Metabolite Genome-Wide Association Study (mGWAS) and Gene-Metabolite Interaction Network Analysis Reveal Potential Biomarkers for Feed Efficiency in Pigs


Abstract
:1. Introduction
- Find significant SNP markers associated with all the metabolites in the metabolomics dataset using mGWAS method and then reveal the biochemical mechanisms underlying genetic variation for porcine feed efficiency using 108 Danish pigs in low and high RFI conditions, genotyped by 68K PorcineSNP80 BeadChip array.
- Annotate identified significant SNP markers to porcine genes.
- Annotate metabolites and identify enriched metabolic pathways and gene-metabolite networks to find the potential biomarkers that were strongly associated with feed efficiency.
2. Results
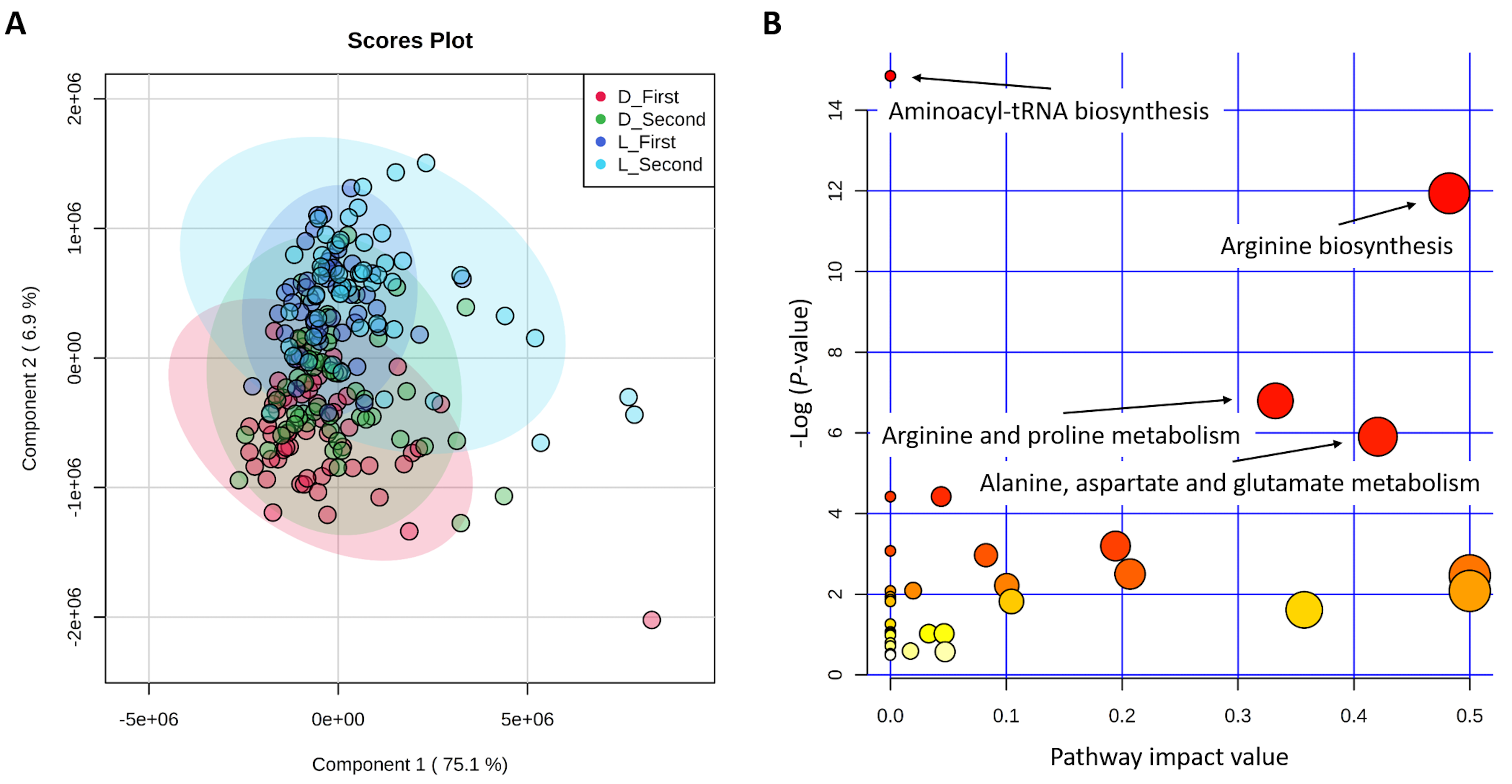
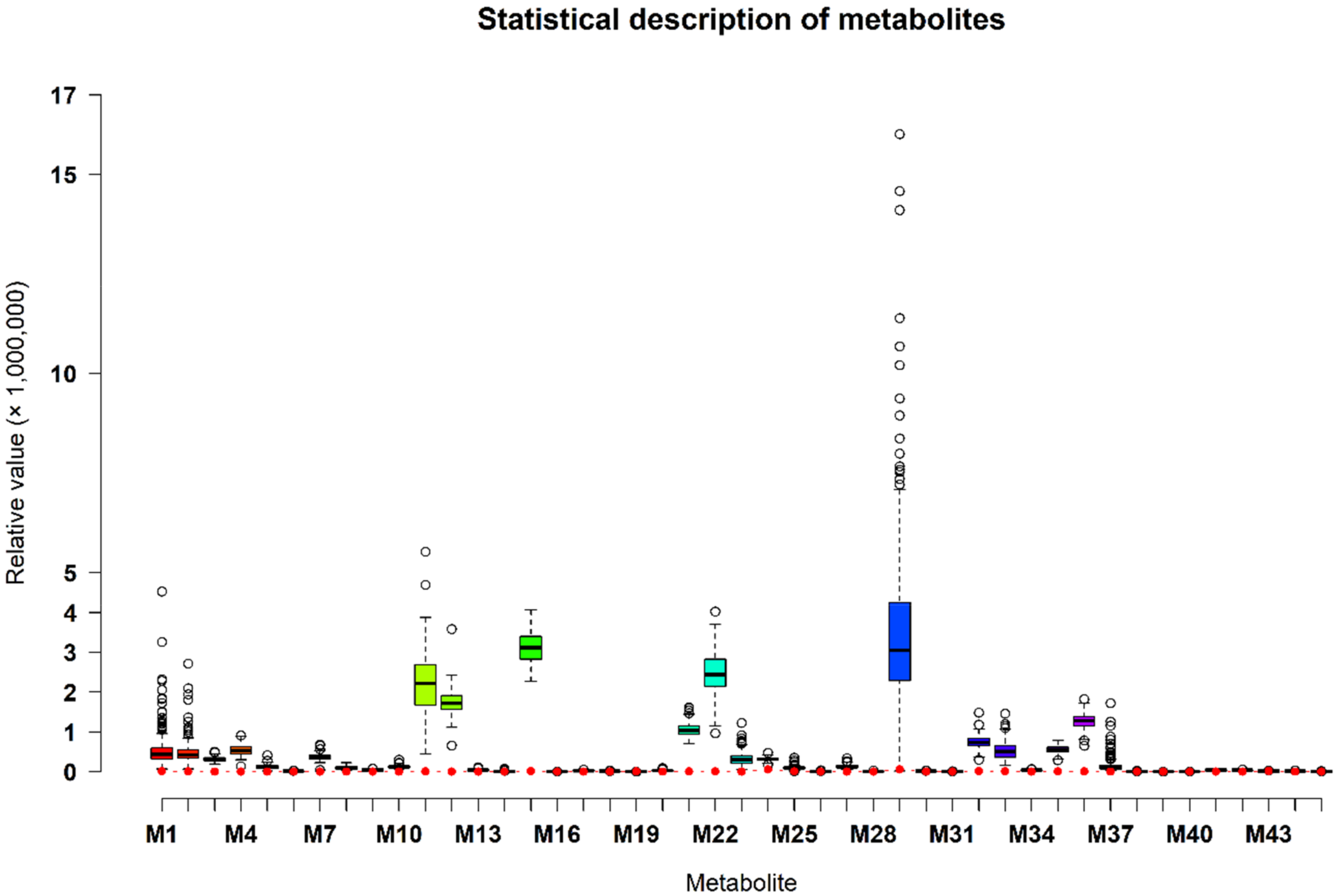
2.1. First Component Score and Significant Metabolic Pathways of 45 Metabolites
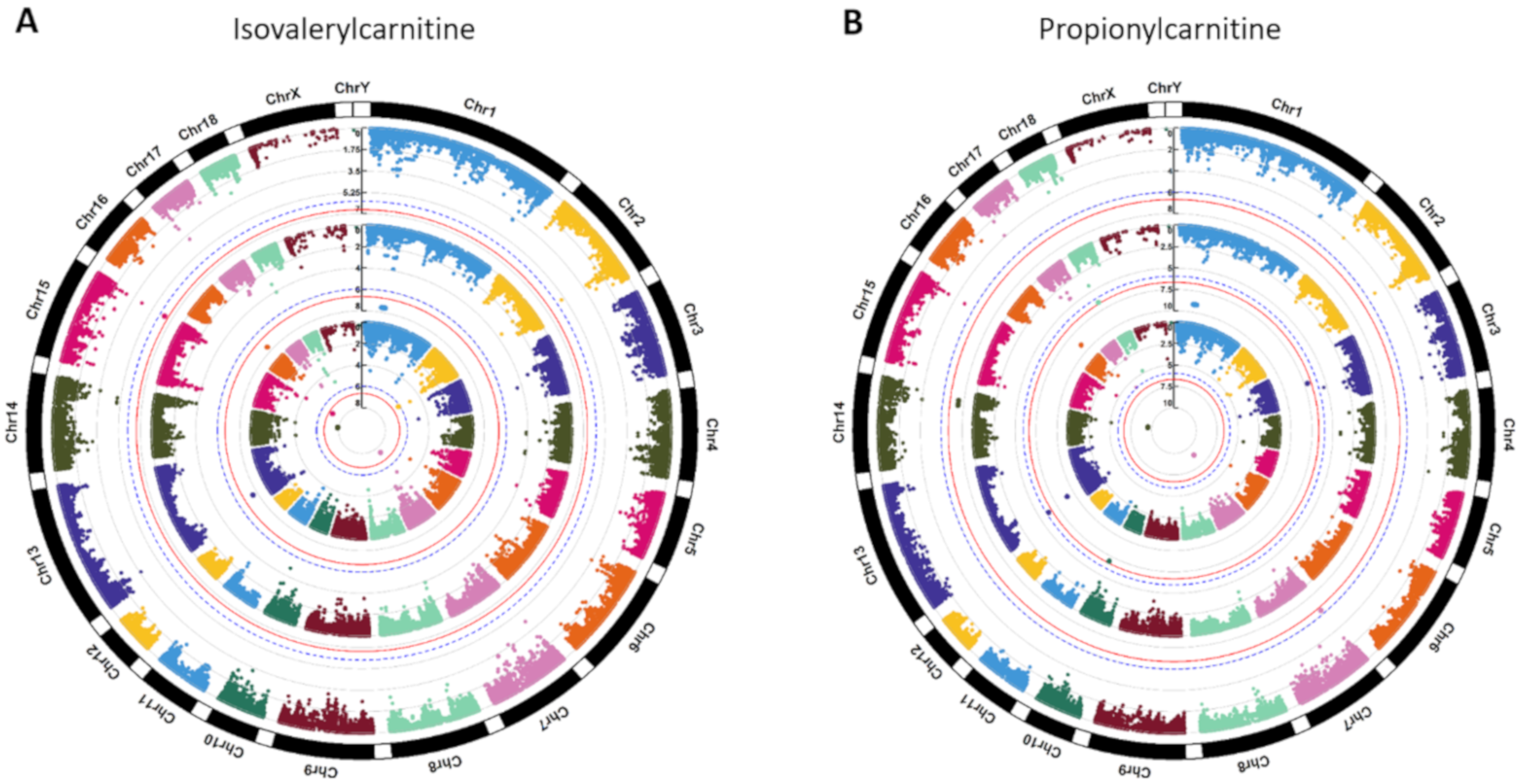
2.2. Genome-Wide Significant SNPs and Gene Annotation
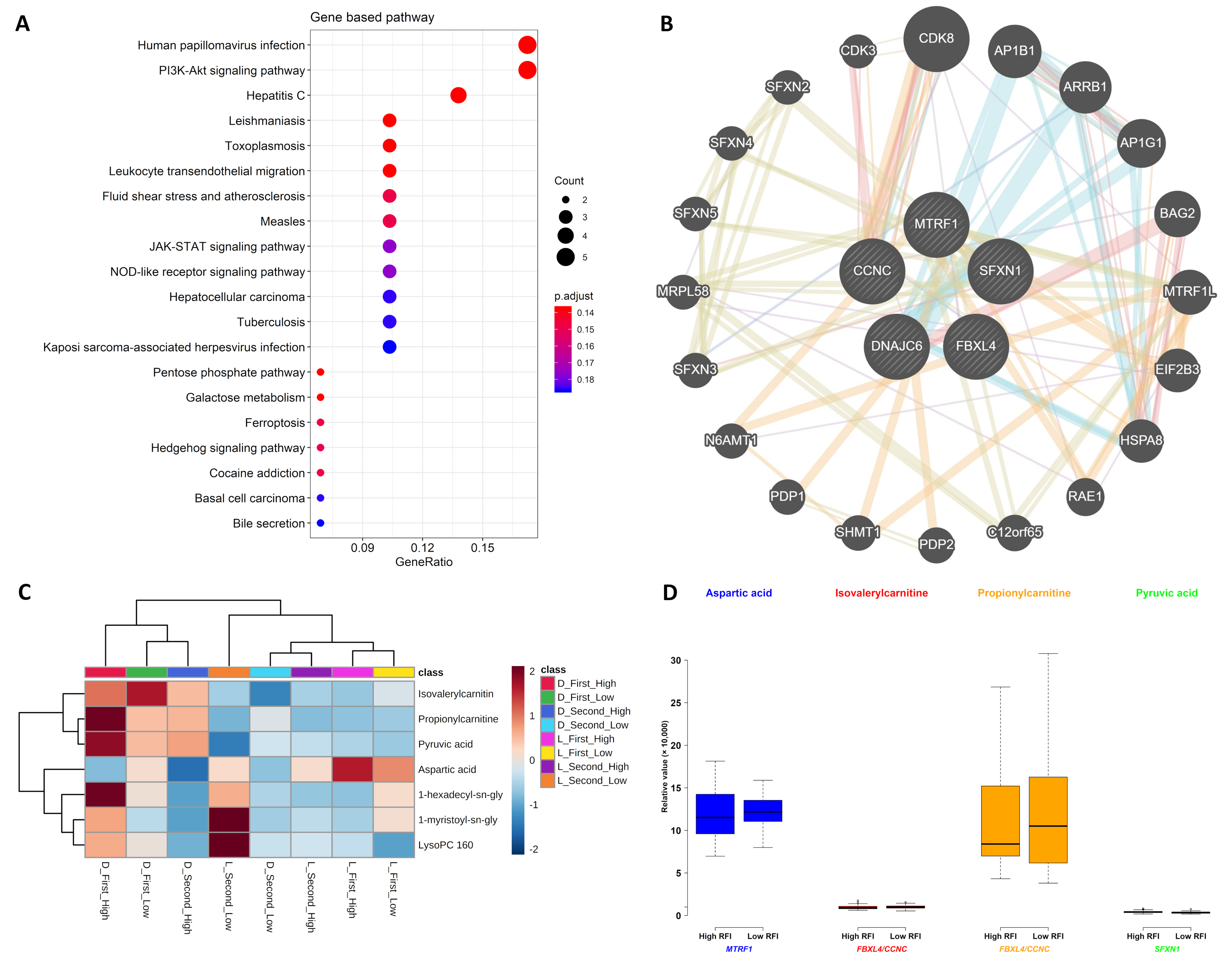
2.3. Gene and Metabolite Interaction Network
3. Discussion
3.1. Metabolites in the PLS-DA and Metabolic Pathways of Pigs
3.2. Genome-Wide Significant SNP-Related Genes Associated with Metabolites
3.3. Gene and Metabolite Interaction Network
3.4. Associations Linking SNP Genotypes, Metabolites, and RFI
4. Materials and Methods
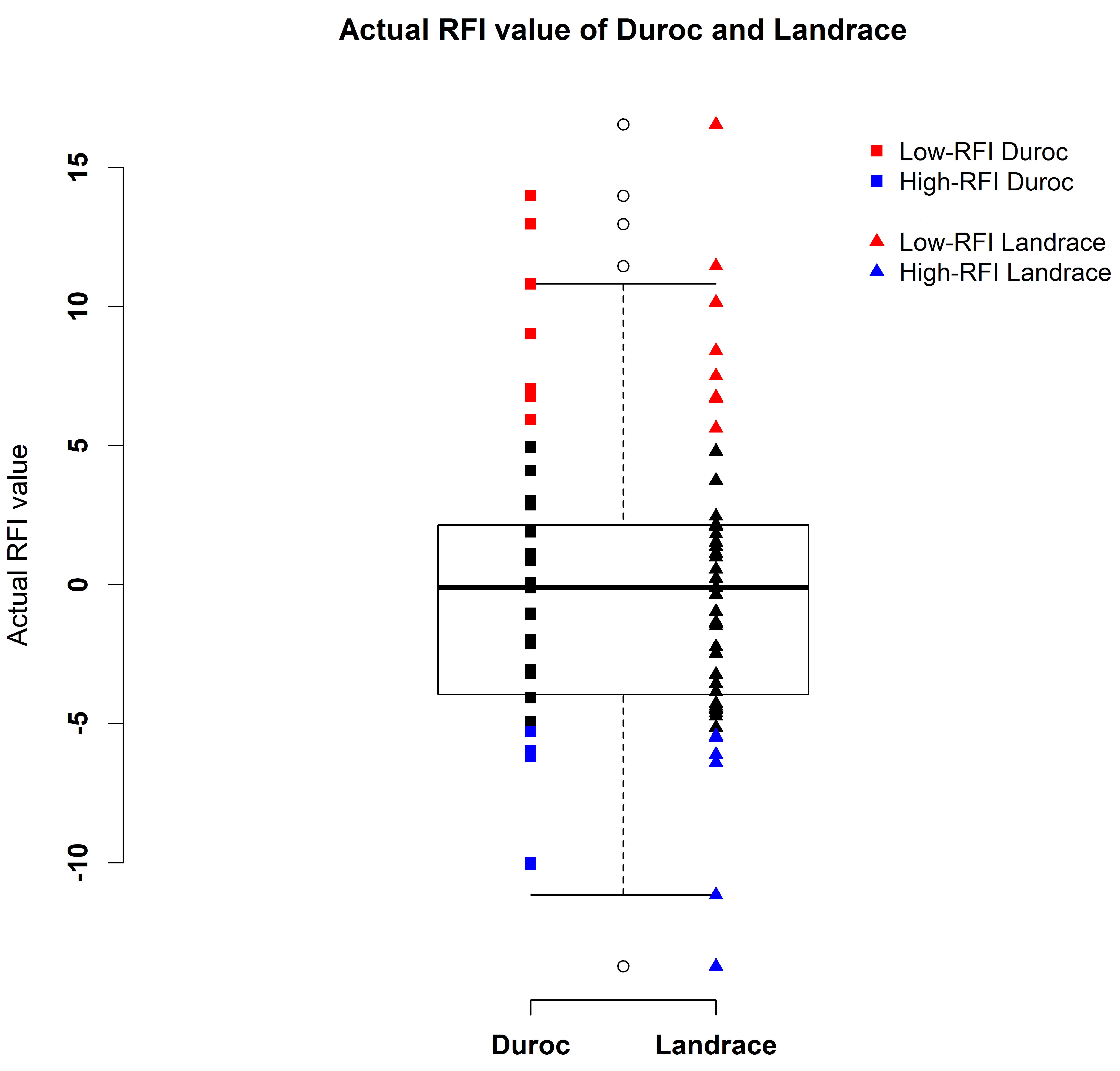
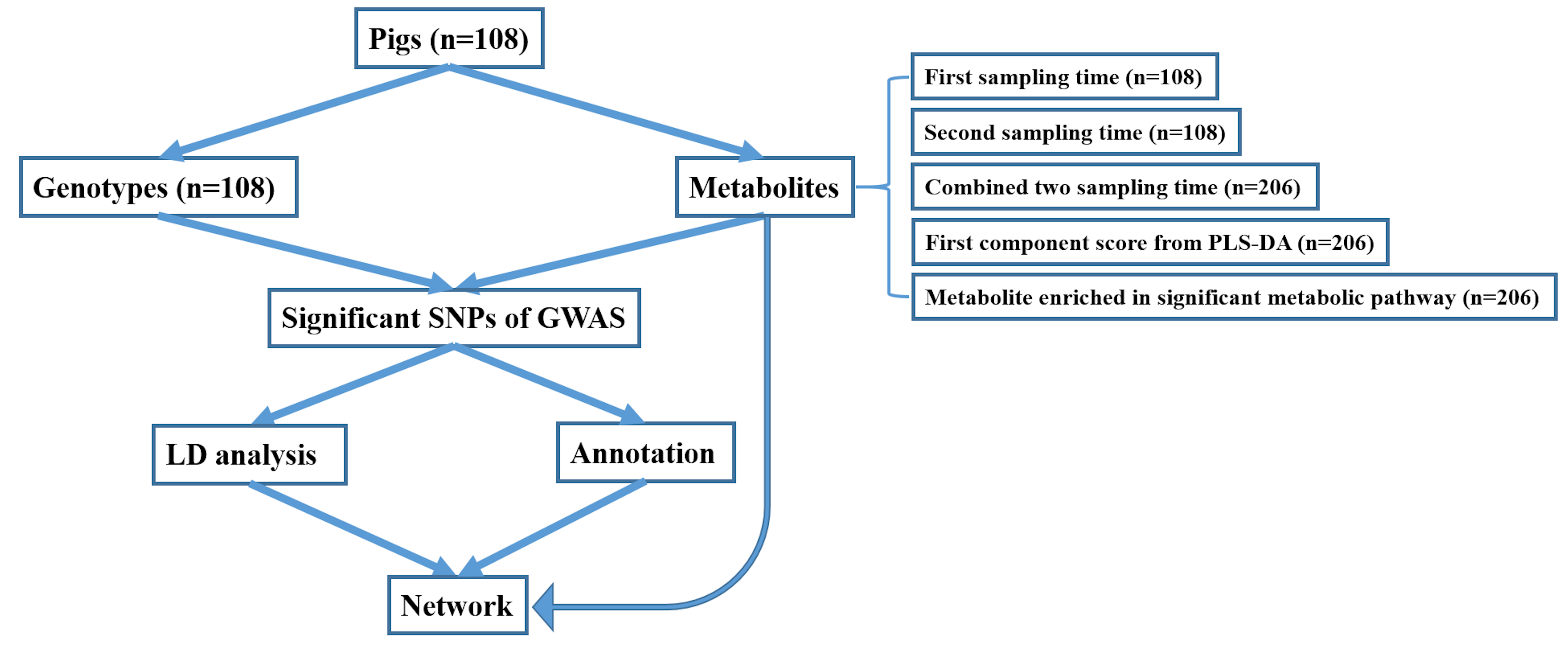
4.1. Animals, Metabolites, and Genotypes
4.2. Partial Least Squares-Discriminant Analysis and Metabolic Pathway Enrichment for 45 Metabolites
4.3. Mixed Linear Model Based Association Analysis
4.4. Significant SNPs and Their Annotated Genes
4.5. Gene-Based Pathway Enrichment Analysis and Gene–Metabolite Interaction Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
Abbreviations
| Component 1 | First component score |
| DFI | Daily feed intake |
| FC | Fold change |
| FDR | False discovery rate |
| LC-MS | Liquid chromatography-mass spectrometry |
| GRM | Genetic relationship matrix |
| GWAS | Genome-wide association study |
| HWE | Hardy–Weinberg equilibrium |
| LD | Linkage disequilibrium |
| LOD | Limit of detection |
| LLOR | Logarithm of likelihood odd ratio |
| MAF | Minor allele frequency |
| mGWAS | Metabolite GWAS |
| NCBI | National Center for Biotechnology Information |
