Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Sampling, and Histopathological Examination
2.2. Experimental Design
3. Results
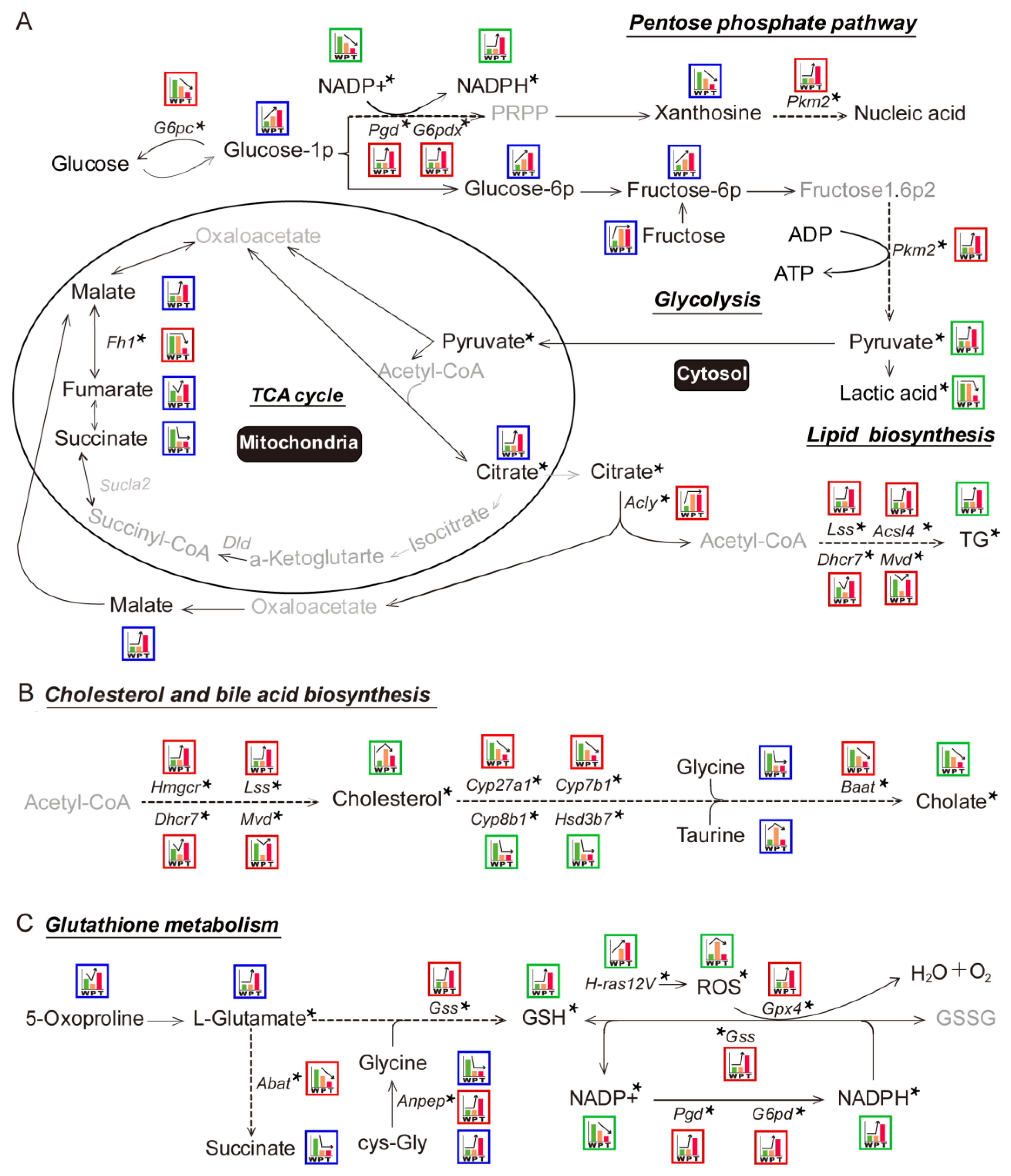
3.1. Differential Metabolites and Pathways Identified by Metabolomic Analysis in Hepatocarcinogenesis
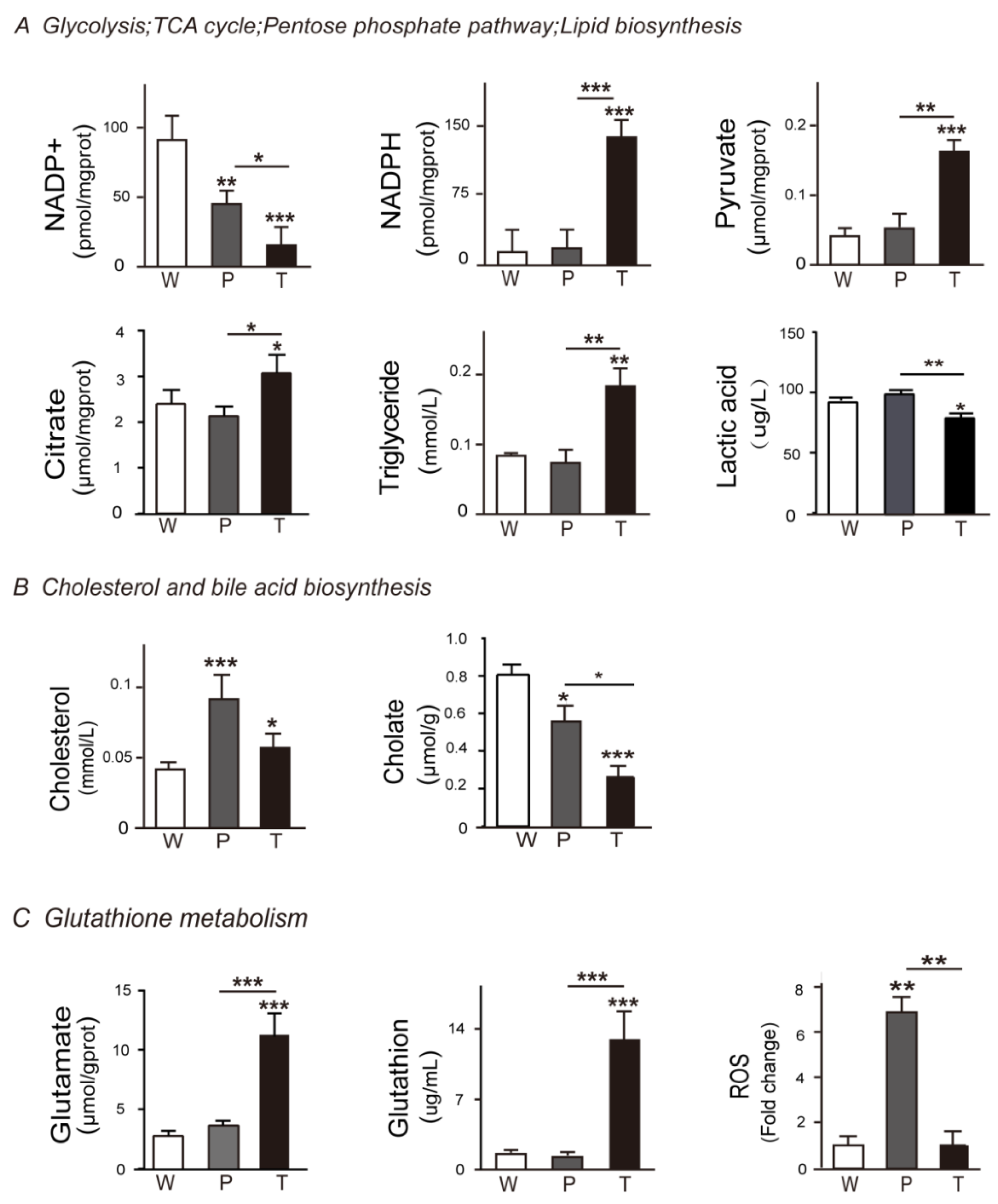
3.2. Variation Tendencies of Metabolites Identified in Hepatocarcinogenesis
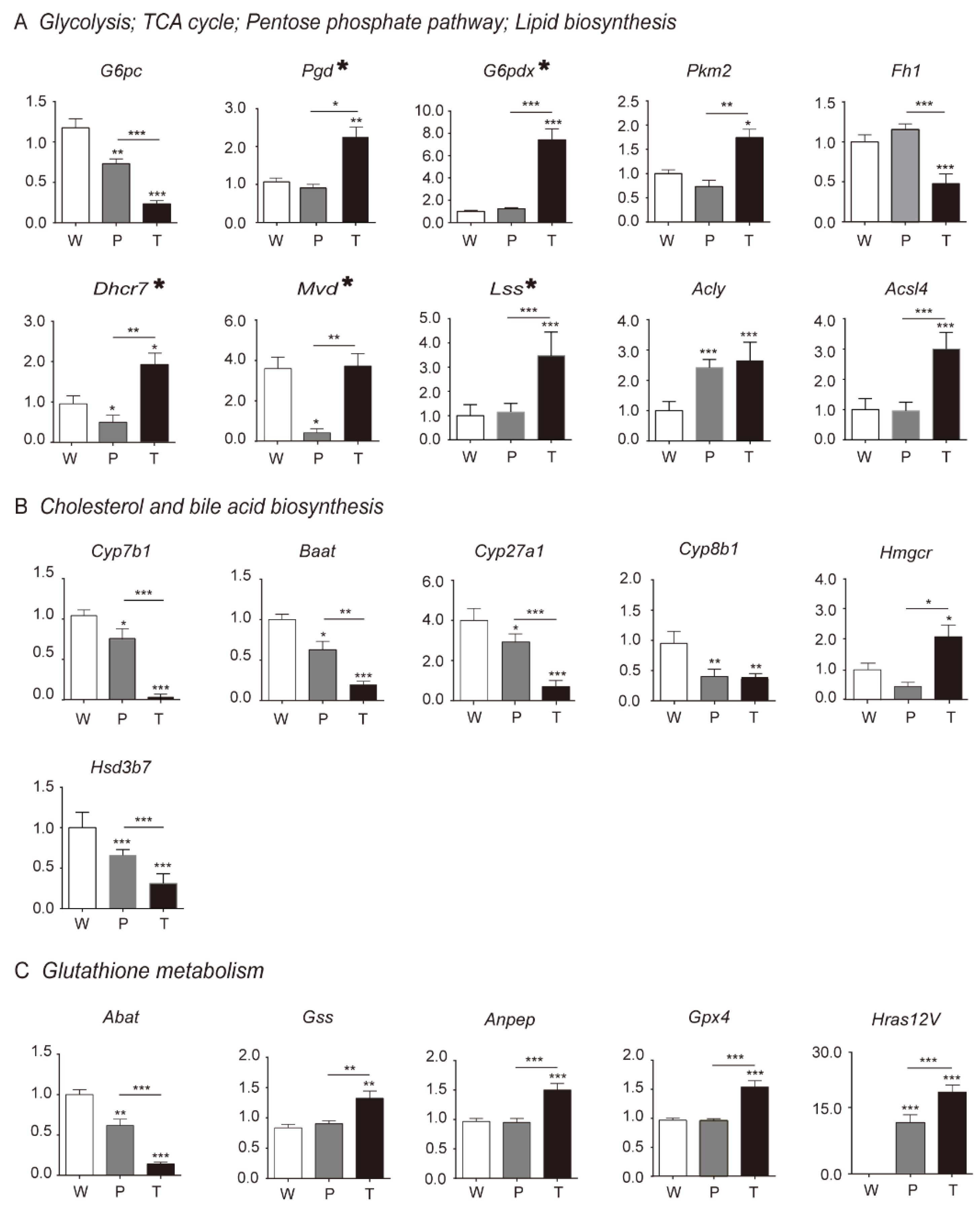
3.3. Differential Genes and Pathways Identification by Transcriptomics Analysis in Hepatocarcinogenesis
3.4. Mitochondrial Citrate Malate Shuttle and Lipid Biosynthesis Enhanced during Hepatocarcinogenesis
3.4.1. Glycolysis
3.4.2. TCA Cycle and Citrate Malate Shuttle
3.4.3. Lipid Biosynthesis
3.5. Alteration of Pentose Phosphate Pathway (PPP), Cholesterol and Bile Acid Metabolism, Glutathione Metabolism during Hepatocarcinogenesis
3.5.1. PPP
3.5.2. Cholesterol and Bile Acid Metabolism
3.5.3. Glutathione Metabolism
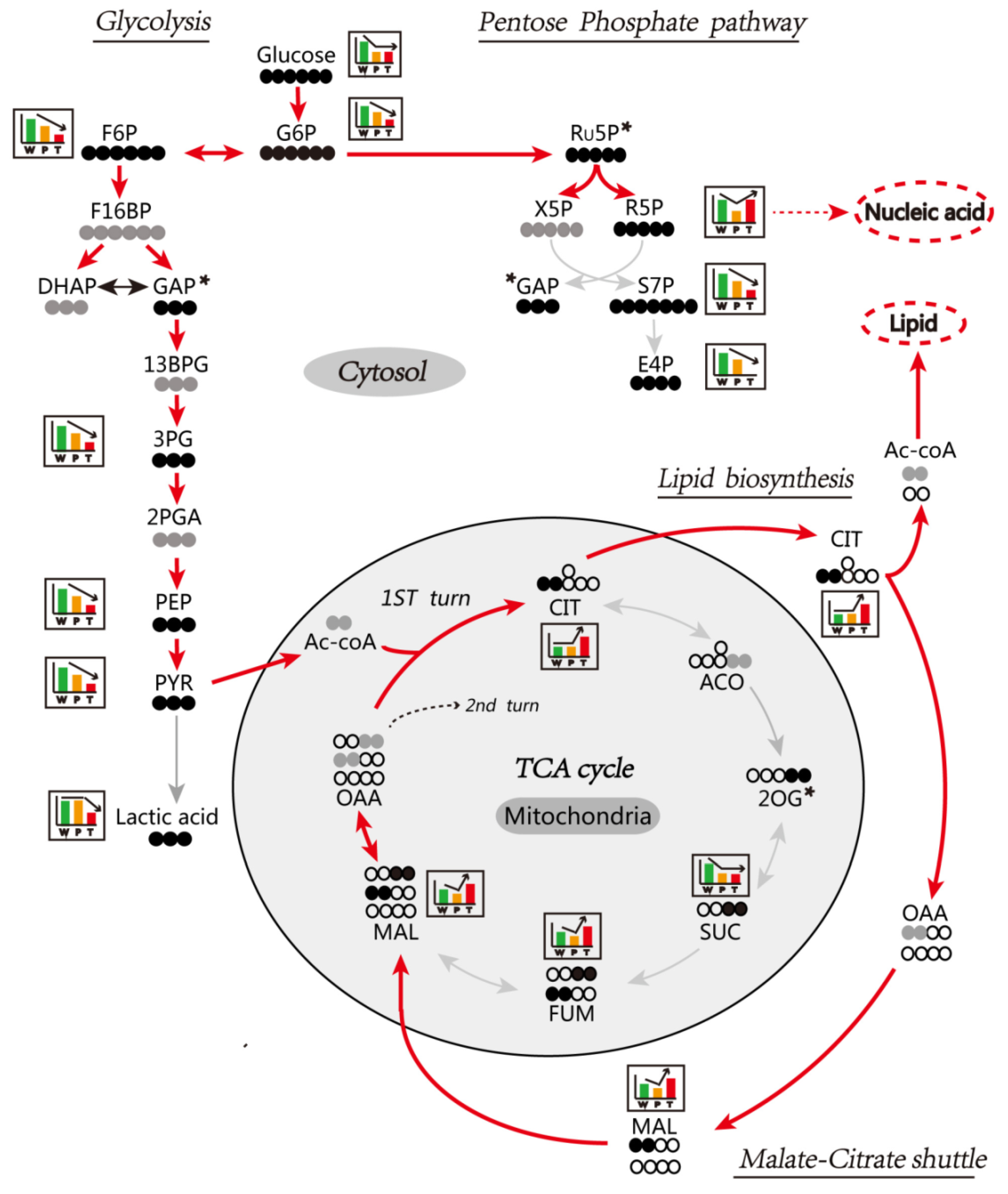
3.6. Enhanced Mitochondrial Citrate-Malate Shuttle and PPP in Hepatocarcinogenesis Confirmed by In Vivo Assay of [U-13C] Glucose Metabolism
3.6.1. Glycolysis
3.6.2. TCA Cycle and Citrate-malate Shuttle
3.6.3. PPP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| DEMs | differentially expressed metabolites |
| LDs | lipid drops |
| MSEA | metabolite set enrichment analysis |
| NGS | next-generation sequencing |
| P | precancerous liver tissues |
| PPP | pentose phosphate pathway |
| T | hepatocellular carcinoma |
| UPLC-MS/MS | ultra-performance liquid chromatography-tandem mass spectrometry |
| DEGs | differentially expressed genes |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MCTs | monocarboxylate transporters |
| NAFLD | nonalcoholic fatty liver disease |
| Non-Tg | C57BL/6J wild-type non-transgenic mice |
| PCA | principal component analysis |
| Ras-Tg | Hras12V transgenic mice |
| TCA | tricarboxylic acid |
| W | normal liver tissues of non-Tg |
References
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef] [Green Version]
- Dekervel, J.; van Pelt, J.; Verslype, C. Advanced unresectable hepatocellular carcinoma: New biologics as fresh ammunition or clues to disease understanding? Curr. Opin. Oncol. 2013, 25, 409–416. [Google Scholar] [CrossRef]
