Enhanced Metabolome Coverage and Evaluation of Matrix Effects by the Use of Experimental-Condition-Matched 13C-Labeled Biological Samples in Isotope-Assisted LC-HRMS Metabolomics

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparison of the Metabolic Composition between Native Control and Toxin Samples
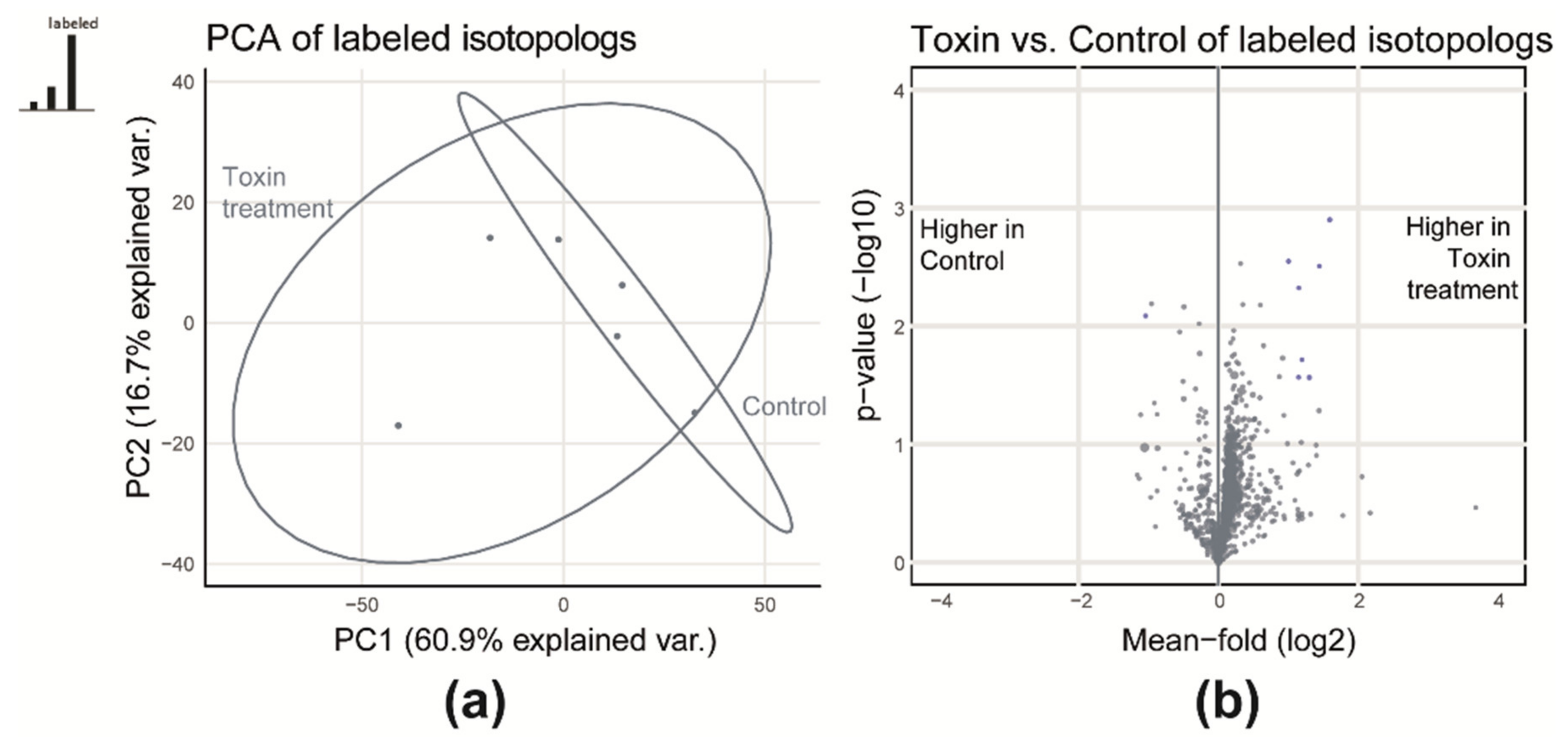
2.1.1. Qualitative Comparison of Control and Toxin Treatment
2.1.2. Comparative Quantification of Control and Toxin Samples
2.2. Investigation of Putative Matrix Effects and Applicability of the Data in Comparative Metabolome Studies
2.2.1. Improvement of Quantification by Internal Standardization
2.2.2. Comparative Statistical Analysis of Native Metabolome Abundances in Toxin and Control Performed with Absolute and Normalized Abundances
2.2.3. Evaluation of Matrix Effects on the Abundances of Labeled Metabolite Signals
3. Materials and Methods
3.1. Chemicals
3.2. Plant Material
3.3. Plant Cultivation
3.4. Sample Preparation for LC-HRMS Analysis
3.5. LC-HRMS Measurement
3.5.1. Liquid Chromatography
3.5.2. Mass Spectrometry
3.6. Data Processing and Statistical Evaluation
3.7. Quality Control
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bueschl, C.; Kluger, B.; Lemmens, M.; Adam, G.; Wiesenberger, G.; Maschietto, V.; Marocco, A.; Strauss, J.; Bodi, S.; Thallinger, G.G.; et al. A novel stable isotope labelling assisted workflow for improved untargeted LC-HRMS based metabolomics research. Metabolomics 2014, 10, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Ouerdane, L.; Meija, J.; Bakirdere, S.; Yang, L.; Mester, Z. Nonlinear signal response in electrospray mass spectrometry: Implications for quantitation of arsenobetaine using stable isotope labeling by liquid chromatography and electrospray Orbitrap mass spectrometry. Anal. Chem. 2012, 84, 3958–3964. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, C.A.; Rubio, V.Y.; Garrett, T.J. Impact of matrix effects and ionization efficiency in non-quantitative untargeted metabolomics. Metabolomics 2019, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.J.; Cho, K.; Nikolskiy, I.; Crawford, P.A.; Patti, G.J. X13CMS: Global tracking of isotopic labels in untargeted metabolomics. Anal. Chem. 2014, 86, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Leeming, M.G.; Isaac, A.P.; Pope, B.J.; Cranswick, N.; Wright, C.E.; Ziogas, J.; O’Hair, R.A.; Donald, W.A. High-resolution twin-ion metabolite extraction (HiTIME) mass spectrometry: Nontargeted detection of unknown drug metabolites by isotope labeling, liquid chromatography mass spectrometry, and automated high-performance computing. Anal. Chem. 2015, 87, 4104–4109. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Capellades, J.; Navarro, M.; Samino, S.; Garcia-Ramirez, M.; Hernandez, C.; Simo, R.; Vinaixa, M.; Yanes, O. geoRge: A Computational Tool To Detect the Presence of Stable Isotope Labeling in LC/MS-Based Untargeted Metabolomics. Anal. Chem. 2016, 88, 621–628. [Google Scholar] [CrossRef]
