
Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases


1
Pharmaceutical Sciences Department, Fakeeh College for Medical Sciences, Jeddah 21461, Saudi Arabia
2
Pharmaceutical Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Cairo 11884, Egypt
Sci. Pharm. 2023, 91(2), 18; https://doi.org/10.3390/scipharm91020018
Submission received: 14 February 2023
/
Revised: 7 March 2023
/
Accepted: 14 March 2023
/
Published: 28 March 2023
(This article belongs to the Special Issue Feature Papers in Scientia Pharmaceutica)
Abstract
:Cancer is a large group of diseases that can affect any organ or body tissue due to the abnormal cellular growth with the unknown reasons. Many of the existing chemotherapeutic agents are highly toxic with a low level of selectivity. Additionally, they lead to development of therapeutic resistance. Hence, the development of targeted chemotherapeutic agents with low side effects and high selectivity is required for cancer treatment. Quinazoline is a vital scaffold well-known to be linked with several biological activities. The anticancer activity is one of the prominent biological activities of this scaffold. Several established anticancer quinazolines work by different mechanisms on the various molecular targets. The aim of this review is to present different features of medicinal chemistry as drug design, structure activity relationship, and mode of action of some targeted anticancer quinazoline derivatives. It gives comprehensive attention on the chemotherapeutic activity of quinazolines in the viewpoint of drug discovery and its development. This review provides panoramic view to the medicinal chemists for supporting their efforts to design and synthesize novel quinazolines as targeted chemotherapeutic agents.
Keywords:
quinazoline; development; discovery; design; synthesis; anticancer; structure activity relationship1. Introduction
Nowadays, cancer is the most terrifying disease affecting the mankind. It is a group of diseases that arise from the abnormal cell growth that has an ability to spread to any part of the body [1]. Research on cancer represents 4% of the whole studies in the world. This area of research expands year after year to include more studies which reflects the global importance of this research area [2]. As cancer is a complicated disorder, there are several difficulties in the treatment process [3]. Many anticancer agents are used alone or in combination with other agents [4]. Understanding the molecular targets, and the cellular proliferation over the past 50 years has triggered the development of over 100 FDA-approved anticancer agents [5]. The earlier anticancer medications as alkylating agents, antifolates, and antimitotic agents were used for treatment of lymphoma and leukemia. These classes of chemotherapeutic agents had a lot of toxic effects with low selectivity and developed resistance [6]. Targeted therapy approach was used to produce more selective, less toxic, and highly effective anticancer agents [7]. Heterocyclic compounds represent the biggest group in the field of medicinal chemistry for treatment of diseases and infections [8,9,10,11]; among this group, quinazoline moiety [12,13,14]. Quinazoline is a heterocyclic system having two aromatic six-membered rings. One of them contains two nitrogen atoms named as pyrimidine ring and this ring is fused to the second aromatic benzene ring [15]. Therefore, quinazoline is a phenyl pyrimidine compound [16]. The quinazoline system can be divided into three members, the 2-quinazolinone containing a carbonyl group at the C-2 (1), the 4-quinazolinone containing a carbonyl group at the C-4 (2), and the 2,4-quinazolinedione containing two carbonyl groups at the C-2 and the C-4 (3). (Figure 1) [17]. Quinazolines have a wide range of pharmacological activities [18,19,20]. They are used as anticancer, antiviral, antibacterial, antitubercular, analgesic, antihypertensive, anti-inflammatory, antidiabetic, sedative-hypnotic, antihistaminic, anticonvulsant, and many other uses [19,20,21,22,23]. The aim of this review is to collect the literatures reported by researchers on quinazoline derivatives, their pharmacological activities, and their structure–activity relationships as chemotherapeutic agents targeting tyrosine kinases.
2. Therapeutic Importance of Quinazolines
Quinazoline molecules form a favored group of multi acting therapeutic agents in the pharmaceutical and biological fields. The easy preparation and the diverse spectrum of pharmacological activities made this scaffold very important among the different therapeutic agents [20,21,22,23]. Placing different substituents at the 4, 6, and 7-positions of the quinazoline system is the basic approach for the development of novel agents having anticancer activity [21,22]. There are many quinazoline derivatives still in the clinical phase for treatment of different diseases. The physicochemical characters of 4-aminoarylquinazoline are shown in the Table 1 [24]. The 4-aminoarylquinazoline (Figure 2) is the core structural feature of many FDA-approved anticancer agents which are illustrated in the Table 2 [23]; among these agents, erlotinib, gefitinib, Afatinib, dacomitinib, and many other agents. These agents are currently used for treatment of different types of cancer such as colon, breast, prostate, and lung cancer. Figure 1 shows some of the pharmacological uses of quinazoline derivatives [23,25].
3. Physicochemical Characters of the Core Structural Feature of Anticancer Quinazolines
4. Methods of Preparation of Quinazolines
Due to the enormous synthetic importance and the various therapeutic activities of quinazoline derivatives, several efforts have been made by many researchers to prepare a library of these molecules. Griess et al. synthesized the first derivative of 2-cyano-quinazolin-4-one in 1869 [27]. Then, Bischler and Lang prepared quinazoline by decarboxylation of quinazoline-2-carbocylic acid [28]. In 1903, Gabriel synthesized quinazoline by reduction of o-nitrobenzylamine to o-aminobenzylamine which condensed with formic acid to yield dihydroquinazoline, additional oxidation gave quinazolin-4-one [29]. Several synthetic strategies have been utilized for preparation of quinazoline molecules [30,31,32,33,34,35,36]. The following synthetic reactions show some common traditional methods for preparation of quinazolines:
- Transition metals-catalyzed method. The nitrobenzamide derivative is reduced by palladium chloride (PdCl2) and iron pentacarbonyl Fe(CO)5 in presence of iodobenzene to give 2-phenyl-4(3H)-quinazolinone. This reaction is performed by a microwave-assisted reaction at 110 °C for 0.5 h with 85% yield (Figure 9) [36].
5. Mode of Action of Quinazolines as Anticancer Agents
Quinazolines constitute a promising group of anticancer agents active against different types of tumors with an effective therapeutic activity [37,38,39,40]. The majority of quinazolines anticancer research focused on the mechanistic pathways of their chemotherapeutic action. Most of the quinazoline derivatives having the anticancer activity were found as protein kinase inhibitors. They cause inhibition of replication and transcription of DNA to prevent a tumor growth. Additionally, some of these anticancer derivatives overcome the breast cancer resistance by inhibition of breast cancer resistant proteins [38]. There are other enzymes inhibited by anticancer quinazolines such as thymidylate synthase, poly ADP-ribose polymeras-1 (PARP), topoisomerase. Therefore, quinazolines exerted their chemotherapeutic activity by various molecular interactions (Figure 13) [40] and mechanistic pathways (Figure 14) [39].
5.1. Crystallographic Studies of Quinazolines
Shewchuk et al. revealed the modes of binding for two members of quinazoline tyrosine kinase inhibitors by X-ray crystallographic study. The two inhibitors were hydroxyaniline-6,7-dimethoxyquinazoline in complex with cyclin-dependent kinase 2 (CDK2) and the methylsulfanylaniline-6,7-dimethoxyquinazoline in complex with the p38 kinase. The 4-anilinoquinazoline moiety in the two inhibitors was attached in the ATP site with the quinazoline ring system oriented along the peptide strand that connects the two domains of the protein and with the anilino substituent projecting into a hydrophobic pocket within the protein interior. In each case, the nitrogen at position-1 of the quinazoline accepted a hydrogen bond from a backbone NH (CDK2, Leu83, p38, Met109) of the domain connector strand, and aromatic hydrogen atoms at C2 and C8 interacted with backbone carbonyl oxygen atoms of the peptide strand. The aniline group of the CDK2-bound compound was basically coplanar with the quinazoline nucleus system and occupied a pocket between Lys33 and Phe80. Regarding the p38-bound inhibitor, the aniline group was pointed out of plane and was located between Lys53 and Thr106 in a mode like that detected for the aryl substituent of the pyridine-imidazole inhibitor (Figure 13) [40].
Cathrine et al. compared the crystal structure of p38 pound to four different compounds of quinazolinone and pyridol-pyrimidine derivatives. They found that binding of these specific molecules is characterized by a peptide flip between Met109 and Gly110 which explain the selectivity of these molecules [41].
5.2. Protein Kinases Inhibitors
Protein kinases are a group of enzymes utilizing ATP as a source of phosphate and phosphorylate certain types of amino acids in various types of proteins. They accomplish some conformational changes in proteins to regulate their biological functions. Human genome includes more than 500 types of protein kinases [42]. In 1980, discovery of the various naturally isolated protein kinase inhibitors anticancer agents such as erbastatin, genistein, quercetin led to the development of novel chemotherapeutic agents working on protein kinase enzymes [43]. Based on the amino acid chain that undergoes phosphorylation, there are three types of protein kinases:
- Tyrosine kinases responsible for phosphorylation of phenolic hydroxyl (OH) group.
- Serine-threonine kinases responsible for phosphorylation of serine and threonine amino acids.
- Histidine-kinases responsible for phosphorylation of nitrogen in histidine residues.
It has been known that mutation in the protein kinases leads to impaired signaling, uncontrolled proliferation of cells, and uncontrolled differentiation of these cells. Accordingly, inhibition of tyrosine kinases could be an important biological target in cancer treatment. Several quinazolines produce their anticancer activity through inhibition of different kinases as shown in Figure 14 [42,43].