- Yin, P.; Wan, D.; Zhao, C.; Chen, J.; Zhao, X.; Wang, W.; Lu, X.; Yang, S.; Gu, J.; Xu, G. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol. Biosyst. 2009, 5, 868–876. [Google Scholar] [CrossRef]
- Shariff, M.I.; Gomaa, A.I.; Cox, I.J.; Patel, M.; Williams, H.R.; Crossey, M.M.; Thillainayagam, A.V.; Thomas, H.C.; Waked, I.; Khan, S.A.; et al. Urinary metabolic biomarkers of hepatocellular carcinoma in an Egyptian population: A validation study. J. Proteome Res. 2011, 10, 1828–1836. [Google Scholar] [CrossRef]
- Bruntz, R.C.; Lane, A.N.; Higashi, R.M.; Fan, T.W. Exploring cancer metabolism using stable isotope-resolved metabolomics (SIRM). J. Biol. Chem. 2017, 292, 11601–11609. [Google Scholar] [CrossRef] [Green Version]
- Lerche, M.H.; Karlsson, M.; Ardenkjaer-Larsen, J.H.; Jensen, P.R. Targeted Metabolomics with Quantitative Dissolution Dynamic Nuclear Polarization. Methods Mol. Biol. 2019, 2037, 385–393. [Google Scholar]
- Reynolds, S.H.; Stowers, S.J.; Patterson, R.M.; Maronpot, R.R.; Aaronson, S.A.; Anderson, M.W. Activated oncogenes in B6C3F1 mouse liver tumors: Implications for risk assessment. Science 1987, 237, 1309–1316. [Google Scholar] [CrossRef]
- Newell, P.; Toffanin, S.; Villanueva, A.; Chiang, D.Y.; Minguez, B.; Cabellos, L.; Savic, R.; Hoshida, Y.; Lim, K.H.; Melgar-Lesmes, P.; et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 2009, 51, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taketomi, A.; Shirabe, K.; Muto, J.; Yoshiya, S.; Motomura, T.; Mano, Y.; Ikegami, T.; Yoshizumi, T.; Sugio, K.; Maehara, Y. A rare point mutation in the Ras oncogene in hepatocellular carcinoma. Surg. Today 2013, 43, 289–292. [Google Scholar] [CrossRef]
- Delire, B.; Starkel, P. The Ras/MAPK pathway and hepatocarcinoma: Pathogenesis and therapeutic implications. Eur. J. Clin. Investig. 2015, 45, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.G.; Moon, H.B.; Lee, M.R.; Hwang, C.Y.; Kwon, K.S.; Yu, S.L.; Kim, Y.S.; Kim, M.; Kim, J.M.; Kim, S.K.; et al. Gender-dependent hepatic alterations in H-ras12V transgenic mice. J. Hepatol. 