| pDFI | Predicted daily feed intake |
| PLS-DA | Partial least squares-discriminant analysis |
| QC | Quality control |
| QTL | Quantitative trait locus |
| RFI | Residual feed intake |
| SNP | Single nucleotide polymorphism |
| UCSC | University of California Santa Cruz |
References
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suravajhala, P.; Kogelman, L.J.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications in animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beebe, K.; Kennedy, A.D. Sharpening Precision Medicine by a Thorough Interrogation of Metabolic Individuality. Comput. Struct. Biotechnol. J. 2016, 14, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Boil. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Ming, D.; Wang, J.; Li, Z.; Ma, X.; Wang, J.; Van Milgen, J.; Wang, F. Integrative analysis of indirect calorimetry and metabolomics profiling reveals alterations in energy metabolism between fed and fasted pigs. J. Anim. Sci. Biotechnol. 2018, 9, 41. [Google Scholar] [CrossRef]
- Carmelo, V.A.O.; Banerjee, P.; Diniz, W.J.D.S.; Kadarmideen, H.N. Metabolomic networks and pathways associated with feed efficiency and related-traits in Duroc and Landrace pigs. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kenéz, Á.; Dänicke, S.; Rolle-Kampczyk, U.; Von Bergen, M.; Huber, K. A metabolomics approach to characterize phenotypes of metabolic transition from late pregnancy to early lactation in dairy cows. Metabolomics 2016, 12. [Google Scholar] [CrossRef]
- Wang, X.; Kadarmideen, H.N. Metabolomics Analyses in High-Low Feed Efficient Dairy Cows Reveal Novel Biochemical Mechanisms and Predictive Biomarkers. Metabolites 2019, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; De Angelis, M.H.; Kronenberg, F.; Meitinger, T.; Mewes, H.-W.; Wichmann, H.-E.; Weinberger, K.M.; Adamski, J.; et al. Genetics Meets Metabolomics: A Genome-Wide Association Study of Metabolite Profiles in Human Serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Strathe, A.B.; Ostersen, T.; Pant, S.; Kadarmideen, H.N. Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Adriano, V.; Carmelo, O.; Kadarmideen, H.N. Genome-Wide Epistatic Interaction Networks Affecting Feed Efficiency in Duroc and Landrace Pigs. Front Genet. 