- Bueschl, C.; Kluger, B.; Neumann, N.K.N.; Doppler, M.; Maschietto, V.; Thallinger, G.G.; Meng-Reiterer, J.; Krska, R.; Schuhmacher, R. MetExtract II: A Software Suite for Stable Isotope-Assisted Untargeted Metabolomics. Anal. Chem. 2017, 89, 9518–9526. [Google Scholar] [CrossRef] [Green Version]
- Saia, S.; Fragasso, M.; De Vita, P.; Beleggia, R. Metabolomics Provides Valuable Insight for the Study of Durum Wheat: A Review. J. Agric. Food Chem. 2019, 67, 3069–3085. [Google Scholar] [CrossRef]
- Allen, D.K.; Bates, P.D.; Tjellstrom, H. Tracking the metabolic pulse of plant lipid production with isotopic labeling and flux analyses: Past, present and future. Prog. Lipid Res. 2015, 58, 97–120. [Google Scholar] [CrossRef] [Green Version]
- Freund, D.M.; Hegeman, A.D. Recent advances in stable isotope-enabled mass spectrometry-based plant metabolomics. Curr. Opin. Biotechnol. 2017, 43, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef]
- Silvestro, L.; Tarcomnicu, I.; Rizea, S. Matrix Effects in Mass Spectrometry Combined with Separation Methods—Comparison HPLC, GC and Discussion on Methods to Control these Effects; InTechs: Piraeus, Greece, 2013; pp. 3–37. [Google Scholar]
- Cech, N.B.; Enke, C.G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Chang, J.S.; Wong, J.W.; Zhang, K.; Krynitsky, A.J.; Bromirski, M.; Wang, J. Effect of sample dilution on matrix effects in pesticide analysis of several matrices by liquid chromatography-high-resolution mass spectrometry. J. Agric. Food Chem. 2015, 63, 5169–5177. [Google Scholar] [CrossRef]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response?: A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, S.; Wang, P.G. Matrix effects and application of matrix effect factor. Bioanalysis 2017, 9, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.W.; Ji, Q.C.; Arnold, M.E. Identifying, Evaluating, and Controlling Bioanalytical Risks Resulting from Nonuniform Matrix Ion Suppression/Enhancement and Nonlinear Liquid Chromatography-Mass Spectrometry Assay Response. Anal. Chem. 2010, 82, 9671–9677. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Denery, J.R.; Nunes, A.A.; Dickerson, T.J. Characterization of differences between blood sample matrices in untargeted metabolomics. Anal. Chem. 2011, 83, 1040–1047. [Google Scholar] [CrossRef]
- Mashego, M.R.; Wu, L.; Van Dam, J.C.; Ras, C.; Vinke, J.L.; Van Winden, W.A.; Van Gulik, W.M.; Heijnen, J.J. MIRACLE: Mass isotopomer ratio analysis of U-13C-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol. Bioeng. 2004, 85, 620–628. [Google Scholar] [CrossRef]