Tyrosine kinases (TK) include an essential part of oncoproteins associated with the various types of malignancies. Hence, they were selected as a promising biological target for anticancer agents [44]. There are two forms of protein tyrosine kinase, transmembrane receptor linked and non-receptor tyrosine kinase (nRTK). More than 20 types of receptor tyrosine kinases (RTK) were discovered [45]. Binding the ligand with one of these extracellular receptors of tyrosine kinase (RTK) results in dimerization of receptors. This process triggers the cytoplasmic tyrosine kinase to phosphorylate different types of tyrosine residues which in turn initiate various cell signaling pathways such as phosphoinositide 3-kinases (PI3Ks), the mitogen-activated protein kinase (MAPK), and signal transducer activator of transcription 3 (STAT3). On the other hand, the non-receptor tyrosine kinase (nRTK) are enzymes having an important role in regulation of cell growth, differentiation, migration, adhesion, and apoptosis [46].
Quinazoline molecules target the (RTKs) which involve the following receptors [47]:
- Epidermal growth factor receptor (EGFR);
- Platelet derived growth factor receptor (PDGFR);
- Vascular endothelial growth factor receptor (VEGFR);
- Fibroblast growth factor receptor (FGFR).
These RTKs were found to be overexpressed in different types of malignancies such as prostate, colon, breast, lung, stomach, and ovarian cancer [48]. The 4-anilinquinazoline derivatives displayed an ability to inhibit these types of PTKs such as EGFR, VEGFR-2, PDGFR, and FGFR. Accordingly, these derivatives were widely explored as anticancer agents against various types of tumors (Figure 15) [47,48,49].
5.2.1. Epidermal Growth Factor Receptor (EGFR) Inhibitors
Epidermal growth factor receptor (EGFR) is a one of the transmembrane receptors-linked tyrosine kinase (TKRs), and it is an important biological target for many anticancer agents due to its role in controlling cellular proliferation, survival, and anagenesis [50]. It is activated by EGF ligand and transforming growth factor α (TGF-α). When the ligand binds to this receptor, the activation and the dimerization processes are performed through autophosphorylation of Tyr-1068 residue, followed by activation of intracellular signaling series [51]. Overexpression of EGFR characterizes the cancerous cells from the normal cells. Consequently, inhibition of proliferation cancerous cells could be obtained by inhibition of the following two types of EGFR inhibitors [52]:
- Tyrosine kinase inhibitors molecules which act as a competitive inhibitor on EGFR;
- Monoclonal antibodies which interfere with the binding of EGF and TGF-α.
Quinazolines act as small tyrosine kinase inhibitor molecules. Many marketed quinazoline derivatives such as gefitinib, erlotinib, and lapatinib belong to this class of anticancer agents [53]. These compounds bind reversibly with the ATP binding area, interfere with its binding site, and then inhibit the biological activity of EGFR TK [54]. After years of treatment with gefitinib and erlotinib, development of EGFR mutations may lead to progress of the disease. This development results in resistance of the cancerous cells toward these anticancer agents. For this reason, a second generation of irreversibly binding anticancer quinazolines was developed. These molecules bind with EGFR TK binding sites by irreversible covalent bond resulting from the α, β carbonyl groups [55]. Afatinib is an example of these derivatives. It was approved by FDA for treatment of advanced non-small cell lung cancer (NSCLC) including EGFR mutations [54,55,56]. Dacomitinib is another example of the second generation which was approved by FDA. Moreover, canertinib is a quinazoline derivative third-generation anticancer agent, displaying a potent activity with IC50 = 0.8 nM. Structures of the previously mentioned derivatives are shown in (Figure 16) [55,56].
Several studies were performed for discovery of novel quinazoline molecules-based EGFR TK inhibitors. The most modifications were carried out through changing the substituents at the C-6 and the C-7 positions of the anilinoquinazoline moiety. The following literature reports discuss the recent progression in chemotherapeutic activity of quinazoline derivatives as tyrosine kinases inhibitors.
6,7-Substituted-4-anilinoquiazolines as Anticancer Agents
Chilin et al. prepared some dioxygenated derivatives by introducing two dioxo groups at the C-6 and the C-7 positions of the quinazoline nucleus to investigate their antiproliferative activity by their ability to inhibit the EGFR TK. Compound (4) having a dioxane ring and a trifluoromethyl group at the C-3 of the anilinoquinazoline ring showed the highest activity of IC50 = 0.77 μM, and 7.1 μM against A431, and NIH313 cell lines, respectively [38]. Ongoing with this strategy, other derivatives of quinazoline were prepared via replacement of the aniline with the biphenylamino group or by expanding or contracting the deoxygenated ring. Among these derivatives, compounds (5)–(7) gave the highest cytotoxic activity (Figure 17) [38,39].
Conconi et al. designed a group of 6,7-dialcoxy-4-phenylamino-quinazolines to be well-fitted inside the EGFR TK binding site. Compound (8) was found to be the highest active EGFR TK inhibitor. This derivative showed good activity with low bioavailability due to its poor water solubility [39]. A novel series of para-substituted anilinoquinazoline containing 6,7-dialcoxy groups was synthesized by Abouzeid and Shouman to be tested against the human breast cancer cell MCF-7 as an EGFR TK inhibitor. The compound containing thiazolyl sulfanilamide moiety (9) showed high potency (IC50 = 0.13 nM). The molecular modeling study of this compound displayed good binding affinity with the ATB binding site of EGFR-TK like the standard drug lapatinib with an additional hydrogen bond between N of thiazole group and water [42]. Another work by Lu et al. showed the modification of 6,7-dialkoxy-substituted-4-anilinoquinazoline to 3-chloro-4-fluorophenyl-6,7-dimethoxyquinazolin-4-amine (10). This compound showed an activity of IC50 = 3.8 nM against EGFR TK. Placing ethylenediamine at the C-6 instead of the alkoxy group maintained the activity due to the formation of hydrogen bonds between ethylenediamine group and Asp776, Cys773 in the binding site of EGFR [43]. As a continuation for this work, Zahang et al. introduced urea at the C-6 position instead of the alkoxy group as shown in the compounds (11–14). These compounds displayed good inhibitory activity ranging from 0.024–1.715 μM [44]. Acyclic amine substituted derivatives (11 and 12) completely blocked the phosphorylation process of EGFR-TK in the A431 cell line (IC50 = 0.01 μM), while cyclic amine substituted derivatives (13 and 14) blocked the phosphorylation process of EGFR in the NCI-H1975 cell line (IC50 = 10 μM). The structural activity relationship (SAR) study showed that side chain with a heteroatom forms a hydrogen bond. Cyclization of the terminal N decreased the inhibitory activity which may resulted from the long side chain that had no ability to form hydrogen bonds [44]. The anticancer quinazolines 8–14 are shown in (Figure 18) [39,40,41,42,43,44].
The 2-nitroimidazole moiety was used as a substituent at the position 7 from the anilinoquinazoline system (15). The resulted compound showed a strong EGFR-TK inhibitory activity (IC50 = 0.47 nM). This excellent activity was noticed in case of some tumors associated with hypoxia against A549 cell line (IC50 = 0.77 and 0.18 μM), HT-29 cell line (IC50 = 1.13 and 0.18 μM) compared to the standard erlotinib against A549 cell line (IC50 = 7.59 and 9.1 μM), HT-29 cell line (IC50 = 2.98 and 4.37 μM) [45].
The literatures reported development of resistance in the most of cancer patients after one year from treating with gefitinib. There are some mechanisms that tried to explain this process, but the exact reason is still unknown. Emergence of secondary mutations resulted in deviation in the downstream signals and generation of alternative pathways. As a trial to overcome this resistance, Yu et al. designed novel anilinoquinazoline derivatives having a hydrophobic group at the C-4 position of the quinazoline ring (16). All the newly synthesized compounds strongly inhibited EGFR type (wt) with IC50 ranging from 1.12–15.4 nM compared to the standard gefitinib of 15.5 nM. The compound containing N-adamantly benzamide ring displayed a potent inhibition against the resistant cell line H1975 and A431 with IC50 = 5.89 and 2.06 μM, respectively. Additionally, it produced a strong inhibitory activity against the EGFR-TK mutation L858R/T790M (IC50 = 4.62 μM). The modeling study revealed that the 1-adamantyl moiety at the para position occupied the hydrophobic region in the binding pocket then, the aniline part became closer to the Met790 amino acid which supported the binding process with the EGFR [46].
Other novel analogues of gefitinib were synthesized to be investigated as EGFR inhibitors. Among these analogues, 4-benzothienylamino quinazoline derivatives showed different secondary amino propoxy side chain at the C-6 and the C-7 of the quinazoline moiety. These derivatives gave a good anticancer activity against six human cancer cell lines, but their activity decreased compared to the standards gefitinib and erlotinib. Compound (17) produced a cytotoxic activity with IC50 = 1.32 μM and induced an apoptosis in the MiaPaCa2 cell line. The 7-amino propoxy side chain derivatives were more active than the 6-amino propoxy side chain derivatives [47]. The anticancer quinazolines 15–17 are shown in (Figure 19) [45,46,47].
Another way for the development of EGFR inhibitors was performed by Zhao et al. through placing the heterocyclic ring azaspirocycle or azetidine instead of the morpholine ring in the anticancer drug gefitinib. The newly synthesized molecules were tested against EGFR, HCC827, and A549 cancer cell lines using the standard gefitinib. The compound containing azaspirocycle (18) showed an excellent activity with IC50 = 15 nM and 28 nM against EGFR and HCC827 respectively, while it showed no activity against A459. The biological results showed that the inhibitory activity of EGFR was retained even by introduction of the four-membered heterocyclic ring instead of the morpholine ring of gefitinib. Additionally, these heterocyclic rings at the 6-position improved the water solubility [48].