2020, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Helke, K.L.; Nelson, K.N.; Sargeant, A.M.; Jacob, B.; McKeag, S.; Haruna, J.; Vemireddi, V.; Greeley, M.; Brocksmith, D.; Navratil, N.; et al. Pigs in Toxicology: Breed Differences in Metabolism and Background Findings. Toxicol. Pathol. 2016, 44, 575–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picone, G.; Zappaterra, M.; Luise, D.; Trimigno, A.; Capozzi, F.; Motta, V.; Davoli, R.; Costa, L.N.; Bosi, P.; Trevisi, P. Metabolomics characterization of colostrum in three sow breeds and its influences on piglets’ survival and litter growth rates. J. Anim. Sci. Biotechnol. 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundekilde, U.; Frederiksen, P.D.; Clausen, M.R.; Larsen, L.B.; Bertram, H. Relationship between the Metabolite Profile and Technological Properties of Bovine Milk from Two Dairy Breeds Elucidated by NMR-Based Metabolomics. J. Agric. Food Chem. 2011, 59, 7360–7367. [Google Scholar] [CrossRef]
- Goto, T.; Mori, H.; Shiota, S.; Tomonaga, S. Metabolomics Approach Reveals the Effects of Breed and Feed on the Composition of Chicken Eggs. Metabolites 2019, 9, 224. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.D.; Shang, X.G.; Li, D.F.; Wang, F.L.; Guan, Y.F.; Wang, Z.Y. Effects of Dietary Conjugated Linoleic Acid on the Fatty Acid Profile and Cholesterol Content of Egg Yolks from Different Breeds of Layers. Poult. Sci. 2008, 87, 284–290. [Google Scholar] [CrossRef]
- Middleton, R.P.; Lacroix, S.; Scott-Boyer, M.-P.; Dordevic, N.; Kennedy, A.D.; Slusky, A.; Carayol, J.; Petzinger-Germain, C.; Beloshapka, A.; Kaput, J. Metabolic Differences between Dogs of Different Body Sizes. J. Nutr. Metab. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.-Z.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the Fruit Metabolome in Tomato Breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Strathe, A.B.; Jensen, J.; Mark, T.; Kadarmideen, H.N. Genetic parameters for different measures of feed efficiency and related traits in boars of three pig breeds. J. Anim. Sci. 2013, 91, 4069–4079. [Google Scholar] [CrossRef]
- Ding, R.; Yang, M.; Wang, X.; Quan, J.; Zhuang, Z.; Zhou, S.; Li, S.; Xu, Z.; Zheng, E.; Cai, G.; et al. Genetic Architecture of Feeding Behavior and Feed Efficiency in a Duroc Pig Population. Front. Genet. 2018, 9, 220. [Google Scholar] [CrossRef]