- De Jong, F.A.; Beecher, C. Addressing the current bottlenecks of metabolomics: Isotopic Ratio Outlier Analysis, an isotopic-labeling technique for accurate biochemical profiling. Bioanalysis 2012, 4, 2303–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahieu, N.G.; Huang, X.J.; Chen, Y.J.; Patti, G.J. Credentialing Features: A Platform to Benchmark and Optimize Untargeted Metabolomic Methods. Anal. Chem. 2014, 86, 9583–9589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampler, E.; Coman, C.; Hermann, G.; Sickmann, A.; Ahrends, R.; Koellensperger, G. LILY-lipidome isotope labeling of yeast: In vivo synthesis of (13)C labeled reference lipids for quantification by mass spectrometry. Analyst 2017, 142, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Cano, P.M.; Jamin, E.L.; Tadrist, S.; Bourdaud’hui, P.; Pean, M.; Debrauwer, L.; Oswald, I.P.; Delaforge, M.; Puel, O. New untargeted metabolic profiling combining mass spectrometry and isotopic labeling: Application on Aspergillus fumigatus grown on wheat. Anal. Chem. 2013, 85, 8412–8420. [Google Scholar] [CrossRef] [PubMed]
- Boysen, A.K.; Heal, K.R.; Carlson, L.T.; Ingalls, A.E. Best-Matched Internal Standard Normalization in Liquid Chromatography-Mass Spectrometry Metabolomics Applied to Environmental Samples. Anal. Chem. 2018, 90, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Karlsen, M.; Oiestad, A.M.; Johansen, J.E.; Liu, H.; Strand, D.H. Evaluation of (1)(3)C- and (2)H-labeled internal standards for the determination of amphetamines in biological samples, by reversed-phase ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2014, 1344, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.I.G.; Rodriguez-González, P. Isotope Dilution Mass Spectrometry; Royal Society of Chemistrys: Cambridge, UK, 2013; pp. 12–156. [Google Scholar]
- Rychlik, M.; Asam, S. Stable isotope dilution assays in mycotoxin analysis. Anal. Bioanal. Chem. 2008, 390, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Szarka, S.; Prokai-Tatrai, K.; Prokai, L. Application of screening experimental designs to assess chromatographic isotope effect upon isotope-coded derivatization for quantitative liquid chromatography-mass spectrometry. Anal. Chem. 2014, 86, 7033–7040. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Mashego, M.R.; van Dam, J.C.; Proell, A.M.; Vinke, J.L.; Ras, C.; van Winden, W.A.; van Gulik, W.M.; Heijnen, J.J. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 2005, 336, 164–171. [Google Scholar] [CrossRef]
- Clendinen, C.S.; Stupp, G.S.; Ajredini, R.; Lee-McMullen, B.; Beecher, C.; Edison, A.S. An overview of methods using (13)C for improved compound identification in metabolomics and natural products. Front Plant Sci. 2015, 6, 611. [Google Scholar] [CrossRef] [Green Version]