2005, 43, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Fan, T.; Li, H.; Li, J.; Wang, K.; Wang, X.; Dong, J.; Chen, J.; Wang, F.; Wang, J.; et al. Differential Proteomic Analysis of Gender-dependent Hepatic Tumorigenesis in Hras12V Transgenic Mice. Mol. Cell Proteom. 2017, 16, 1475–1490. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.; Rong, Z.; Dong, J.; Li, J.; Wang, K.; Wang, X.; Li, H.; Chen, J.; Wang, F.; Wang, J.; et al. Metabolomic and transcriptomic profiling of hepatocellular carcinomas in Hras12V transgenic mice. Cancer Med. 2017, 6, 2370–2384. [Google Scholar] [CrossRef] [Green Version]
- Frith, C.H.; Ward, J.M. A morphologic classification of proliferative and neoplastic hepatic lesions in mice. J. Environ. Pathol. Toxicol. 1979, 3, 329–351. [Google Scholar]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef]
- Nezami Ranjbar, M.R.; Luo, Y.; Di Poto, C.; Varghese, R.S.; Ferrarini, A.; Zhang, C.; Sarhan, N.I.; Soliman, H.; Tadesse, M.G.; Ziada, D.H.; et al. GC-MS Based Plasma Metabolomics for Identification of Candidate Biomarkers for Hepatocellular Carcinoma in Egyptian Cohort. PLoS ONE 2015, 10, e0127299. [Google Scholar] [CrossRef]
- Hatab, H.M.; Abdel Hamid, F.F.; Soliman, A.F.; Al-Shafie, T.A.; Ismail, Y.M.; El-Houseini, M.E. A combined treatment of curcumin, piperine, and taurine alters the circulating levels of IL-10 and miR-21 in hepatocellular carcinoma patients: A pilot study. J. Gastrointest. Oncol. 2019, 10, 766–776. [Google Scholar] [CrossRef]
- Bruns, H.; Petrulionis, M.; Schultze, D.; Al Saeedi, M.; Lin, S.; Yamanaka, K.; Ambrazevicius, M.; Strupas, K.; Schemmer, P. Glycine inhibits angiogenic signaling in human hepatocellular carcinoma cells. Amino Acids 2014, 46, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, K.B.; Lee, M.J.; Bae, S.C.; Jang, J.J. Nicotinamide inhibits the early stage of carcinogen-induced hepatocarcinogenesis in mice and suppresses human hepatocellular carcinoma cell growth. J. Cell Physiol. 2012, 227, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.F.; Li, G.P.; Su, J.D.; Pu, Z.J.; Feng, J.L.; Ye, Y.Q.; Wei, B.L. Involvement of NF-kappaB activation in the apoptosis induced by extracellular adenosine in human hepatocellular carcinoma HepG2 cells. Biochem. Cell Biol. 2010, 88, 705–714. [Google Scholar] [CrossRef]