- Dekaney, C.M.; Wu, G.; Jaeger, L.A. Gene expression and activity of enzymes in the arginine biosynthetic pathway in porcine fetal small intestine. Pediatr. Res. 2003, 53, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.M.; Grunewald, T.G.P.; Fendt, S.-M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.; Riquet, J.; Gruand, J.; Billon, Y.; Fève, K.; Sellier, P.; Noblet, J.; Bidanel, J.-P. Detecting QTL for feed intake traits and other performance traits in growing pigs in a Piétrain–Large White backcross. Animal 2010, 4, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Shirali, M.; Duthie, C.-A.; Doeschl-Wilson, A.; Knap, P.W.; Kanis, E.; Van Arendonk, J.; Roehe, R. Novel insight into the genomic architecture of feed and nitrogen efficiency measured by residual energy intake and nitrogen excretion in growing pigs. BMC Genet. 2013, 14, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, M.P.; Tribout, T.; Iannuccelli, N.; Bouffaud, M.; Servin, B.; Tenghe, A.; Déhais, P.; Muller, N.; Del Schneider, M.P.; Mercat, M.-J.; et al. A genome-wide association study of production traits in a commercial population of Large White pigs: Evidence of haplotypes affecting meat quality. Genet. Sel. Evol. 2014, 46, 12. [Google Scholar] [CrossRef] [Green Version]
- Onteru, S.K.; Gorbach, D.M.; Young, J.M.; Garrick, D.J.; Dekkers, J.C.M.; Rothschild, M.F. Whole Genome Association Studies of Residual Feed Intake and Related Traits in the Pig. PLoS ONE 2013, 8, e61756. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Ostersen, T.; Strathe, A.B.; Mark, T.; Jensen, J.; Kadarmideen, H.N. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014, 15, 27. [Google Scholar] [CrossRef] [Green Version]
- Makowski, L.; Noland, R.C.; Koves, T.R.; Xing, W.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Muoio, D.M. Metabolic profiling of PPARα-/- mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009, 23, 586–604. [Google Scholar] [CrossRef] [Green Version]
- Kerner, J.; Froseth, J.A.; Miller, E.R.; Bieber, L.L. A study of the acylcarnitine content of sows’ colostrum, milk and newborn piglet tissues: Demonstration of high amounts of isovaleryl-carnitine in colostrum and milk. J. Nutr. 1984, 114, 854–861. [Google Scholar] [CrossRef]