To develop water solubility, different hydrophilic amino substituents were introduced at the C-7 of the anilinoquinazoline system. Out of this series, compound (19) having 4-phenylpiperidine moiety via a propyl chain at the C-7 of the anilinoquinazoline system displayed a strong cytotoxic activity against HepG2, A549, MCF-7, DU145, and SH-SY5Y cancer cell line at 5–10 μM. The cytotoxic activity of compound (19) against EGFR was 3.62 nM compared to gefitinib (IC50 = 2.21 nM). The structural activity relationship study (SAR) explained that the ortho and the para-cyano groups on the aniline ring showed higher activity than the meta position due to the formation of a hydrogen bond with the amino acids of the binding pocket [49].
In 2013, Cai et al. designed new derivatives of 4-benzothiazoleaminoquinazolines for biological screening as EGFR inhibitors. Their EGFR inhibitory activity was lower than that of the standard drugs gefitinib and erlotinib, but their inhibitory activity against SCR and ABI was considerable (SCr = 91.8% and AB1 = 82.3%) at 1 μM. The SAR study showed that presence of bulky substituents at the 6 or the 7 positions of the quinazoline moiety increased the potency [50]. The anticancer quinazolines 18–20 are shown in (Figure 20) [48,49,50].
Several studies were performed to overcome the resistance resulting from the first-generation EGFR inhibitors. Afatinib anticancer emerged as a lead compound for synthesis of the irreversible second-generation EGFR inhibitors.
The irreversible binding resulted from the formation of a covalent bond between the Michael acceptor moiety and the cysteine amino acid in EGFR binding site. Among these irreversible EGFR inhibitors, compounds (21) and (22) displayed a strong inhibition against EGFR resistant cells H197, IC50 = 10.2, and 16.1 nM, respectively. The SAR study revealed that 3-ethylaniline and 3-chloro-4-fluoroaniline groups are the best substituents for the inhibitory activity [51].
Hou et al. synthesized 4-anilinoquinazoline compounds having benzamide group at the 6-position of the quinazoline moiety. Among these compounds, compound (23) showed a potent activity with IC50 = 5 nM, and it was also effective against cell lines having EGFR mutations. The SAR study displayed that the irreversible covalent bond formed between the benzamide group of this compound and the sulfhydryl group (SH) of the Cys797 residue of EGFR binding site [52].
A novel series of the naproxen-erlotinib conjugates (24) was synthesized to target more than one pathway involved in the development of cancer. Naproxen works as a nonsteroidal anti-inflammatory drug (NSAIDs), while erlotinib is a quinazoline-based anticancer drug. The conjugated compounds were tested against A431 and HCC827 cancer cell lines. All the derivatives showed a promising EGFR inhibitory activity that ranged from IC50 = 0.005–0.88 μM. Pharmacokinetic studies explained that the hydrolysis of this conjugate gave the hydroxylated form of erlotinib with a strong activity IC50 = 0.001 μM. The presence of naproxen at the C-6 position was better than the C-7 position [53]. The anticancer quinazolines 21–24 are shown in (Figure 21) [51,52,53].
Other derivatives of the morpholin-3-one fused quinazoline molecules (25) were prepared by Qin et al. as EGFR inhibitors. Out of these molecules, compound (25) showed the highest activity against EGFR kinase (IC50 = 53.1 nM). Molecular modeling study revealed a good docking of this compound into the binding pocket of EGFR such as the standard drug gefitinib [54].
Another series of propen-phenylamino-quinazoline molecules having similar fragments at the 6 and the 7 positions of the quinazoline moiety was designed and prepared to be investigated as EGFR inhibitors. Among these derivatives, compound (26) showed IC50 = 1.35–8.83 μM against the four tested cell lines A431, A549, NCI-H1975, and SW480. In addition, it showed a strong EGFR inhibitory activity (IC50 = 20.72 nM) better than the standard lapatinib (IC50 = 27.06 nM). The Western blot analysis revealed an inhibition of EGF induced EGFR in the two cell lines A549 and NCI-H1975 [55].
These results encouraged them to synthesize a novel series of 6,7-dimorpholinoalkyl quinazoline compounds. Compound (27) having 3-chloro-4-(3-fluorophenyl)aniline produced an excellent EGFR inhibitory activity with IC50 = 7, 9.3 nM against the two cell lines wt and T790M, respectively [55]. The anticancer quinazolines 25–27 are shown in (Figure 22) [54,55].
6-Substituted-4-anilinoquinazoline Derivatives
The mono substituted anilinoquinazoline derivatives at the 6-position of the quinazoline system were designed and synthesized as EGFR inhibitors. Among this series, compound 28 has an alkynyl group at the 6-position from the anilinoquinazoline system. This compound showed a potent inhibitory activity with IC50 = 14.1 nM higher than the reference gefitinib (IC50 = 39 nM) [56].
Another series of the 4-anilinoquinazolines containing dioxygenated rings at the 6-position was synthesized by Liu et al. The dioxygenated group is attached to the 4-anilinoquinazoline system via an amide linkage. Additionally, there are different substituents at the 3-position such as chlorine or bromine substituents. The compound (29) having dioxepine ring at the 6-position and bromine at the 3-position showed a significant EGFR inhibitory activity of IC50 = 0.098 μM. It also showed IC50 = 2.77, and 5.02 μM against A431 and MCF-7 cancer cell lines, respectively. The Western blot analysis at 100 nM displayed a complete inhibition of the EGFR autophosphorylation [57].
Continuing with this strategy, Li et al. prepared benzylamino-substituted-4-anilinoquinazoline having hydroxy group (OH) at 4-position (30). This compound gave strong cytotoxic activity with IC50 = 0.28 μM against Hep G2, and 0.59 μM against A16-F10 cell line. It also gave significant EGFR inhibitory activity with IC50 = 0.87 μM. The SAR study explained the essential role of meta-bromo-substituted-4-anilino moiety. The para substitution on the benzyl rig was more active than ortho substitution. The hydroxy substitution of benzyl ring was more active than the methoxy substituted form [58]. The anticancer quinazolines 28–30 are shown in (Figure 23) [56,57,58].
Later in 2011, new derivatives of cinnamic acid-substituted anilinoquinazoline were designed as EGFR inhibitors. Out of these derivatives, compounds (31) and (32) have a bromo or a chloro substitution at the 3-position of the aniline ring, producing a potent EGFR inhibitory activity of IC50 = 0.12 and 0.19 μM, respectively. The activity of the standard erlotinib was IC50 = 0.03 μM [59]. They also showed IC50 = 0.33, and 0.49 μM against A431 cancer cell line. The SAR study revealed that the meta or the ortho substituted rings were more potent than the para substituted derivatives [60]. In addition, compound (33) with a 4-chloro-6-ureidoquinazoline moiety displayed a potent EGFR inhibitory activity against EKVX, NCI-H322M, A498, TK-10, and MDA-MB-468 cell lines ranging from 0.37–1 μM [61]. Compound (34) was synthesized by Zhang et al. It is an anilinoquinazoline derivative with the 4-aryl-amino-6-(furan-2-yl) substitution. The EGFR inhibitory activity of this compound was IC50 = 5.06 nM. Molecular modeling study revealed the binding interaction of this derivative was like that of the standard erlotinib [62]. The anticancer quinazolines 31–34 are shown in (Figure 24) [59,60,61,62].
Structural activity relationship studies (SAR)
- The 4-anilinoquinazoline with substitution at the C-6 and/or the C-7 positions is the general pharmacophoric group required for the EGFR inhibitory activity. These structural requirements are shown by the common tyrosine-kinase inhibitors such as gefitinib, erlotinib, and other anticancer pharmaceutically marketed products.
- The electron-withdrawing groups such as fluoro, bromo, chloro, and ethylene at the aniline ring is advantageous for the antiproliferative activity.
- The 3-bromo substituted quinazoline molecules displayed potent activity.
- The 3-chloro-4-fluoro-aniline substituted quinazoline molecules showed strong activity.
- Changing the aniline moiety at the 4-position with other groups decreased the activity.
- The electron donating groups at the 6 and/or the 7-positions improved the binding activity of N1 and N3 of quinazoline system with the binding pocket.
- The propoxy linker at the C-6 and/or the C-7 of quinazoline moiety showed stronger activity than the methoxy group.
- Dioxygenated groups at the 6 and the 7 positions of quinazoline moiety improved the cytotoxic activity.
- The Michael addition group at 6-position of quinazoline leads to irreversible binding with the receptor-site.
5.2.2. Vascular Endothelial Growth Factor Receptors (VEGFR) Inhibitors
The VEGFR are a group of vascular endothelial growth factor receptors. They are associated with the angiogenesis process which include formation of new blood vessels (neovascularization) [63]. It is also responsible for some important physiological and pathological stages [64]. They are produced by the vascular endothelial growth factor (VEGF) and required for the growth of the different tumors. The EGFR stimulation and hypoxia stimulate the VEGFR production by cancer cells [65]. The binding site of the VEGF is the VEGFR-2 tyrosine kinase. When the binding process happen, it activates the autophosphorylation process involved in the proliferation of cancer cells [66]. The VEGF overexpression is associated with the progression of tumors in several types of cancer. Therefore, targeting the VEGF is a main strategy of second-generation anticancer agents which have a dual activity through an inhibition of the EGFR TK and the VEGFR TK like vandetanib. Cediranib is another quinazoline derivative working as VEGF inhibitor [67]. Cediranib is a quinazoline-based marketed anticancer agent having an indole-ether moiety joined to the quinazoline system at the 4-position. It inhibits VEGFR-TK [68]. The anticancer quinazolines vandetanib and cediranib are shown in (Figure 26) [67,68].
The inhibitors having urea and thiourea based 4-arylquinazolines work as dual inhibitors for both EGFR and VEGFR [68]. Figure 27 [69] illustrates the effect of each group on the biological activity based on the SAR study of the dual acting quinazolines.
Garofalo et al. designed novel derivatives of quinazoline VEGFR inhibitors by introduction of aryloxy, aniline, and N-methylaniline fragments at the 4-position of the quinazoline system. The urea-substituted aryloxyquinazolines were 100-fold more active than the anilinoquinazoline against the VEGFR-2. Among these derivatives, naphthyl-substituted derivative (37) showed activity with IC50 = 30 nM [69]. The same research group carried out further modification by placing methyl or halogen substituents on the aryloxy ring to see their activity against the VEGFR-2. A remarkable inhibition was observed by the methyl derivative of the compound (37) which gave IC50 = 2 nM [70]. Some new derivatives of the 7-aminoalkoxy-4-aryloxy-quinazolines were synthesized as multi-tyrosine kinase inhibitors. The piperidinopropoxy substituted derivative (38) produced a potent activity with IC50 = 4.09 μM, 1, 5.02 μM, and 0.33 μM against PC3, MCF7, HT29, and HUVEC cancer cell lines, respectively [71].