- Ciou, S.C.; Chou, Y.T.; Liu, Y.L.; Nieh, Y.C.; Lu, J.W.; Huang, S.F.; Cheng, L.H.; Lo, J.F.; Chen, M.J.; Yang, M.C.; et al. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int. J. Cancer 2015, 137, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, O.; Kan, M.; Zhang, M.; Shao, D.; Pan, Y.; Zheng, H.; Zhang, X.; Chen, L.; Liu, S. Berberine induces apoptosis by suppressing the arachidonic acid metabolic pathway in hepatocellular carcinoma. Mol. Med. Rep. 2015, 12, 4572–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, D.J.; Nakamura, S.; Amachi, R.; Hiasa, M.; Oda, A.; Tsuji, D.; Itoh, K.; Harada, T.; Horikawa, K.; Teramachi, J.; et al. Effective impairment of myeloma cells and their progenitors by blockade of monocarboxylate transportation. Oncotarget 2015, 6, 33568–33586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, V.A.; Pinheiro, C.; Morais-Santos, F.; Felipe-Silva, A.; Longatto-Filho, A.; Baltazar, F. Characterization of monocarboxylate transporter activity in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 11780–11787. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Song, X.; Che, L.; Li, X.; Pilo, M.G.; Vidili, G.; Porcu, A.; Solinas, A.; Cigliano, A.; Pes, G.M.; et al. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int. 2017, 37, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Kanematsu, M.; Kondo, H.; Goshima, S.; Tsuge, Y.; Watanabe, H. Magnetic resonance imaging of hepatocellular carcinoma. Oncology 2008, 75 (Suppl. S1), 65–71. [Google Scholar] [CrossRef]
- Kutami, R.; Nakashima, Y.; Nakashima, O.; Shiota, K.; Kojiro, M. Pathomorphologic study on the mechanism of fatty change in small hepatocellular carcinoma of humans. J. Hepatol. 2000, 33, 282–289. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhutter, H.G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers (Basel) 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrero, J.A.; Fontana, R.J.; Su, G.L.; Conjeevaram, H.S.; Emick, D.M.; Lok, A.S. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology 2002, 36, 1349–1354. [Google Scholar] [CrossRef]
- Wang, A.G.; Moon, H.B.; Chae, J.I.; Kim, J.M.; Kim, Y.E.; Yu, D.Y.; Lee, D.S. Steatosis induced by the accumulation of apolipoprotein A-I and elevated ROS levels in H-ras12V transgenic mice contributes to hepatic lesions. Biochem. Biophys. Res. Commun. 2011, 409, 532–538. [Google Scholar] [CrossRef]