- Bartels, G.L.; Holwerda, K.J.; Kruijssen, D.A.C.M.; Remme, W.J. Anti-ischaemic efficacy of L-propionylcarnitine—A promising novel metabolic approach to ischaemia? Eur. Heart J. 1996, 17, 414–420. [Google Scholar] [CrossRef]
- Lew, D.J.; Dulić, V.; Reed, S.I. Isolation of three novel human cyclins by rescue of G1 cyclin (cln) function in yeast. Cell 1991, 66, 1197–1206. [Google Scholar] [CrossRef]
- Arai, E.; Sakamoto, H.; Ichikawa, H.; Totsuka, H.; Chiku, S.; Gotoh, M.; Mori, T.; Nakatani, T.; Ohnami, S.; Nakagawa, T.; et al. Multilayer-omics analysis of renal cell carcinoma, including the whole exome, methylome and transcriptome. Int. J. Cancer 2014, 135, 1330–1342. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Liu, Y.; Jankovic, V.; Sashida, G.; Lee, J.M.; Shieh, J.H.; Naoe, T.; Moore, M.; Nimer, S.D. Cyclin C regulates human hematopoietic stem/progenitor sell quiescence. Stem Cells 2010, 28, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Bondi, J.; Husdal, A.; Bukholm, G.; Nesland, J.M.; Bakka, A.; Bukholm, I.R.K. Expression and gene amplification of primary (A, B1, D1, D3, and E) and secondary (C and H) cyclins in colon adenocarcinomas and correlation with patient outcome. J. Clin. Pathol. 2005, 58, 509–514. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Dai, H.; Almannai, M.; Wang, J.; Faqeih, E.A.; Al Asmari, A.; Saleh, M.A.M.; Elamin, M.A.O.; Alfadhel, M.; Alkuraya, F.S.; et al. Molecular and clinical spectra of FBXL4 deficiency. Hum. Mutat. 2017, 38, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Ballout, R.A.; Al Alam, C.; Bonnen, P.E.; Huemer, M.; El-Hattab, A.W.; Shbarou, R. FBXL4-Related Mitochondrial DNA Depletion Syndrome 13 (MTDPS13): A Case Report With a Comprehensive Mutation Review. Front. Genet. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.M.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 2013, 93, 471–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankiewicz, E.; Mao, X.; Mangham, D.C.; Xu, L.; Yeste-Velasco, M.; Fisher, G.; North, B.; Chaplin, T.; Young, B.; Wang, Y.; et al. Identification of FBXL4 as a Metastasis Associated Gene in Prostate Cancer. Sci. Rep. 2017, 7, 5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, Y.; Wang, X.; Qi, J.; Jin, X.; Tong, H.; Zhou, Z.; Zhang, Z.C.; Han, J. Fbxl4 Serves as a Clock Output Molecule that Regulates Sleep through Promotion of Rhythmic Degradation of the GABAA Receptor. Curr. Biol. 2017, 27, 3616–3625.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, S.L.; Tang, Z.L.; Cui, W.T.; Mu, Y.L.; Chu, M.X.; Zhao, S.H.; Wu, Z.F.; Li, K.; Peng, K.M. Expression and SNP association analysis of porcine FBXL4 gene. Mol. Biol. Rep. 2009, 37, 579–585. [Google Scholar] [CrossRef]