In 2018, Sun et al. prepared substituted thiourea quinazoline-based derivatives as dual inhibitors of EGFR and VEGFR-2. Among these derivatives, compound (39) having thiourea linked by ether linkage at the position-4 of the quinazoline scaffold gave the highest potency of IC50 = 0.02 and 0.05 μM against EGFR and VEGFR-2, respectively. The compound 40 also displayed IC50 = 0.01 and 0.08 μM against the EGFR and the VEGFR-2, respectively. In addition, these two compounds showed potent cytotoxicity against other cancer cell lines such as HCT-116, MCF-7, and B16. The SAR study revealed the importance of the two electron withdrawing groups at the terminal phenyl ring 3-CF3, 4-Cl of (39) and 3-CF3, 4-Br of the compound 40 [72]. The anticancer quinazolines 37–40 are shown (Figure 28) [72].
The dual inhibition of the two enzymes EGFR and FGFR-2 is a very useful strategy for treatment of resistant cancer cases since they are targeting different signaling pathways to inhibit the tumor growth.
Barbosa et al. synthesized the 2-chloro-4-substituted-anilinoquinazoline derivatives containing a hydrogen bond donor and acceptor groups at the 4-position of the terminal phenyl ring to interact with the suitable target. Compound 41 gave a strong potency with IC50 = 1.63, 0.85 μM against the EGFR and the VEGFR, respectively [73].
Other derivatives of the diarylamide-substituted-4-anilinoquinazoline were prepared to be investigated as dual EGFR and FEGFR-2 inhibitors. The antiproliferative activity of this compound was assessed by MTT assay against HT-29, MCF-7 and H460 cell lines. The compounds 42 and 43 having a methyl piperazine substituent at the position-7 of quinazoline displayed the highest activity of IC50 = 0.13, 0.56 μM against HT-29, MCF-7 for compound 42, IC50 = 0.15, 1.81 μM against HT-29, MCF-7 for compound 43 [74]. The anticancer quinazolines 41–43 are shown in (Figure 29) [73,74].
In search of new dual inhibitors, some derivatives having diarylurea group at the 4-position of the terminal phenyl ring attached to the quinazoline moiety were designed and tested as a dual inhibitor against the EGFR and the VEGFR. It was found that the derivative with meta and para dimethyl fragments on the terminal phenyl ring and chloro substituent ortho to the urea group (44) produced an excellent activity with IC50 = 1 nM against EGFR, and 79 nM against the VEGFR. The compound 45 showed IC50 = 51 nM against EGFR, and 14 nM against VEGFR compared to the reference drug vandetanib IC50 = 11, 15 nM against EGFR, and VEGFR, respectively. The SAR study revealed that the derivatives possessing the 4-methylpiperazine group at the position-7 from the quinazoline moiety with a diaryl urea substituent displayed a better antiproliferative activity than the derivatives possessing morpholine or piperidine groups [75].
The substitution at the 5-position of the quinazoline scaffold instead of the 4-position was studied by Xi et al. He synthesized the 5-anilino-8-nitroquinazoline derivatives to be investigated for their antiproliferative activity against the EGFR and the VEGFR-2. The compound (47) showed a strong and a selective inhibitory activity with IC50 = 12 nM against the EGFR kinase enzyme, and IC50 = 1.8 μM against HUVEC cell line [76].
Hybrid derivatives of quinazoline-indazole were synthesized by Elsayed et al. as VEGFR-2 inhibitors. The molecule (46) displayed a potent cytotoxic activity with IC50 = 5.4 nM against the VEGFR-2 kinase enzyme. It also showed 130% growth inhibition on the full NCI panel of cancer cell lines when exposed to in vitro antiproliferative assay. In addition, a 99.6% was shown against HUVEC cancer cell line at 10 μM [77]. The anticancer quinazolines 44–47 are shown in (Figure 30) [75,76,77].
SAR of VEGFR Inhibitors
- Quinazolin-4-aniloino or quinazoline-4-oxyaryl scaffold is required for VEGFR inhibitory activity.
- Substitution at the 6-position of the quinazoline moiety with an electron-releasing group enhances the activity.
- Substitution at the 7-position of the quinazoline with an aminoalkoxy group increases the activity.
- Aromatic spacer between urea or thiourea and N or O at the 4-position of quinazoline is necessary for the activity.
5.2.3. PDGFR Inhibitors
PDGFR is a type of cell surface TK receptor functions as the other types of tyrosine kinases. It has an important role in the cell proliferation, growth, and differentiation. The uncontrolled cellular growth resulted from the hyperactivity of PDGFR resulted in different diseases like pulmonary fibrosis, restenosis, and cancer [78]. In 2012, Shepard et al. discovered the 4-piperazino-substituted quinazoline derivative (48) as a PDGFR inhibitor. It showed a strong activity against FLT-3, c-KIT [79]. The anticancer quinazolines 48 is shown in (Figure 31) [79].
5.2.4. Serine-Threonine Kinase Inhibitors
Serine-threonine kinas is a type of TK that performs the autophosphorylation process at the oxygen atom of serine or threonine amino acid residues. It also plays an important role in regulation of cell proliferation, differentiation, and apoptosis. There are two types of this kinase [80]:
- Serine-threonine receptor type kinase (TGFBR).
- Serine-threonine non-receptor type kinas (aurora kinases, CDK, and PI3K).
Aurora Kinase Inhibitors
Aurora kinase plays an essential role in cell division processes such as chromosomes segregation, centrosomes maturation, and cytokinesis. There are three types of this kinase A, B, and C. These types are overexpressed in many types of tumors as breast, ovarian, colon, pancreatic, and thyroid cancers. The overexpression of this kinase is also associated with genetic characters and treated by aurora kinase inhibitors.
The 4-aminoquinazoline molecule ZM447439 (49) was revealed by AstraZeneca in 2003 as dual aurora kinase inhibitor of A and B types with IC50 = 0.1 μM [81]. When the phenyl ring attached to the 4-position of the quinazoline moiety is replaced by the six-membered pyrimidine ring (50), a compound with a higher affinity to aurora A and B was obtained. The activity of this compound was 0.011 and 0.025 μM against aurora A and B, respectively. Substitution of the compound (50) with the morpholinopropoxy group at the 7-position of the quinazoline system showed a potent inhibitory activity with IC50 = 0.003 and 0.001 μM. Moreover, substitution of the terminal phenyl ring (benzamido group) with a small lipophilic group as chloro substituent (51) displayed an excellent activity of less than 0.1 nM [82].
Introduction of a small five-membered heterocyclic ring such as thiazole and thiophene instead of the central phenyl ring of the aniline moiety resulted in an excellent inhibitory activity against aurora kinase due to the strong binding affinity. Additionally, placing a methylene group between the amide group and the five-membered ring (52) considerably increased the cellular potency [83]. The anticancer quinazolines 49–52 are shown in (Figure 32) [81,82,83].
A novel class of the selective aurora kinase inhibitors containing pyrazolo-substituted-quinazoline scaffold was discovered [84]. Among this class, barasertib, or AZD1152 (53) is a phosphate-based antineoplastic drug showed a 1000-fold greater potency against aurora kinase B (IC50 = 0.37 nM) than A [85]. It is an FDA-approved antineoplastic agent for treatment of hematologic cancer and solid malignant tumors [86]. Moreover, another acetanilide-aminopyrazole-substituted quinazoline (54) which was designed by a group of researchers in AstraZeneca, was found to be more potent and selective than the 3-aminopyrazole derivative with IC50 less than 1 nM against aurora kinase B [87]. It also inhibited other kinases PDGFR a, B, CSF-1R, and c-KIT with IC50 values 0.069, 0.006, 0.036, and 0.009 μM, respectively [88,89]. The compound (55), which have a structural similarity with this class, displayed high selectivity toward aurora A with IC50 = 0.038 μM [90]. Continuing in this direction, Cai et al. designed some urea derivatives of the 4-anilinoquinazoline scaffold to be investigated as cytotoxic agents against aurora A and B kinases. The 2-methylpiperidine analogue (56) produced a strong activity (IC50 = 0.9–3.1 μM) against solid tumors. Additionally, it showed selectivity with aurora A (IC50 = 61 nM) while aurora B was IC50 = 172 nM [91]. Similarly, Hsu et al. discovered BPR1K871 (57) as a quinazoline derivative multikinase inhibitor. It displayed a strong cytotoxic activity against MOLM-13, MV4-11 AML, aurora kinase A, and FLT-3 equal to 5, 5, 22, and 19 nM, respectively [92]. The anticancer quinazolines 53–57 are shown in (Figure 33) [84,85,86,87,88,89,90,91,92].
SAR of Aurora Kinase Inhibitors
- Quinazoline with an aminoalkyl or an aminoaryl moiety at the 4-position of the quinazoline is required for the anticancer activity.
- Substitution at the 5 and the 6-positions of the quinazoline with an electron releasing group increases the activity.
- A lipophilic aromatic group attached to the 4-aminoaryl group increases the activity.
Cyclin-Dependent Kinase (CDK) Inhibitors
Cyclin-dependent kinase (CDK) is a type of serine-threonine kinases. It plays an important role in the protein phosphorylation process. It also regulates transcription, mRNA processing, metabolism, and cells differentiation [93]. Cyclin is a regulatory protein binds with CDK to form cyclin-CDK complex that phosphorylates specific substrates in the cell cycle [94]. In many types of cancer, CDKs become overreactive or CDK inhibiting proteins are not working. Therefore, CDKs display a target to inhibit the uncontrolled cellular growth by blocking this uncontrolled activity [95]. The X-ray analysis revealed the mode of binding of anilinoquinazoline derivatives with CDKs [96].
In a recent study, Sielecki et al. designed a group of 2, 4, and 6 substituted quinazolines as CDK inhibitors. Among these derivatives, compound (58) formed extensive hydrogen bonds with ATP pocket of CDK2 enzyme. It showed a potent inhibitory activity with IC50 = 0.6 μM [97].