- Cho, E.S.; Choi, J.Y. MRI features of hepatocellular carcinoma related to biologic behavior. Korean J. Radiol. 2015, 16, 449–464. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar] [CrossRef]
- Yan, X.; Yao, M.; Wen, X.; Zhu, Y.; Zhao, E.; Qian, X.; Chen, X.; Lu, W.; Lv, Q.; Zhang, L.; et al. Elevated apolipoprotein B predicts poor postsurgery prognosis in patients with hepatocellular carcinoma. Oncol. Targets Ther. 2019, 12, 1957–1964. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Retention Time (min) | Liver Tissues | Fold Change/p-Value | ||||
|---|---|---|---|---|---|---|---|
| W Mean | P Mean | T Mean | W vs. P | P vs. T | W vs. T | ||
| Glycolysis; TCA cycle; Pentose phosphate pathway; Lipid biosynthesis | |||||||
| Glucose-1p (glucose-1-phosphate) | 16.7179 | 1.09 × 10−1 | 1.48 × 10−1 | 2.14 × 10−1 | 0.74/1.38 × 10−2 | 0.69/1.97 × 10−2 | 0.51/2.81 × 10−3 |
| Xanthosine | 24.2838 | 1.73 × 100 | 1.10 × 100 | 4.54 × 10−1 | 1.57/2.81 × 10−3 | 2.43/9.13 × 10−4 | 3.81/7.11 × 10−5 |
| Glucose-6p (glucose-6-phosphate) | 21.4070 | 1.69 × 10−2 | 4.56 × 10−2 | 1.68 × 10−1 | 0.37/6.57 × 10−3 | 0.27/3.77 × 10−6 | 0.10/1.50 × 10−5 |
| Fructose-6p (fructose-6-phosphate) | 21.2896 | 2.67 × 10−2 | 7.82 × 10−2 | 2.39 × 10−1 | 0.34/1.15 × 10−3 | 0.33/1.15 × 10−2 | 0.11/9.13 × 10−4 |
| Fructose | 17.8171 | 9.71 × 10−1 | 3.36 × 100 | 3.30 × 100 | 0.29/1.12 × 10−2 | 1.02/9.37 × 10−1 | 0.29/2.45 × 10−2 |
| Citrate (citric acid) | 17.2200 | 1.00 × 10−2 | 1.21 × 10−2 | 4.55 × 10−2 | 0.83/6.84 × 10−1 | 0.27/3.91 × 10−2 | 0.22/3.18 × 10−2 |
| Succinate (succinic acid) | 11.8994 | 1.40 × 10−1 | 6.65 × 10−2 | 6.21 × 10−2 | 2.11/4.43 × 10−3 | 1.07/7.65 × 10−1 | 2.26/2.41 × 10−3 |
| Fumarate (fumaric acid) | 12.2976 | 1.56 × 10−1 | 1.33 × 10−1 | 2.29 × 10−1 | 1.17/3.28 × 10−2 | 0.53/7.11 × 10−5 | 0.68/5.66 × 10−4 |
| Malate (L-Malic acid) | 13.8313 | 2.08 × 100 | 1.81 × 100 | 3.39 × 100 | 1.15/9.40 × 10−2 | 0.53/2.68 × 10−4 | 0.61/1.45 × 10−3 |
| Cholesterol and bile acid biosynthesis | |||||||
| Glycine | 11.8509 | 2.22 × 100 | 1.47 × 100 | 1.63 × 100 | 1.51/3.34 × 10−5 | 0.90/5.04 × 10−1 | 1.37/2.26 × 10−2 |