- A Cabrera, R.; Usry, J.L.; Arellano, C.; Nogueira, E.T.; Kutschenko, M.; Moeser, A.; Odle, J. Effects of creep feeding and supplemental glutamine or glutamine plus glutamate (Aminogut) on pre- and post-weaning growth performance and intestinal health of piglets. J. Anim. Sci. Biotechnol. 2013, 4, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.B.; Lee, J.W.; Huang, H.J.; Wang, C.H.; Lee, T.T.; Yen, H.T.; Yu, B. Effects of Supplemental Glutamine on Growth Performance, Plasma Parameters and LPS-induced Immune Response of Weaned Barrows after Castration. Asian Australas. J. Anim. Sci. 2012, 25, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; McPhee, C.P.; Daniels, L.J.; Wade, C.M. Selection for growth rate in pigs on restricted feeding: Genetic parameters and correlated responses in residual feed intake. Proc. Assoc. Advmt. Anim. Breed. Genet. 2001, 14, 219–222. [Google Scholar]
- Wang, X.; Ma, P.; Liu, J.-F.; Zhang, Q.; Zhang, Y.; Ding, X.; Jiang, L.; Wang, Y.; Zhang, Y.; Sun, D.; et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. 2015, 16, 111. [Google Scholar] [CrossRef]
- Sargolzaei, M.; Chesnais, J.; Schenkel, F. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, G.; Hao, D.; Lund, M.S.; Kadarmideen, H.N. Comparisons of improved genomic predictions generated by different imputation methods for genotyping by sequencing data in livestock populations. J. Anim. Sci. Biotechnol. 2020, 11, 3–12. [Google Scholar] [CrossRef]
- Purcell, S.M.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Boil. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.H., Jr. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Match Status | Involved Metabolites | p-Value | −Log (p-Value) | False Discovery Rate (FDR) | Pathway Impact Value |
|---|---|---|---|---|---|---|
| Aminoacyl-tRNA biosynthesis (ssc00970) | 9/48 | Alanine, Arginine, Aspartic acid, Glutamic acid, Isoleucine, Methionine, Phenylalanine, Proline, Threonine | 3.55 × 10−7 | 14.85 | 2.98 × 10−5 | 0 |