Bathini et al. prepared a series of 2,7,8-trisubstituted quinazolines for testing as CDK inhibitors. It was found that substitution at the 4-position of the aniline ring of the quinazoline system influences CDK-4 inhibition. The piperazine quinazoline-based derivative (59) showed the highest activity among these derivatives (IC50 = 0.007 μM) [98]. Placing of cyclopentyl group at the 8-position from the quinazoline system resulted in a higher CDK-4 inhibitory activity in the compound (60) (IC50 = 0.001 μM) [99]. The molecular modeling study of compound (61) showed that the pyrazole ring attached to the quinazoline moiety should have electron withdrawing group for a good activity [99]. A new quinazoline molecule (62) containing Michael acceptor at the 6-position of the quinazoline system was prepared by Shi et al. and showed a strong activity against A549 cell line. The anticancer activity of this molecule was explained based on accumulation of the reactive oxygen species which down-regulated the cyclin B1 regulatory proteins resulting in apoptosis and cell cycle arrest [100]. The anticancer quinazolines 58–62 are shown in (Figure 34) [97,98,99,100].
Phosphoinositid-3-Kinase (PI3K) Inhibitors
Phosphoinositid-3-kinase (PI3K) performs essential functions in many cellular processes such as proliferation, differentiation, and migration. It is activated by a wide range of receptor tyrosine kinases. The transduction system is activated by several types of tyrosine kinases which generate another messenger called phosphatidylinositol 3,4,5 triphosphate (PIP3). This messenger controls the cellular function by phosphorylating effectors and adaptors. There are different isoforms of PI3Ks such as PI3Kα, PI3Kβ, PI3Kδ, and PI3Kϒ. The inhibitors of PI3K work by inhibition of one or more of the activator kinases enzymes. The mutations and the dysregulation of PI3K occur in many types of tumors which make it an important target for the anticancer agents [101].
Idelalisib (63, Zydelgi®) [102] is a highly effective quinazoline derivative selective PI3Kδ inhibitor (IC50 = 2.5 nM). It is an FDA-approved antineoplastic medication for treatment of hematologic malignancies and used as a second line of choice for treatment of patients having relapse from chronic lymphocytic leukemia [102].
Copanilisin (64, Aliqopa®) [103] is another PI3K inhibitor. It is an imidazoquinazoline molecule that inhibits two types of PI3K including PI3Kα and PI3Kδ isoforms. The inhibition of these enzymes leads to inhibition of growth and proliferation of the malignant B cells. It also controls tumor cell death by apoptosis. This medication was approved by the FDA for treatment of patients having relapsed follicular lymphoma [103]. The anticancer quinazolines 63 and 64 are shown in (Figure 35) [102,103]
Shao et al. designed 4,6-disubstituted quinazoline as PI3K inhibitors. These derivatives were tested against A549, HCT-116, U-87 MG, and KB cell lines using MTT assay method. Out of these derivatives, compound (65) displayed a potent cytotoxic activity with IC50 = 4.32, 0.54, 1.37, and 4.45 μM, respectively. Furthermore, this compound was found as an inhibitor of PI3K, mTOR, and AKT. It also showed tumor growth inhibition in the nude mice U-87 MG model [104].
A hybrid derivative of chalcone-quinazoline (66) was designed by Wani et al. to be investigated for the cytotoxic activity against PI3K, AKT, and mTOR cancer cell lines. This derivative inhibited proliferation of the cancer cells lines in micromolar range. Additionally, it inhibited in vivo tumor growth in the animal models [105].
Another hybrid derivative of a hydroxamic-quinazoline molecule was designed as a dual inhibitor of PI3K and HDAC. The HDAC is the histone deacetylase enzyme which allow wrapping the DNA before expression. Disorientation of HDAC changes gene expression and cell phenotype, which results in cancer incidence. Therefore, combination of HDAC inhibitor and quinazoline-based PI3K inhibitor targeted more than one target and led to a potent cytotoxic activity. Compound (67) showed IC50 = 2.98, and 50 nM against PI3K, and HDAC, respectively. This compound was also tested for the in vitro cytotoxicity on human colon carcinoma HCT116, leukemia (K562), and T lymphocyte (Hut78). It displayed IC50 = 0.33, 0.095, 0.062 μM, respectively [106]. The anticancer quinazolines 65–67 are shown in (Figure 36) [104,105,106].
Xin et al. synthesized some derivatives of 4-phenylquinazoline having benzamide moiety at the 6-position to be investigated for their PI3K inhibitory activity. Among these analogues, compound (68) displayed the highest cytotoxic activity with IC50 = 17 nM. A further extension for this work by preparing different derivatives with different substituents at the 6-position, produced the compound (69) having the trifluoromethyl benzamide group. This derivative displayed PI3K inhibitory activity with IC50 = 9.7 nM, and antiproliferative activity IC50 = 1 μM against the RPMI-8226 human B cell line [107].
Another derivative containing 4-pyrrolidineamino group instead of 4-anilino group was designed by Xin et al. and tested as PI3Kδ inhibitor. It was found that the compound having tetrahydo-2H-pyran group at the 4-position (70) increased PI3K binding affinity and produced a strong inhibition (IC50 = 2.7 nM) [108]. A novel series of 2-pyridin-substituted-quinazoline-4-one was synthesized to be investigated as PI3K inhibitors. Of these newly synthesized derivatives, compound (71), containing oxo-benzothiazine group attached to the benzamide moiety at the 3-position from quinazoline-4-one system, showed IC50 = 60.29 μM against the human hepatic carcinoma HepPG-2 cell line compared to the reference doxorubicin (IC50 = 69.6 μM). It also displayed IC50 of 31.92 μM against PI3K which was also better than the standard LY294002 (IC50 = 57.3 μM) [109]. The anticancer quinazolines 68–71 are shown in (Figure 37) [107,108,109].
6. Pharmaceutical Marketed Anticancer Quinazolines
7. Conclusions
Discovery of novel anticancer agents has advanced from a period of toxic non-selective agents to less toxic selective agents. Deficiency of selectivity and high toxicity are major drawbacks of the current chemotherapeutic drugs. Hence, the targeted therapy acting on a specific biological target is required for the treatment of these life-threatening diseases. Continuous research efforts in the field of drug discovery have resulted in exploration of quinazoline and its derivatives as a targeted anticancer agent. Quinazolines were used as tyrosine kinases inhibitors for treatment of cancer. A huge frame of scientific experiments proved the efficacy of quinazolines as targeted anticancer agents. Several molecules of the anticancer quinazolines were approved by the FDA and are available in the market for treatment of different types of cancer diseases. Most modifications performed on quinazolines were carried out via changing the substituents at C-4, C-6, and C-7 from the quinazoline system to develop the anticancer activity. Small molecular changes in the quinazoline system led to dramatic changes in the selectivity, the potency, and the anticancer activity. Regardless of the availability of quinazoline molecules, an abundant potential remains in this promising system to be discovered for preparing useful chemotherapeutic agents. Additional structural variations in the quinazoline system might lead to novel derivatives with more activity than the current available drugs. The previously mentioned literature survey displayed the importance of structural modifications of quinazolines on their anticancer activity. This review presents a clear perspective and a good understanding to scientists and chemists aiming toward development of quinazolines as targeted chemotherapeutic agents.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The author would like to extend his thanks to the Unit of Scientific Research, Fakeeh College for Medical Sciences, Jeddah, Saudi Arabia for providing valuable suggestions, and moral support.
Conflicts of Interest
The author declares no conflict of interest, financial or otherwise.
References
- Zayed, M.F.; Ahmed, H.E.A.; Ihmaid, S.K.; Omar, A.M.; Abdelrahim, A.S. Synthesis and screening of some new fluorinated quinazolinone–sulphonamide hybrids as anticancer agents. J. Taibah Univ. Sci. 2015, 10, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Malhotra, A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur. J. Med. Chem. 2021, 211, 113016. [Google Scholar] [CrossRef]
- Zayed, M.F.; Rateb, H.; Ahmed, S.; Khaled, O.; Ibrahim, S. Quinazolinone-amino acid hybrids as Dual Inhibitors of EGFR Kinase and Tubulin Polymerization. Molecules 2018, 23, 1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia; Ishtiaq, M.; Khan, K.M. Quinazoline and quinazolinone as important medicinal scaffolds: A comparative patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ahmed, S.; Ihmaid, S.; Ahmed, H.E.; Rateb, H.; Ibrahim, S. Design, Synthesis, Cytotoxic Evaluation and Molecular Docking of New Fluoroquinazolinones as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Int. J. Mol. Sci. 2018, 19, 1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, M.F.; Ahmed, H.E.A.; Ihmaid, S.; El-Adl, K.; Asiri, A.; Omar, A.M. Modelling and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules 2017, 22, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zayed, M.F.; Hassan, M.H. Design, Synthesis and Biological Evaluation Studies of Novel Quinazoline Derivatives as Cytotoxic Agents. Drug Res. 2013, 63, 210–215. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Abel, A.; Maruca, A.; Montalbano, A.; Raimondi, V.M.; Tarantelli, C.; Gaudio, E.; et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas. Eur. J. Med. Chem. 2022, 243, 114744. [Google Scholar] [CrossRef]
- Barreca, M.; Ingarra, M.A.; Raimondi, V.M.; Spanò, V.; Piccionello, P.A.; Franco, D.M.; Menilli, L.; Gandin, V.; Miolo, G.; Barraja, P.; et al. New tricyclic systems as photosensitizers towards triple negative breast cancer cells. Arch. Pharm. Res. 2022, 45, 806–821. [Google Scholar] [CrossRef]
- Duró, C.; Jernei, T.; Szekeres, K.J.; Láng, G.G.; Oláh-Szabó, R.; Bősze, S.; Szabó, I.; Hudecz, F.; Csámpai, A. Synthesis and SAR Analysis of Novel 4-Hydroxytamoxifen Analogues Based on Their Cytotoxic Activity and Electron-Donor Character. Molecules 2022, 27, 6758. [Google Scholar] [CrossRef]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)quinazolin-4(3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Med. Chem. Commun. 2017, 8, 871–885. [Google Scholar] [CrossRef]
- Haider, K.; Das, S.; Joseph, A.; Yar, M.S. An appraisal of anticancer activity with structure-activity relationship of quinazoline and quinazolinone analogues through EGFR and VEGFR inhibition: A review. Drug Dev. Res. 2022, 83, 859–890. [Google Scholar] [CrossRef] [PubMed]
- Auti, P.S.; George, G.; Paul, A.T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 2020, 10, 41353–41392. [Google Scholar] [CrossRef]
- Sunil Kumar, A.; Kudva, J.; Lahtinen, M.; Peuronen, A.; Sadashiva, R.; Naral, D. Synthesis, characterization, crystal structures and biological screening of 4-amino quinazoline sulfonamide derivatives. J. Mol. Struct. 2019, 1190, 29–36. [Google Scholar] [CrossRef]