| Taurine | 15.9588 | 4.25 × 10−1 | 7.66 × 10−1 | 3.44 × 10−1 | 0.55/4.97 × 10−2 | 2.23/3.18 × 10−2 | 1.24/4.32 × 10−1 |
| Glutathione metabolism | |||||||
| 5-oxoproline | 14.4595 | 1.49 × 10−1 | 1.10 × 10−1 | 3.16 × 10−1 | 1.35/3.48 × 10−3 | 0.34/1.60 × 10−4 | 0.47/1.97 × 10−2 |
| L-glutamate (glutamic acid) | 15.2758 | 6.10 × 10−3 | 5.40 × 10−3 | 1.55 × 10−2 | 1.13/4.75 × 10−1 | 0.35/1.47 × 10−2 | 0.39/2.02 × 10−2 |
| Succinate (succinic acid) | 11.8994 | 1.40 × 10−1 | 6.65 × 10−2 | 6.21 × 10−2 | 2.11/4.43 × 10−3 | 1.07/7.65 × 10−1 | 2.26/2.41 × 10−3 |
| Glycine | 11.8509 | 2.22 × 100 | 1.47 × 100 | 1.63 × 100 | 1.51/3.34 × 10−5 | 0.90/5.04 × 10−1 | 1.37/2.26 × 10−2 |
| cys-Gly (L-cysteinylglycine) | 17.2563 | 5.49 × 10−2 | 4.74 × 10−2 | 8.88 × 10−2 | 1.16/5.06 × 10−1 | 0.53/7.09 × 10−3 | 0.62/1.79 × 10−2 |
| mRNA Accession No. | Gene Symbol (Full Name) | Fragments Per Kilobase Million (FPKM) | Fold Change/p-Value | ||||
|---|---|---|---|---|---|---|---|
| W | P | T | W vs. P | P vs. T | W vs. T | ||
| Glycolysis; TCA cycle; Pentose phosphate pathway; Lipid biosynthesis | |||||||
| 001199296 | Acly (ATP-citrate lyase, transcript variant 1) | 2.54 × 101 | 4.57 × 101 | 4.98 × 101 | 0.56/2.45 × 10−2 | 1.09/9.89 × 10−2 | 0.51/1.09 × 10−4 |
| 010209 | Fh1 (fumarate hydratase 1) | 1.37 × 102 | 1.24 × 102 | 5.65 × 101 | 1.10/4.58 × 10−1 | 0.45/9.75 × 10−4 | 2.42/6.23 × 10−5 |
| 008061 | G6pc (glucose-6-phosphatase) | 2.18 × 102 | 9.61 × 101 | 1.13 × 101 | 2.27/5.01 × 10−12 | 0.12/3.62 × 10−13 | 19.33/1.98 × 10−39 |
| 008062 | G6pdx (glucose-6-phosphate dehydrogenase X-linked) | 1.42 × 100 | 3.65 × 100 | 1.69 × 101 | 0.39/6.88 × 10−1 | 0.22/6.03 × 10−4 | 0.08/6.17 × 10−5 |
| 001081274 | Pgd (6-phosphogluconate dehydrogenase) | 7.74 × 100 | 1.28 × 101 | 1.00 × 101 | 1.02/1.00 × 100 | 3.43/8.58 × 10−10 | 0.30/9.60 × 10−10 |
| 011099 | Pkm (pyruvate kinase, muscle) | 5.32 × 100 | 7.12 × 100 | 2.58 × 101 | 0.75/7.91 × 10−1 | 3.62/1.66 × 10−4 | 0.21/1.76 × 10−5 |
| 146006 | Lss (lanosterol synthase) | 2.29 × 101 | 1.35 × 101 | 5.84 × 101 | 1.69/1.42 × 10−1 | 4.32/9.60 × 10−11 | 0.39/2.61 × 10−7 |
| 007856 | Dhcr7 (7-dehydrocholesterol reductase) | 4.91 × 101 | 1.79 × 101 | 6.65 × 101 | 2.74/8.36 × 10−5 | 3.72/3.19 × 10−11 | 0.74/2.80 × 10−3 |