| Arginine biosynthesis (ssc00220) | 5/14 | Arginine, Aspartic acid, Citrulline, Glutamic acid, Ornithine | 6.53 × 10−6 | 11.94 | 2.74 × 10−4 | 0.48 |
| Arginine and proline metabolism (ssc00330) | 5/38 | Arginine, Glutamic acid, Ornithine, Proline, Pyruvic acid | 1.12 × 10−3 | 6.79 | 0.031 | 0.33 |
| Alanine, aspartate and glutamate metabolism (ssc00250) | 4/28 | Alanine, Aspartic acid, Glutamic acid, Pyruvic acid | 2.72 × 10−3 | 5.91 | 0.057 | 0.42 |
| Significant SNP Name | Associated Metabolite Number | Metabolite from First Sampling Time | Metabolite from Second Sampling Time | Metabolite from Combined Two Sampling Times |
|---|---|---|---|---|
| ALGA0003891, ALGA0003900, ALGA0003935, ALGA0003952, ALGA0003953, ALGA0003995, ALGA0004000, ALGA0004002, ALGA0004005, ALGA0004006, ALGA0004024, ALGA0004041, ALGA0004042, ALGA0004046, ALGA0004048, ALGA0004073, ALGA0004090, ALGA0004093, ALGA0004143, ALGA0004148, ALGA0004169, ALGA0004173, ALGA0004177, ASGA0003182, ASGA0003194, ASGA0003235, ASGA0003288, ASGA0003312, ASGA0003314, ASGA0003315, ASGA0003317, ASGA0003333, ASGA0003335, ASGA0057312, ASGA0083304, DRGA0000994, DRGA0001072, DRGA0001073, H3GA0001865, H3GA0001937, H3GA0001949, H3GA0001956, H3GA0001966, H3GA0046845, INRA0002726, INRA0002819, INRA0002820, INRA0002823, MARC0021047, MARC0027518, MARC0034307, MARC0050325, MARC0059407, MARC0063106, MARC0068954, MARC0075306, SIRI0000655 | 2 | NA | Isovalerylcarnitine, Propionylcarnitine | NA |
| MARC0080116 | 2 | Pyruvic acid | NA | Citrulline |
| ALGA0038416, ALGA0081238, DRGA0014486, WU_10.2_14_132246191 | 3 | Isovalerylcarnitine, Propionylcarnitine | NA | Propionylcarnitine |
| ASGA0093565, H3GA0053559, WU_10.2_6_136216429, WU_10.2_6_136863547, WU_10.2_6_136876717, WU_10.2_6_136972846 | 3 | 1-hexadecyl-sn-glycero-3-phosphocholine, 1-myristoyl-sn-glycero-3-phosphocholine, LysoPC(16:0) | NA | NA |
| M1GA0016778, WU_10.2_X_114649203 | 3 | Pyruvic acid | NA | Citrulline, Pyruvic acid |
| ALGA0099866, WU_10.2_X_105559450 | 4 | 1-hexadecyl-sn-glycero-3-phosphocholine, 1-myristoyl-sn-glycero-3-phosphocholine, LysoPC(16:0), Pyruvic acid | NA | NA |
| ASGA0018324 | 4 | Citrulline, Pyruvic acid | NA | Citrulline, Pyruvic acid |
| ASGA0081223, INRA0003881, MARC0046138, WU_10.2_X_103597980, WU_10.2_X_103653646, WU_10.2_X_104796075, WU_10.2_X_104910069, WU_10.2_X_104956283, WU_10.2_X_104980830, WU_10.2_X_105583738 | 4 | 1-hexadecyl-sn-glycero-3-phosphocholine, 1-myristoyl-sn-glycero-3-phosphocholine, LysoPC(16:0) | NA | 1-hexadecyl-sn-glycero-3-phosphocholine |
