Smad4 reduces hepatic inflammation and fibrogenesis during nonalcoholic steatohepatitis progression. J. Dig. Dis. 2018, 19, 301–313. [Google Scholar] [CrossRef]
- Valenti, L.; Mendoza, R.M.; Rametta, R.; Maggioni, M.; Kitajewski, C.; Shawber, C.J.; Pajvani, U.B. Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes 2013, 62, 4052–4062. [Google Scholar] [CrossRef] [Green Version]
- Liang, N.; Damdimopoulos, A.; Goni, S.; Huang, Z.; Vedin, L.L.; Jakobsson, T.; Giudici, M.; Ahmed, O.; Pedrelli, M.; Barilla, S.; et al. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARα. Nat. Commun. 2019, 10, 1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porteiro, B.; Fondevila, M.F.; Delgado, T.C.; Iglesias, C.; Imbernon, M.; Iruzubieta, P.; Crespo, J.; Zabala-Letona, A.; Ferno, J.; Gonzalez-Teran, B.; et al. Hepatic p63 regulates steatosis via IKKβ/ER stress. Nat. Commun. 2017, 8, 15111. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, P.; Wu, J.; Tao, F.; Zhou, J. Transcriptional regulation of early growth response gene-1 (EGR1) is associated with progression of nonalcoholic fatty liver disease (NAFLD) in patients with insulin resistance. Med. Sci. Monit. 2019, 25, 2293–3004. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Deng, X.; Xu, K. Increased expression of sterol regulatory element binding protein-2 alleviates autophagic dysfunction in NAFLD. Int. J. Mol. Med. 2018, 41, 1877–1886. [Google Scholar] [CrossRef]
- Kabbani, M.; Michailidis, E.; Steensels, S.; Zou, C.H.; Zeck, B.; Inna, R.L.; Fulmer, C.G.; Quirk, C.; Ashbrook, A.W.; Belkaya, S.; et al. PNPLA3-148M Overexpression in primary human hepatocytes exacerbates steatosis in tissue culture and chimeric mouse models of NAFLD. Hepatology 2019, 70, 1325A–1326A. [Google Scholar]

{kind=link}
| Factor | Model | Pathway | Regulation | Reference |
|---|---|---|---|---|
| PPARα | Humans, mice | Lipid metabolism, inflammation, fibrosis | Upregulation | [20] |
| PPARγ | Humans, mice | Lipid metabolism, inflammation, fibrosis | Upregulation | [20] |
| SREBP Family | Humans, mice | Lipid metabolism | Genetic variations increase risk of NAFLD | [21] |
| ChREBP | Humans, mice | Lipid metabolism | Upregulation | [22] |
| CAR | Humans, mice | Lipid metabolism, inflammation | Upregulation | [23] |
| LXR | Humans | Lipid metabolism, inflammation | Upregulation | [24] |
| FXR | Humans | Lipid metabolism | Downregulation | [25] |
| STAT5 | Humans | Lipid metabolism | Upregulated | [26] |
| C/EBPα | Mice | Lipid metabolism | Upregulation | [27] |
| PGC1α | Mice | Glucose homeostasis | Downregulation | [28] |
| FoxO | Humans | Glucose homeostasis | Upregulation | [29] |
| HNF4α | Humans | Central regulator, Glucose homeostasis | Downregulation | [25] |
| NF-κB | Humans, mice | Inflammation | Upregulation | [30] |
| IRFs | Mice | Inflammation | Upregulation | [31] |
| STAT1/3 | Mice | Inflammation | Upregulation | [32] |
| AP-1 and c-Jun | Humans, mice | Inflammation, fibrosis | Upregulation | [30,33] |
| SHP | Humans, mice | Inflammation | Downregulation | [34] |
| Nrf2 | Mice | Inflammation | Upregulation | [35] |
| Runx2 | Mice | Inflammation | Upregulation | [36] |
| C/EBPβ | Inflammation | |||
| IRE1α | Human | Metabolic stress | Upregulation | [37] |
| Xbp1 | Mice | Metabolic stress | Upregulation | [38] |
| eIF2α | Mice | Metabolic stress | Upregulation | [39] |
| ATF4 | Humans | Metabolic stress | Upregulation | [40] |
| ATF6 | Humans | Metabolic stress | Upregulation | [41] |
| Smad | Humans, mice | Fibrosis | Upregulation | [42] |
| TGFβ | Humans, mice | Fibrosis | Upregulation | [42] |
| AEBP1 | Humans, mice | Fibrosis | Upregulation | [43] |
| AATF/che-1 | Humans, mice | Fibrosis | Upregulation | [44] |
| YAP | Humans, mice | Fibrosis | Upregulation | [45] |