| 138656 | Mvd (mevalonate kinase) | 1.48 × 101 | 3.57 × 100 | 1.96 × 101 | 4.14/1.16 × 10−2 | 5.50/9.23 × 10−5 | 0.75/1.25 × 10−1 |
| 50790 | Acsl4 (acyl-CoA synthetase long-chain family member 4) | 8.57 × 100 | 1.22 × 101 | 3.44 × 101 | 0.70/2.01 × 10−1 | 0.36/5.76 × 10−6 | 0.25/9.19 × 10−4 |
| Cholesterol and bile acid biosynthesis | |||||||
| 007519 | Baat (bile acid-CoA: amino acid acyltransferase) | 1.50 × 102 | 1.09 × 102 | 3.50 × 101 | 1.37/1.30 × 10−2 | 0.32/3.45 × 10−6 | 4.28/6.06 × 10−12 |
| 024264 | Cyp27a1 (sterol 26-hydroxylase) | 1.94 × 102 | 1.13 × 102 | 2.69 × 101 | 1.71/4.56 × 10−6 | 0.24/2.09 × 10−9 | 7.21/1.04 × 10−23 |
| 007825 | Cyp7b1 (25-hydroxycholesterol 7-alpha-hydroxylase) | 3.64 × 102 | 1.91 × 102 | 7.32 × 100 | 1.90/1.38 × 10−13 | 0.04/4.09 × 10−37 | 49.71/7.72 × 10−78 |
| 010012 | Cyp8b1 (7-alpha-hydroxycholest-4-en-3-one) | 1.40 × 102 | 1.06 × 101 | 2.08 × 100 | 13.17/4.69 × 10−30 | 0.20/1.13 × 10−1 | 67.17/1.61 × 10−30 |
| 133943 | Hsd3b7 (3 beta-hydroxysteroid dehydrogenase type 7) | 2.79 × 102 | 7.90 × 101 | 4.26 × 101 | 3.53/1.30 × 10−27 | 0.54/6.85 × 10−2 | 5.47/2.05 × 10−31 |
| 008255 | Hmgcr (3-hydroxy-3-methylglutaryl-coenzyme A reductase) | 3.18 × 101 | 2.31 × 101 | 8.69 × 101 | 1.38/2.88 × 10−1 | 3.76/6.22 × 10−14 | 0.37/8.95 × 10−11 |
| Glutathione metabolism | |||||||
| 172961 | Abat (4-aminobutyrate aminotransferase) | 8.37 × 101 | 4.87 × 101 | 1.67 × 101 | 1.72/2.31 × 10−3 | 0.34/4.02 × 10−3 | 5.02/1.61 × 10−8 |
| 008162 | Gpx4 (phospholipid hydroperoxide glutathione) | 1.83 × 102 | 1.67 × 102 | 3.12 × 102 | 1.10/3.64 × 10−1 | 1.87/5.35 × 10−22 | 0.59/1.86 × 10−18 |
| 008486 | Anpep(alanyl (membrane) aminopeptidase) | 2.57 × 101 | 2.93 × 101 | 5.17 × 101 | 0.88/6.89 × 10−1 | 1.76/3.11 × 10−4 | 0.50/4.85 × 10−5 |
| 008180 | Gss (glutathione synthetase) | 2.87 × 101 | 3.21 × 101 | 5.37 × 101 | 0.90/7.04 × 10−1 | 1.68/4.86 × 10−4 | 0.53/8.80 × 10−5 |
| Metabolites | Liver Tissues | p-Value | ||||
|---|---|---|---|---|---|---|
| W Mean | P Mean | T Mean | P vs. W | W vs. T | P vs. T | |
| Phosphoenolpyruvic acid-M3 | 2.19 × 10−1 | 1.49 × 10−1 | 9.82 × 10−2 | 2.32 × 10−4 | 3.10 × 10−7 | 1.35 × 10−3 |