| Chromosome | Position | Significant SNP Name | Gene Component | Gene | Gene Description | Metabolite from First Sampling Time (p-Value) | Metabolite from Second Sampling Time (p-Value) | Metabolite from Combined Two Sampling Times (p-Value) |
|---|---|---|---|---|---|---|---|---|
| 1 | 74467285 | ALGA0004000 | 6th intron | FBXL4 (NM_001171752) | F-box and leucine rich repeat protein 4 | NA | Isovalerylcarnitine (2.79 × 10−8), Propionylcarnitine (8.32 × 10−10) | NA |
| 1 | 75151870 | ALGA0004041 | 1st intron | CCNC (NM_001190160) | Cyclin C | NA | Isovalerylcarnitine (2.79 × 10−8), Propionylcarnitine (8.32 × 10−10) | NA |
| 1 | 75167426 | ALGA0004042 | 9th intron/8th intron # | CCNC (NM_001190160) | Cyclin C | NA | Isovalerylcarnitine (2.79 × 10−8), Propionylcarnitine (8.32 × 10−10) | NA |
| 2 | 83663964 | MARC0110390 | 7th intron | SFXN1 (NM_001098602) | Sideroflexin 1 | Pyruvic acid (1.25 × 10−7) | NA | NA |
| 6 | 135424176 | ASGA0093565 | 8th intron | DNAJC6 (NM_001145378) | DnaJ heat shock protein family (Hsp40) member C6 | 1-hexadecyl-sn-glycero-3-phosphocholine (2.78 × 10−9), 1-myristoyl-sn-glycero-3-phosphocholine (1.35 × 10−8), LysoPC (16:0) (1.22 × 10−7) | NA | NA |
| 11 | 26591544 | ALGA0061605 | 5th intron/9th intron # | MTRF1 (NM_001243580) | Mitochondrial translation release factor 1 | NA | Aspartic acid (2.29 × 10−7) | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Kadarmideen, H.N. Metabolite Genome-Wide Association Study (mGWAS) and Gene-Metabolite Interaction Network Analysis Reveal Potential Biomarkers for Feed Efficiency in Pigs. Metabolites 2020, 10, 201. https://doi.org/10.3390/metabo10050201
Wang X, Kadarmideen HN. Metabolite Genome-Wide Association Study (mGWAS) and Gene-Metabolite Interaction Network Analysis Reveal Potential Biomarkers for Feed Efficiency in Pigs. Metabolites. 2020; 10(5):201. https://doi.org/10.3390/metabo10050201
Chicago/Turabian StyleWang, Xiao, and Haja N. Kadarmideen. 2020. "Metabolite Genome-Wide Association Study (mGWAS) and Gene-Metabolite Interaction Network Analysis Reveal Potential Biomarkers for Feed Efficiency in Pigs" Metabolites 10, no. 5: 201. https://doi.org/10.3390/metabo10050201