- El-Zahabia, M.A.; Bamanie, H.F.; Ghareeb, S.; Alshaeri, K.H.; Alasmari, M.M.; Muostafa, M.; Al-Marzoki, Z.; Zayed, M.F. Design, Synthesis, Molecular Modeling andAnti-Hyperglycemic Evaluation of Quinazoline-Sulfonylurea Hybrids as Peroxisome Proliferator-Activated Receptor Gamma (PPAR) and Sulfonylurea Receptor (SUR) Agonists. Int. J. Mol. Sci. 2022, 23, 9605. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ibrahim, S.; Habib, E.E.; Hassan, M.H.; Ahmed, S.; Rateb, H.S. Design, synthesis, antimicrobial and anti-biofilm evaluation, and molecular docking of new substituted fluoroquinazolinones. J. Med. Chem. 2019, 15, 657–673. [Google Scholar]
- Zhang, J.; Liu, J.; Ma, Y.; Ren, D.; Cheng, P.; Zhao, J.; Zhang, F.; Yao, Y. One-pot synthesis and antifungal activity against plant pathogens of quinazolinone derivatives containing an amide moiety. Bioorg. Med. Chem. Lett. 2016, 26, 2273–2277. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ballardo, L.; García-Páez, F.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Calderón-Zamora, L.; Rendón-Maldonado, G.; Osuna-Martínez, U.; Sarmiento-Sánchez, J.I. Synthesis, biological evaluation and molecular docking of 3-substituted quinazoline-2,4(1H, 3H)-diones. J. Chem. Sci. 2020, 132, 100. [Google Scholar] [CrossRef]
- Jain, R.K.; Kashaw, V. Design, synthesis and evaluation of novel 2,3-disubstituted-4-(3H) quinazolinone derivatives. Asian J. Pharm. Pharmacol. 2018, 4, 644–656. [Google Scholar] [CrossRef]
- Hricoviniova, J.; Hricoviniova, Z.; Kozica, K. Antioxidant, Cytotoxic, Genotoxic, and DNA Protective Potential of 2,3-Substituted Quinazolinones: Structure—Activity Relationship Study. Int. J. Mol. Sci. 2021, 22, 610. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F. New fluorinated quinazolinone derivatives as anticonvulsant agents. J. Taibah Univ. Sci. 2014, 9, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Ghorab, M.M.; Abdel-Kader, M.S.; Alqahtani, A.S.; Soliman, A.M. Synthesis of some quinazolinones inspired from the natural alkaloid L-norephedrine as EGFR inhibitors and radiosensitizers. J. Enzym. Inhib. Med. Chem. 2021, 36, 218–237. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) Chemical Computing Group. Available online: http://www.chemcomp.com (accessed on 10 May 2019).
- Zayed, M.F.; Hassan, M.H. Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and anti-inflammatory agents. Saudi Pharm. J. 2014, 22, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Swiss Institute of Bioinformatics (SwissADME). Available online: http://www.swissADME.ch (accessed on 10 November 2022).
- Zayed, M.F.; Ahmed, E.A.; Omar, A.M.; Abdelrahim, A.S.; El-Adl, K. Design, synthesis, and biological evaluation studies of novel quinazolinone derivatives as anticonvulsant agents. Med. Chem. Res. 2013, 22, 5823–5831. [Google Scholar] [CrossRef]
- Wang, D.; Gao, F. Quinazoline derivatives: Synthesis and bioactivities. Chem. Cent. J. 2013, 7, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, D.J.; Cusack, D.; O’Sullivan, T.P.; Guiry, P.J. Synthesis of quinazolinones and quinazolines. Tetrahedron 2005, 61, 10153–10202. [Google Scholar] [CrossRef]
- Meyer, J.F.; Wagner, E.C. The Niementowski reaction. The use of methyl anthranilate or isatoic anhydride with substituted amides or amidines in the formation of 3-substituted-4-keto-3,4-dihydroquinazolines. The course of the reaction. J. Org. Chem. 1943, 8, 239–252. [Google Scholar] [CrossRef]
- Asif, M. Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 2014, 395637. [Google Scholar] [CrossRef]
- Gupta, T.; Rohilla, A.; Pathak, A.; Akhtar, M.J.; Haider, M.R.; Yar, M.S. Current perspectives on quinazolines with potent biological activities: A review. Synth. Commun. 2018, 48, 1099–1127. [Google Scholar] [CrossRef]
- Bisht, A.S.; Negi, J.S.; Sharma, D.K. Chemistry and activity of quinazoline moiety: A systematic review study. Int. J. Pharm. Chem. Anal. 2020, 7, 61–65. [Google Scholar] [CrossRef]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-one-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, K.T.P.; Yi, S.C. Synthesis of Quinazoline and Quinazolinone Derivatives via Ligand-Promoted Ruthenium-Catalyzed Dehydrogenative and Deaminative Coupling Reaction of 2-Aminophenyl Ketones and 2-Aminobenzamides with Amines. Org. Lett. 2019, 21, 3337–3341. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.M.; Sivaramakrishna, A. Remarkably flexible quinazolinones—Synthesis and biological applications. J. Heterocycl. Chem. 2019, 57, 942–954. [Google Scholar] [CrossRef]
- Zayed, M.F. Medicinal Chemistry of Quinazolines as Analgesic and Anti-Inflammatory Agents. ChemEngineering 2022, 6, 94. [Google Scholar] [CrossRef]
- Chilin, A.; Conconi, M.T.; Marzaro, G.; Guiotto, A.; Urbani, L.; Tonus, F.; Parnigotto, P. Exploring epidermal growth factor receptor (EGFR) inhibitor features: The role of fused dioxygenated rings on the quinazoline scaffold. J. Med. Chem. 2010, 53, 1862–1866. [Google Scholar] [CrossRef]
- Conconi, M.T.; Marzaro, G.; Urbani, L.; Zanusso, I.; Di, L.R.; Castagliuolo, I.; Brun, P.; Tonus, F.; Ferrarese, A.; Guiotto, A.; et al. Quinazoline-based multi-tyrosine kinase inhibitors: Synthesis, modeling, antitumor and antiangiogenic properties. Eur. J. Med. Chem. 2013, 67, 373–383. [Google Scholar] [CrossRef]
- Kumar, D.; Mariappan, G.; Husain, A.; Monga, J.; Kumar, S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab. J. Chem. 2017, 10, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, E.C.; Patel, B.S.; Becker, W.J.; Cameron, M.P.; Zaller, D.; Pikounis, B.V.; O’Keefe, J.S.; Scapin, G. Structural basis for p38α MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat. Struct. Mol. Biol. 2003, 10, 764–769. [Google Scholar] [CrossRef]
- Abouzid, K.; Shouman, S. Design, synthesis and in vitro antitumor activity of 4-aminoquinoline and 4-aminoquinazoline derivatives targeting EGFR tyrosine kinase. Bioorg. Med. Chem. 2008, 16, 7543–7551. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zheng, W.; Ji, L.; Luo, Q.; Hao, X.; Li, X.; Wang, F. Synthesis, characterization, screening and docking analysis of 4-anilinoquinazoline derivatives as tyrosine kinase inhibitors. Eur. J. Med. Chem. 2013, 61, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, T.; Ji, X.; Li, J.; Tong, L.; Li, Z.; Yang, W.; Xu, Y.; Li, M.; Ding, J. Design, synthesis and biological evaluation of novel 4-anilinoquinazolines with C-6 urea-linked side chains as inhibitors of the epidermal growth factor receptor. Bioorg. Med. Chem. 2013, 21, 7988–7998. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yuan, Y.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y. Identification of novel 4-anilinoquinazoline derivatives as potent EGFR inhibitors both under normoxia and hypoxia. Bioorg. Med. Chem. 2014, 22, 6796–6805. [Google Scholar] [CrossRef]
- Yu, H.; Li, Y.; Ge, Y.; Song, Z.; Wang, C.; Huang, S.; Jin, Y.; Han, X.; Zhen, Y.; Liu, K.; et al. Novel 4-anilinoquinazoline derivatives featuring an 1-adamantyl moiety as potent EGFR inhibitors with enhanced activity against NSCLC cell lines. Eur. J. Med. Chem. 2016, 110, 195–203. [Google Scholar] [CrossRef]
- Wu, X.; Li, M.; Qu, Y.; Tang, W.; Zheng, Y.; Lian, J.; Ji, M.; Xu, L. Design and synthesis of novel gefitinib analogues with improved anti-tumor activity. Bioorg. Med. Chem. 2010, 18, 3812–3822. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Lin, Z.; Wang, F.; Zhao, W.; Dong, X. Four-membered heterocycles-containing 4-anilino-quinazoline derivative s as epidermal growth factor receptor (EGFR) kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5385–5388. [Google Scholar] [CrossRef]
- Chang, J.; Ren, H.; Zho, M.; Chong, Y.; Zhao, Y.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a se ies of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation f anticancer activities in vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef]
- Cai, J.; Sun, M.; Wu, X.; Chen, P.; Wang, X.; Zong, M.J. Design and synthesis of novel 4-benzothiazole amino quinazolines dasatinib derivatives as potential anti-tumor agents. Eur. J. Med. Chem. 2013, 63, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Y.; Zhou, H.; Zheng, Q.; Li, Y.; Zheng, S.; Zhao, S.; Chen, D.; Fan, C. Structure-activity study of quinazoline derivatives leading to the discovery of potent EGFR-T790M inhibitors. Eur. J. Med. Chem. 2015, 102, 445–463. [Google Scholar] [CrossRef]
- Hou, W.; Ren, Y.; Zhang, Z.; Sun, H.; Ma, Y.; Yan, B. Novel quinazoline derivatives bearing various 6-benzamide moieties as highly selective and potent EGFR inhibitors. Bioorg. Med. Chem. 2018, 26, 1740–1750. [Google Scholar] [CrossRef]
- Zhang, Y.; Tortorella, M.D.; Liao, J.; Qin, X.; Chen, T.; Luo, J.; Guan, J.; Talley, J.J.; Tu, Z. Synthesis and evaluation of novel erlotinib-NSAID conjugates as more comprehensive anticancer agents. ACS Med. Chem. Lett. 2015, 6, 1086–1090. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Lv, Y.; Liu, P.; Li, Z.; Hu, L.; Zeng, C.; Yang, L. Novel morpholin-3-one fused quinazoline derivatives as EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1571–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, Y.; Liu, J.; Wang, W.; Li, X.; Zhao, L.; Wang, W.; Li, B. Novel 4-arylaminoquinazoline derivatives with (E)-propen-1-yl moiety as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur. J. Med. Chem. 2017, 138, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, L.; Xu, H.; Li, X.; Zhao, L.; Wang, W.; Li, B.; Zhang, X. 6,7-Dimorpholinoalkoxy quinazoline derivatives as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur. J. Med. Chem. 2017, 147, 77–89. [Google Scholar] [CrossRef]
- Liu, L.T.; Yuan, T.T.; Liu, H.H.; Chen, S.F.; Wu, Y.T. Synthesis and biological evaluationJournalofsubstituted6-kynyl -4-anilinoquinazoline derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6373–6377. [Google Scholar] [CrossRef]
- Li, D.D.; Fang, F.; Li, R.; Du, Q.R.; Sun, J.; Gong, H.B.; Zhu, H.L. Discovery of 6-substituted 4-anilin qinazolines with dioxygenated rings as novel EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5870–5875. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Li, D.D.; Lu, X.; Xu, Y.Y.; Zhu, H.L. Design and synthesis of 4,6-substituted-(diphenylamino)quinazolines as potent EGFR inhibitors with antitumor activity. Bioorg. Med. Chem. 2012, 20, 317–323. [Google Scholar] [CrossRef]
- Li, D.D.; Lv, P.C.; Zhang, H.; Zhang, H.J.; Hou, Y.P.; Liu, K.; Ye, Y.H.; Zhu, H.L. The combination of 4-anilinoquinazoline and cinnamic acid: A novel mode of binding to the epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2011, 19, 5012–5022. [Google Scholar] [CrossRef]