| Lactic acid-M3 | 1.48 × 10−1 | 1.27 × 10−1 | 8.77 × 10−2 | 7.31 × 10−2 | 3.51 × 10−5 | 1.02 × 10−3 |
| Citric acid-M2 | 1.47 × 10−1 | 1.44 × 10−1 | 1.54 × 10−1 | 2.71 × 10−1 | 1.06 × 10−2 | 5.26 × 10−3 |
| Succinic acid-M2 | 1.68 × 10−1 | 1.14 × 10−1 | 1.21 × 10−1 | 2.45 × 10−6 | 6.11 × 10−6 | 7.27 × 10−2 |
| Fumaric acid-M2 | 1.03 × 10−1 | 9.71 × 10−2 | 1.16 × 10−1 | 8.87 × 10−3 | 2.19 × 10−5 | 6.08 × 10−8 |
| Malic acid-M2 | 1.30 × 10−1 | 1.23 × 10−1 | 1.37 × 10−1 | 1.06 × 10−3 | 2.98 × 10−3 | 7.25 × 10−7 |
| Glyceraldehyde 3-phosphate-M3 | 7.55 × 10−2 | 6.31 × 10−2 | 5.79 × 10−2 | 1.77 × 10−1 | 6.51 × 10−2 | 5.70 × 10−1 |
| Erythrose 4-phosphate -M4 | 7.02 × 10−3 | 1.64 × 10−3 | 0.00 × 100 | 1.07 × 10−3 | 8.30 × 10−5 | 2.00 × 10−5 |
| Ribulose-5-phosphate -M5 | 2.78 × 10−3 | 2.00 × 10−3 | 1.83 × 10−3 | 4.34 × 10−1 | 2.52 × 10−1 | 8.79 × 10−1 |
| Glucose 6-phosphate -M6 | 1.99 × 10−1 | 1.22 × 10−1 | 4.87 × 10−2 | 5.31 × 10−3 | 5.67 × 10−7 | 1.21 × 10−3 |
| Sedoheptulose 7-phosphate -M7 | 7.26 × 10−3 | 4.71 × 10−3 | 1.20 × 10−3 | 2.29 × 10−2 | 2.42 × 10−6 | 8.75 × 10−5 |
| Glucose-M6 | 2.65 × 10−1 | 1.63 × 10−1 | 1.36 × 10−1 | 2.60 × 10−5 | 3.79 × 10−6 | 2.04 × 10−1 |
| Ribose 5-phosphate -M5 | 5.05 × 10−3 | 2.75 × 10−3 | 5.11 × 10−3 | 7.39 × 10−3 | 9.44 × 10−1 | 2.17 × 10−2 |
| Fructose 6-phosphate -M6 | 1.70 × 10−1 | 1.01 × 10−1 | 3.78 × 10−2 | 1.11 × 10−4 | 2.66 × 10−11 | 1.85 × 10−4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, C.; Chen, J.; Li, H.; Fan, T.; Zheng, X.; Wang, H.; Zhang, N.; Liu, Y.; Luo, X.; Wang, J.; et al. Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis. Metabolites 2020, 10, 193. https://doi.org/10.3390/metabo10050193
Lei C, Chen J, Li H, Fan T, Zheng X, Wang H, Zhang N, Liu Y, Luo X, Wang J, et al. Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis. Metabolites. 2020; 10(5):193. https://doi.org/10.3390/metabo10050193
Chicago/Turabian StyleLei, Chuanyi, Jun Chen, Huiling Li, Tingting Fan, Xu Zheng, Hong Wang, Nan Zhang, Yang Liu, Xiaoqin Luo, Jingyu Wang, and et al. 2020. "Role of the Mitochondrial Citrate-malate Shuttle in Hras12V-Induced Hepatocarcinogenesis: A Metabolomics-Based Analysis" Metabolites 10, no. 5: 193. https://doi.org/10.3390/metabo10050193