| Human | Mouse Model | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Steatosis | Fibrosis | HCC | Steatosis | NASH | ||||||||||||
| Regulation in Human Fibrosis | Transcription Factor/Regulator | c Non-fibrotic NAFLD vs. Healthy | e Steatotic vs. Healthy | a Fibrotic vs. Healthy | b Fibrotic vs. Non-fibrotic NAFLD | d NASH vs. Healthy | f External IPA NASH | g External IPA HCC | h HFD | i WD | j MCD | k NASH Diet | l NASH Diet + CCl4 | n CCl4 | m WD + CCl4 | o STAM |
| Consistent Activation (2 ≥ datasets) | FOXO1 * | |||||||||||||||
| IRF1 * | ||||||||||||||||
| IRF3 * | ||||||||||||||||
| JUN * | ||||||||||||||||
| NFκB * | ||||||||||||||||
| RELA * | ||||||||||||||||
| SP1 * | ||||||||||||||||
| SREBP1 * | ||||||||||||||||
| STAT1 * | ||||||||||||||||
| C/EBPβ † | ||||||||||||||||
| CTNNB1 † | ||||||||||||||||
| SMAD3 † | ||||||||||||||||
| Activation (1 dataset) | CREB | |||||||||||||||
| EGR1 | ||||||||||||||||
| ESR2 | ||||||||||||||||
| IRF7 | ||||||||||||||||
| LXR | ||||||||||||||||
| NFAT | ||||||||||||||||
| NRF2 | ||||||||||||||||
| RARα | ||||||||||||||||
| RUNX2 | ||||||||||||||||
| SPI1 | ||||||||||||||||
| STAT2 | ||||||||||||||||
| Consistent Inhibition (2 ≥ datasets) | PPARα * | |||||||||||||||
| PPARγ * | ||||||||||||||||
| RXRα * | ||||||||||||||||
| HNF4α † | ||||||||||||||||
| SMAD7 † | ||||||||||||||||
| Inhibition (1 dataset) | AHR | |||||||||||||||
| HDAC1 | ||||||||||||||||
| HNF1α | ||||||||||||||||
| NR1h | ||||||||||||||||
| NR3C1 | ||||||||||||||||
| NR5A2 | ||||||||||||||||
| RBL1 | ||||||||||||||||
| STAT5a | ||||||||||||||||
| THRβ | ||||||||||||||||
| Inconsistent Regulation | STAT3 | |||||||||||||||
| MYC | ||||||||||||||||
| p53 | ||||||||||||||||
| IRF8 | ||||||||||||||||
| ESR1 | ||||||||||||||||
| j MCD | k NASH Diet | l NASH Diet + CCl4 | n CCl4 | m WD + CCl4 | Association with Human Fibrosis | |
|---|---|---|---|---|---|---|
| SMAD4 | NASH | |||||
| SMAD2 | NASH | |||||
| YAP1 | NASH | |||||
| NOTCH1 | NASH | |||||
| EP300 | NASH | |||||
| NCOR | NASH | |||||
| p63 | NASH, steatosis | |||||
| SREBP2 | Steatosis | |||||
| CAR | Steatosis | |||||
| FOS | Steatosis, insulin resistance | |||||
| PGC1α | Steatosis, insulin resistance | |||||
| PPARδ | N/A | |||||
| HIF1α | N/A | |||||
| MED1 | N/A | |||||
| NCOA1 | N/A | |||||
| SMARCA4 | N/A | |||||
| NCOA2 | N/A | |||||
| FOXO3 | N/A | |||||
| HDAC2 | N/A | |||||
| STAT5b | N/A | |||||
| STAT6 | N/A |
| j MCD | k NASH Diet | l NASH Diet + CCl4 | n CCl4 | m WD + CCl4 | |
|---|---|---|---|---|---|
| AR | |||||
| ARNTL | |||||
| CDKN2A | |||||
| C/EBPα | |||||
| ChREBP | |||||
| CIITA | |||||
| E2F | |||||
| FOXO4 | |||||
| HNF1α | |||||
| HSF1 | |||||
| IRF9 | |||||
| KLF4 | |||||
| MAX | |||||
| MYB | |||||
| PGR | |||||
| RARB | |||||
| RB1 | |||||
| RORA | |||||
| SNAI | |||||
| SP3 | |||||
| STAT4 | |||||
| TCF7L2 | |||||
| THRα | |||||
| VDR | |||||
| WT1 | |||||
| Ybx1 | |||||
| ZNFn1a1 |
| a Fibrotic vs. Healthy | b Fibrotic vs. Non-Fibrotic NAFLD | d NASH vs. Healthy | f External IPA NASH | |
|---|---|---|---|---|
| CCND1 | ||||
| CCNE1 | ||||
| HMGB1 | ||||
| IRF2 | ||||
| IRF5 | ||||
| KLF2 | ||||
| NRIP1 | ||||
| SOX2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steensels, S.; Qiao, J.; Ersoy, B.A. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites 2020, 10, 283. https://doi.org/10.3390/metabo10070283
Steensels S, Qiao J, Ersoy BA. Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease. Metabolites. 2020; 10(7):283. https://doi.org/10.3390/metabo10070283
Chicago/Turabian StyleSteensels, Sandra, Jixuan Qiao, and Baran A. Ersoy. 2020. "Transcriptional Regulation in Non-Alcoholic Fatty Liver Disease" Metabolites 10, no. 7: 283. https://doi.org/10.3390/metabo10070283