- Mowafy, S.; Farag, N.A.; Abouzid, K.A.M. Design, synthesis and in vitro anti-proliferative activity of 4,6-quinazolinediamines as potent EGFR-TK inhibitors. Eur. J. Med. Chem. 2013, 61, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Chen, L.; Zhao, L.; Li, B.; Wang, W. Synthesis and in vitro biological evaluation of novel quinazoline derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef]
- Musumeci, F.; Radi, M.; Brullo, C.; Schenone, S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 2012, 55, 10797–10822. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. The role of hypoxia-induced factorsproofintumorprogression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Manley, P.W.; Bold, G.; Bruggen, J.; Fendrich, G.; Fuet, P.; Mestan, J.; Schnell, C.; Stolz, B.; Meyer, T.B.; Meyhack, B.; et al. Advances in the structural biology, design and clinical d v lopmentVEGF-R kinase inhibitors for the treatment of angiogenesis. Biochim. Biophys. Acta 2004, 1697, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, P.; Castellana, M.; Virili, C.; Giorgino, F.; Giovanella, L. Efficacy of vandetanib in treating locally advanced or metastatic medullary thyroid carcinoma according to RECIST criteri: A systematic review and meta-analysis. Front. Endocrinol. 2018, 9, 224. [Google Scholar] [CrossRef]
- Batchelr, T.T.; Lholland, P.M.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, A.; Goossens, L.; Six, P.; Lemoine, A.; Ravez, S.; Farce, A.; Depreux, P. Impact of aryloxy-linked quinazolines: A novel series of selective VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2106–2112. [Google Scholar] [CrossRef]
- Garofalo, A.; Farce, A.; Ravez, S.; Lemoine, A.; Six, P.; Chavatte, P.; Goossens, L.; Depreux, P. Synthesis and structure-activity relationships of (aryloxy)quinazoline ureas as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors. J. Med. Chem. 2012, 55, 1189–1204. [Google Scholar] [CrossRef]
- Ravez, S.; Barczyk, A.; Six, P.; Cagnon, A.; Garofalo, A.; Goossens, L.; Depreux, P. Inhibition of tumor cell growth and angiogenesis by 7-aminoalkoxy-4-aryloxy-quinazoline ureas, a novel series of multi-tyrosine kinase inhibitors. Eur. J. Med. Chem. 2014, 79, 369–381. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, J.; Wang, N.; Kong, X.; Fu, F.; Wang, H.; Yao, J. Design and discovery of quinazoline- and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules 2018, 23, 24. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.L.; Lima, L.M.; Tesch, R.; Sant’Anna, C.M.; Totzke, F.; Kubbutat, M.H.; Schächtele, C.; Laufer, S.A.; Barreiro, E.J. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2014, 71, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Gong, F.H.; Li, C.G.; Zhang, C.; Wang, Y.J.; Xu, Y.G.; Sun, L.P. Design and discovery of 4-anilinoquinazoline-acylamino derivatives as EGFR and VEGFR-2 dual TK inhibitors. Eur. J. Med. Chem. 2016, 109, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Gong, F.H.; Ye, J.Q.; Zhang, C.; Yue, X.H.; Li, C.G.; Xu, Y.G.; Sun, L.P. Design an discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2. Eur. Med. Chem. 2017, 125, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.; Zhang, J.Q.; Liu, Z.C.; Zhang, J.H.; Yan, J.F.; Jin, Y.; Lin, J. Novel 5-anilinoquinaz line-8-nitro derivatives as inhibitors of VEGFR-2 tyrosine kinase: Synthesis, biological evaluation and molecular docking. Org. Biomol. Chem. 2013, 11, 4367–4378. [Google Scholar] [CrossRef]
- Elsayed, N.M.Y.; Serya, R.A.T.; Tolba, M.F.; Ahmed, M.; Barakat, K.; Abou El Ella, D.A.; Abouzid, K.A.M. Design, synthesis, biological evaluation and dynamics simulation of indazole derivatives with antiangiogenic and antiproliferative anticancer activity. Bioorg. Chem. 2019, 82, 340–359. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 11, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Shepard, D.R.; Cooney, M.M.; Elson, P.; Bukowski, R.M.; Dreicer, R.; Rini, B.J.; Garcia, J.A. A phase II study of tandutinib (MLN518), a selective inhibitor of type III tyrosine receptor kinases, in patients with metastaticrenal cell carcinoma. Invest. New Drugs 2012, 30, 364–367. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signaling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.D. Aurora kinases: Shining lights on the therapeutic horizon. Oncogene 2005, 24, 5005–5015. [Google Scholar] [CrossRef] [Green Version]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Gadea, B.B.; Ruderman, J.V. Aurora kinase inhibit ZM447439 blocks chromosome-induced spindle assembly, the completion of ch m s me condensation, and the establishment of the spindle integrity check oint in Xenopus egg extracts. Mol. Biol. Cell. 2005, 16, 1305–1318. [Google Scholar] [CrossRef] [Green Version]
- Heron, N.M.; Anderson, M.; Blows, D.P.; Bred, J.; Eden, J.M.; Green, S.; Hill, G.B.; Johnson, T.; Jung, F.H.; McMiken, H.H.J.; et al. SAR and inhibitor complex structure determination of a novel class of potent specific Aurora kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1320–1323. [Google Scholar] [CrossRef]
- Jung, F.H.; Pasqet, G.; Brempt, C.L.; Lohmann, J.J.M.; Warin, N.; Renaud, F.; Germain, H.; De Savi, C.; Roberts, N.; Johnston, T.; et al. Discovery of novel and potent thiazoloquinazolines as selective aurora A and B kinase inhibitors. J. Med. Chem. 2006, 49, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.; Seedhouse, C.; Shang, S.; Richardson, J.; Russell, N.; Pallis, M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152-HQPA in acute myelogenous leukemia cells. Mol. Cancer Ther. 2010, 9, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortlock, A.A.; Foote, K.M.; Heron, N.M.; Jung, F.H.; Pasquet, G.; Lohmann, J.J.M.; Warin, N.; Renaud, F.; De Savi, C.; Roberts, N.J.; et al. Discovery, synthesis and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J. Med. Chem. 2007, 50, 2213–2324. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Mortlock, A.A.; Heron, N.M.; Jung, F.H.; Hill, G.B.; Pasquet, G.; Brady, M.C.; Green, S.; Heaton, S.P.; Kearney, S.; et al. Synthesis and SAR of 1-acetanilide-4-aminopyrazole-substituted quinazolines: Selective inhibitors of aurora B kinase with potent anti-tumor activity. Bioorg. Med. Chem. Lett. 2008, 18, 1904–1909. [Google Scholar] [CrossRef]
- Long, L.; Wang, Y.H.; Zhuo, J.X.; Tu, Z.C.; Wu, R.; Yan, M.; Liu, Q.; Lu, G. Structure-based drug design: Synthesis and biological evaluation of quinazolin-4-amine derivatives as selective aurora A kinase inhibitors. Eur. J. Med. Chem. 2018, 157, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Li, L.; Hong, K.H.; Wu, X.; Chen, J.; Wang, P.; Cao, M.; Zong, X.; Ji, M. Discovery of 4-aminoquinazoline-u rea derivatives as aurora kinase inhibitors with antiproliferative activity. Bioorg. Med. Chem. 2014, 22, 5813–5823. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Coumar, M.S.; Wang, W.C.; Shiao, H.Y.; Ke, Y.Y.; Lin, W.H.; Kuo, C.C.; Chang, C.W.; Kuo, F.M.; Chen, P.Y.; et al. Discovery of BPR1K871, a quinazoline based, multi-kinase inhibit f r the treatment of AML and solid tumors: Rational design, synthesis, in vitro and in vivo evaluation. Oncotarget 2016, 7, 86239–86256. [Google Scholar]
- Malumbres, M.; Barbacid, M.; Mammalian, M. Cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cycle regulators and beyond. Dev. Cell. 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; Dios, A.D. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.; Kuyper, L.F. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J. Med. Chem. 2000, 43, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sielecki, T.M.; Johnson, T.L.; Liu, J.; Muckelbauer, J.K.; Grafstrom, R.H.; Cox, S.; Boylan, J.; Burton, C.R.; Chen, H.; Smallwood, A.; et al. Quinazolines as cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1157–1160. [Google Scholar] [CrossRef]
- Bathini, Y.; Singh, I.; Harvey, P.J.; Keller, P.R.; Singh, R.; Micetich, R.G.; Fry, D.W.; Dobrusin, E.M.; Toogood, P.L. 2-Aminoquinazoline inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2005, 15, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Wang, S.T.; Sun, P.H.; Song, F.J. Molecular modeling studies of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as otent CDK2/cyclin A inhibitors using 3D-QSAR and docking. Int. J. Mol. Sci. 2010, 11, 3705–3724. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Li, Y.; Ren, X.; Zhang, Y.; Yang, Z.; Qi, C. A novel quinazoline-based analog induces G2/M cell cycle arrest and apoptosis in human A549 lung cancer cells via a ROS-dependentJournalmechanism. Biochem. Biophys. Res. Commun. 2017, 486, 314–320. [Google Scholar] [CrossRef]
- Marone, R.; Cmiljaovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef]
- Markham, A. Idelalisib: First global approval. Drugs 2014, 74, 1701–1707. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppa, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, P.; et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Wang, J.; Chen, J.G.; Wang, X.M.; Li, H.; Li, Y.P.; Li, Y.; Yang, G.D.; Mei, Q.B.; Zhang, S.Q. Discovery of 2-methoxy-3-phenylsulfonamido-5-(quinazolin-6-yl or quinolin-6-yl)benzamides as novel PI3K inhibitors and anticancer agents by bioisostere. Eur. J. Med. Chem. 2014, 75, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Wani, Z.A.; Guru, S.K.; Rao, A.V.; Sharma, S.; Mahajan, G.; Behl, A.; Kumar, A.; Sharma, P.R.; Kamal, A.; Bhushan, S.; et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food Chem. Toxicol. 2016, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dai, W.; Chen, X.; Geng, A.; Chen, Y.; Lu, T.; Zhu, Y. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-c]quinazoline derivatives as novel phosphatidylinositol 3-kinase and histone deacetylase dual inhibitors. RSC Adv. 2017, 7, 52180–52186. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Hei, Y.Y.; Zhang, H.; Xie, X.X.; Mao, S.; Zu, S.J.; Zhang, S.Q. Discovery of 6-benzamide containing 4-phenylquinazoline de ivatives as novel PI3Kδ inhibitors. Lett. Drug Des. Dis. 2017, 14, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Hei, Y.Y.; Zhang, H.; Shen, Y.; Zhang, S.Q. Design and synthesis of novel 6-aryl substituted 4-anilinequinazoline de ivatives as potential PI3Kδ inhibitors. Bioorg. Med. Chem. Lett. 2017, 14, 1972–1977. [Google Scholar] [CrossRef]
- Xin, M.; Duan, W.; Feng, Y.; Hei, Y.; Zhang, H.; Shen, Y.; Zhao, H.Y.; Mao, S.; Zhang, S.Q. Novel 6- yl substituted 4-pyrrolidineaminoquinazoline derivatives as potent phosphoinositide 3-kinase delta (PI3Kδ) inhibitors. Bioorg. Med. Chem. 2018, 26, 2028–2040. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 25 January 2023).
Figure 1.
Quinazolines (1–3) and some of their pharmacological activities.