- Vlahakis, C.; Hazebroek, J.; Beecher, C.; de Jong, F. Isotopic ratio outlier analysis improves metabolomics prediction of nitrogen treatment in maize. Phytochemistry 2019, 164, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Del Carmen Piqueras, M.; Raskind, A.; de Jong, F.A.; Beecher, C.; Bhattacharya, S.K.; Banerjee, S. Quantitative Metabolomics Using Isotope Residue Outlier Analysis (IROA(®)) with Internal Standards. Methods Mol. Biol. 2019, 1996, 41–46. [Google Scholar] [CrossRef]
- Myer, C.; Perez, J.; Abdelrahman, L.; Mendez, R.; Khattri, R.B.; Junk, A.K.; Bhattacharya, S.K. Differentiation of soluble aqueous humor metabolites in primary open angle glaucoma and controls. Exp. Eye Res. 2020, 194, 108024. [Google Scholar] [CrossRef] [PubMed]
- Stupp, G.S.; Clendinen, C.S.; Ajredini, R.; Szewc, M.A.; Garrett, T.; Menger, R.F.; Yost, R.A.; Beecher, C.; Edison, A.S. Isotopic ratio outlier analysis global metabolomics of Caenorhabditis elegans. Anal. Chem. 2013, 85, 11858–11865. [Google Scholar] [CrossRef] [Green Version]
- Ceranic, A.; Doppler, M.; Buschl, C.; Parich, A.; Xu, K.; Koutnik, A.; Burstmayr, H.; Lemmens, M.; Schuhmacher, R. Preparation of uniformly labelled (13)C- and (15)N-plants using customised growth chambers. Plant Methods 2020, 16, 46. [Google Scholar] [CrossRef]
- Doppler, M.; Kluger, B.; Bueschl, C.; Steiner, B.; Buerstmayr, H.; Lemmens, M.; Krska, R.; Adam, G.; Schuhmacher, R. Stable Isotope-Assisted Plant Metabolomics: Investigation of Phenylalanine-Related Metabolic Response in Wheat Upon Treatment With the Fusarium Virulence Factor Deoxynivalenol. Front Plant Sci. 2019, 10, 1137. [Google Scholar] [CrossRef]
- Doppler, M.; Bueschl, C.; Kluger, B.; Koutnik, A.; Lemmens, M.; Buerstmayr, H.; Rechthaler, J.; Krska, R.; Adam, G.; Schuhmacher, R. Stable Isotope-Assisted Plant Metabolomics: Combination of Global and Tracer-Based Labeling for Enhanced Untargeted Profiling and Compound Annotation. Front Plant Sci. 2019, 10, 1366. [Google Scholar] [CrossRef] [Green Version]
- Warth, B.; Parich, A.; Bueschl, C.; Schoefbeck, D.; Neumann, N.K.; Kluger, B.; Schuster, K.; Krska, R.; Adam, G.; Lemmens, M.; et al. GC-MS based targeted metabolic profiling identifies changes in the wheat metabolome following deoxynivalenol treatment. Metabolomics 2015, 11, 722–738. [Google Scholar] [CrossRef] [Green Version]
- Blazenovic, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Macoy, D.M.; Kim, W.Y.; Lee, S.Y.; Kim, M.G. Biotic stress related functions of hydroxycinnamic acid amide in plants. J. Plant Biol. 2015, 58, 156–163. [Google Scholar] [CrossRef]
- Kikot, G.E.; Hours, R.A.; Alconada, T.M. Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: A review. J. Basic Microbiol. 2009, 49, 231–241. [Google Scholar] [CrossRef]
- Contreras, M.D.M.; Yepez, A.J.M.; Martinez, R.R. Determination of deoxynivalenol (DON) in wheat, barley and corn and its relationship with the levels of total molds, Fusarium spp., infestation percentage, and water activity. Arch. Latinoam. Nutr. 2000, 50, 183–186. [Google Scholar]
- Mizuno, H.; Ueda, K.; Kobayashi, Y.; Tsuyama, N.; Todoroki, K.; Min, J.Z.; Toyo’oka, T. The great importance of normalization of LC-MS data for highly-accurate non-targeted metabolomics. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef]
- Chen, D.; Han, W.; Su, X.; Li, L.; Li, L. Overcoming Sample Matrix Effect in Quantitative Blood Metabolomics Using Chemical Isotope Labeling Liquid Chromatography Mass Spectrometry. Anal. Chem. 2017, 89, 9424–9431. [Google Scholar] [CrossRef]
- Han, W.; Li, L. Matrix effect on chemical isotope labeling and its implication in metabolomic sample preparation for quantitative metabolomics. Metabolomics 2015, 11, 1733–1742. [Google Scholar] [CrossRef]
- Doppler, M.; Kluger, B.; Bueschl, C.; Schneider, C.; Krska, R.; Delcambre, S.; Hiller, K.; Lemmens, M.; Schuhmacher, R. Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants. Int. J. Mol. Sci. 2016, 17, 1017. [Google Scholar] [CrossRef] [Green Version]
- Redestig, H.; Kobayashi, M.; Saito, K.; Kusano, M. Exploring matrix effects and quantification performance in metabolomics experiments using artificial biological gradients. Anal. Chem. 2011, 83, 5645–5651. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ćeranić, A.; Bueschl, C.; Doppler, M.; Parich, A.; Xu, K.; Lemmens, M.; Buerstmayr, H.; Schuhmacher, R. Enhanced Metabolome Coverage and Evaluation of Matrix Effects by the Use of Experimental-Condition-Matched 13C-Labeled Biological Samples in Isotope-Assisted LC-HRMS Metabolomics. Metabolites 2020, 10, 434. https://doi.org/10.3390/metabo10110434
Ćeranić A, Bueschl C, Doppler M, Parich A, Xu K, Lemmens M, Buerstmayr H, Schuhmacher R. Enhanced Metabolome Coverage and Evaluation of Matrix Effects by the Use of Experimental-Condition-Matched 13C-Labeled Biological Samples in Isotope-Assisted LC-HRMS Metabolomics. Metabolites. 2020; 10(11):434. https://doi.org/10.3390/metabo10110434
Chicago/Turabian StyleĆeranić, Asja, Christoph Bueschl, Maria Doppler, Alexandra Parich, Kangkang Xu, Marc Lemmens, Hermann Buerstmayr, and Rainer Schuhmacher. 2020. "Enhanced Metabolome Coverage and Evaluation of Matrix Effects by the Use of Experimental-Condition-Matched 13C-Labeled Biological Samples in Isotope-Assisted LC-HRMS Metabolomics" Metabolites 10, no. 11: 434. https://doi.org/10.3390/metabo10110434