Figure 2.
Surface map for interactions of the 4-aminoarylquinazoline. The pink color shows hydrogen bonding area, the green color indicates hydrophobic area, and the blue color indicates a mild polar area [24].
Figure 2.
Surface map for interactions of the 4-aminoarylquinazoline. The pink color shows hydrogen bonding area, the green color indicates hydrophobic area, and the blue color indicates a mild polar area [24].

Figure 3.
Synthesis of 4(3H)-quinazolinone by Niemtowski technique.

Figure 4.
Synthesis of 2-methyl-3-phenyl-4-quinazolinone by Morgan technique.

Figure 5.
Synthesis of 4(3H)-quinazolinone by amination of isatoic anhydride.

Figure 6.
Synthesis of 2-methyl-5-nitro-4(3H)-quinazolinone by amination of the benzoxazin-4-one derivative.
Figure 6.
Synthesis of 2-methyl-5-nitro-4(3H)-quinazolinone by amination of the benzoxazin-4-one derivative.

Figure 7.
Synthesis of 2,4-quinazolindione.

Figure 8.
Synthesis of 2-phenyl-4(3H)-quinazolinone.

Figure 9.
Synthesis of 2-phenyl-4(3H)-quinazolinone by the transition metal catalyzed method.

Figure 10.
Synthesis of 2,4-diamino-6-iodo-quinazoline.

Figure 11.
Synthesis of 2-substituted-4-aminoquinazoline.

Figure 12.
Synthesis of 2-substituted-4-methylquinazoline.

Figure 13.
4-anilinoquinazoline derivative in a complex with CDK2 [40].
Figure 13.
4-anilinoquinazoline derivative in a complex with CDK2 [40].

Figure 14.
Modes of action of anticancer quinazolines.

Figure 15.
Receptor tyrosine kinases which are targeted by the anticancer quinazolines.

Figure 16.
Examples for the marketed quinazolines anticancer.

Figure 17.
The anticancer quinazolines 4–7.

Figure 18.
The anticancer quinazolines 8–14.

Figure 19.
The anticancer quinazolines 15–17.

Figure 20.
The anticancer quinazolines 18–20.

Figure 21.
The anticancer quinazolines 21–24.

Figure 22.
The anticancer quinazolines 25–27.

Figure 23.
The anticancer quinazolines 28–30.

Figure 24.
The anticancer quinazolines 31–34.

Figure 25.
The SAR of quinazolines as EGFR inhibitors.

Figure 26.
The anticancer quinazolines vandetanib and cediranib.

Figure 27.
The general structural model for the quinazoline-based dual EGFR and VEGFR inhibitors.

Figure 28.
The anticancer quinazolines 37–40.

Figure 29.
The anticancer quinazolines 41–43.

Figure 30.
The anticancer quinazolines 44–47.

Figure 31.
The anticancer quinazolines 48.

Figure 32.
The anticancer quinazolines 49–52.

Figure 33.
The anticancer quinazolines 53–57.

Figure 34.
The anticancer quinazolines 58–62.

Figure 35.
The anticancer quinazolines 63 and 64.

Figure 36.
The anticancer quinazolines 65–67.

Figure 37.
The anticancer quinazolines 68–71.


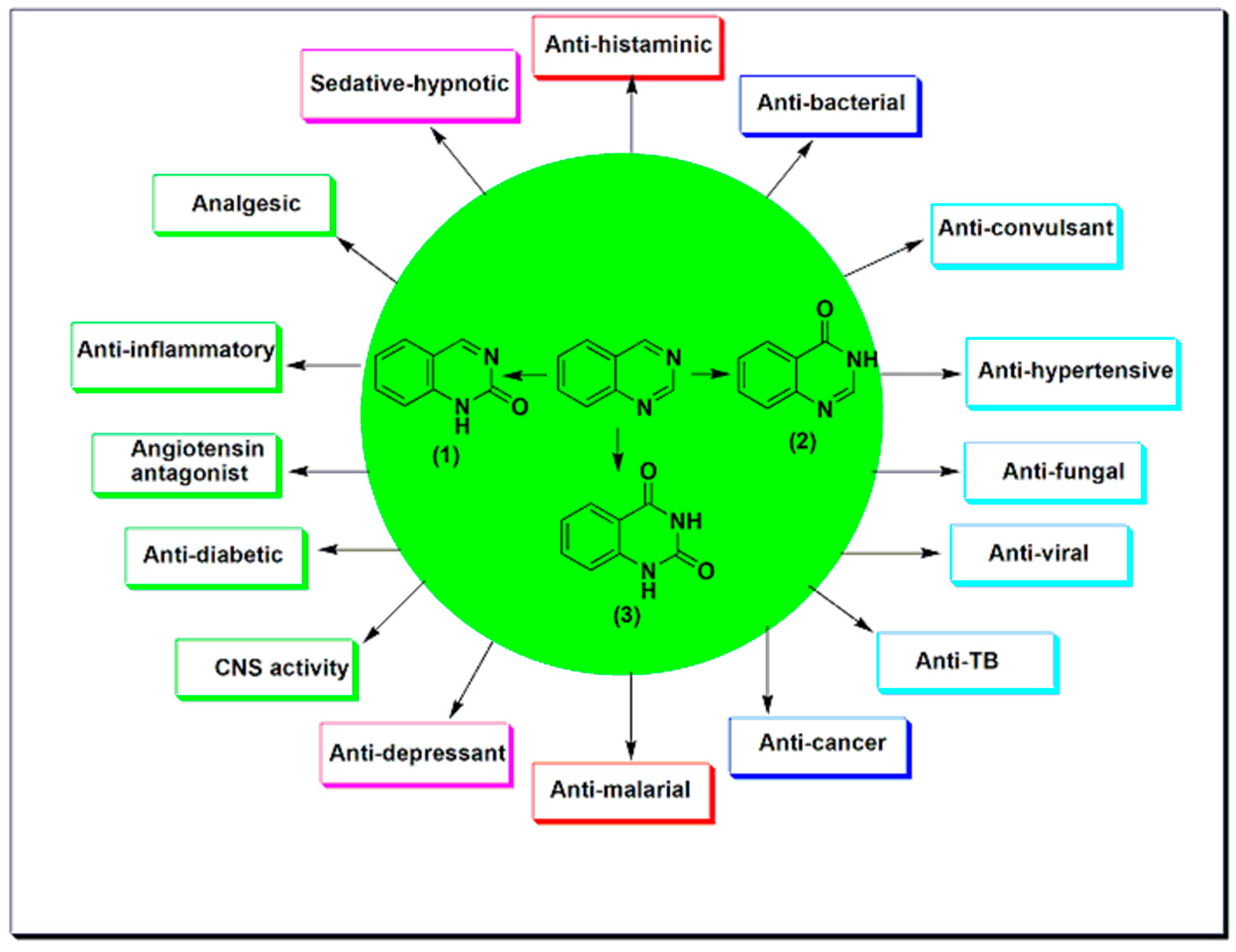{kind=link}
{kind=link}
{kind=link}
{kind=link}
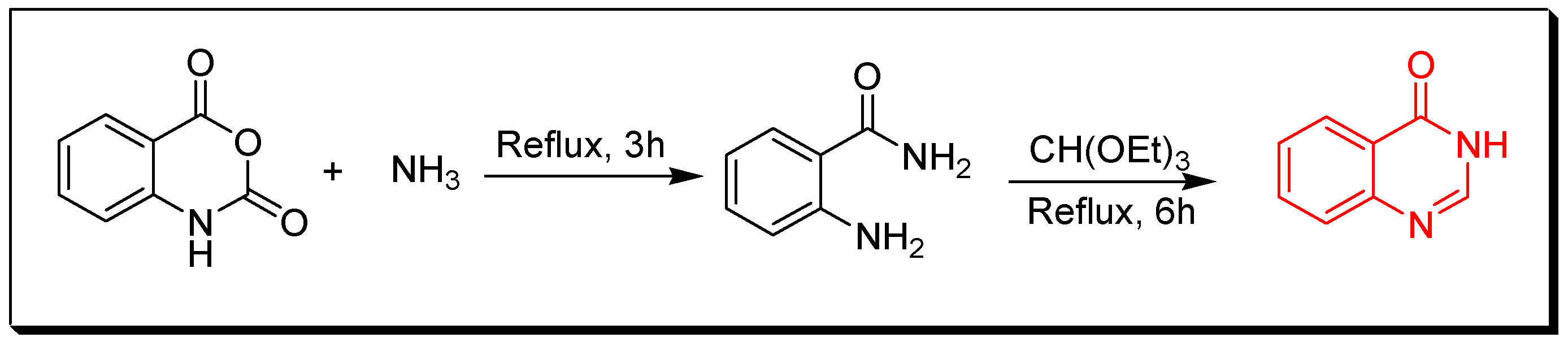{kind=link}
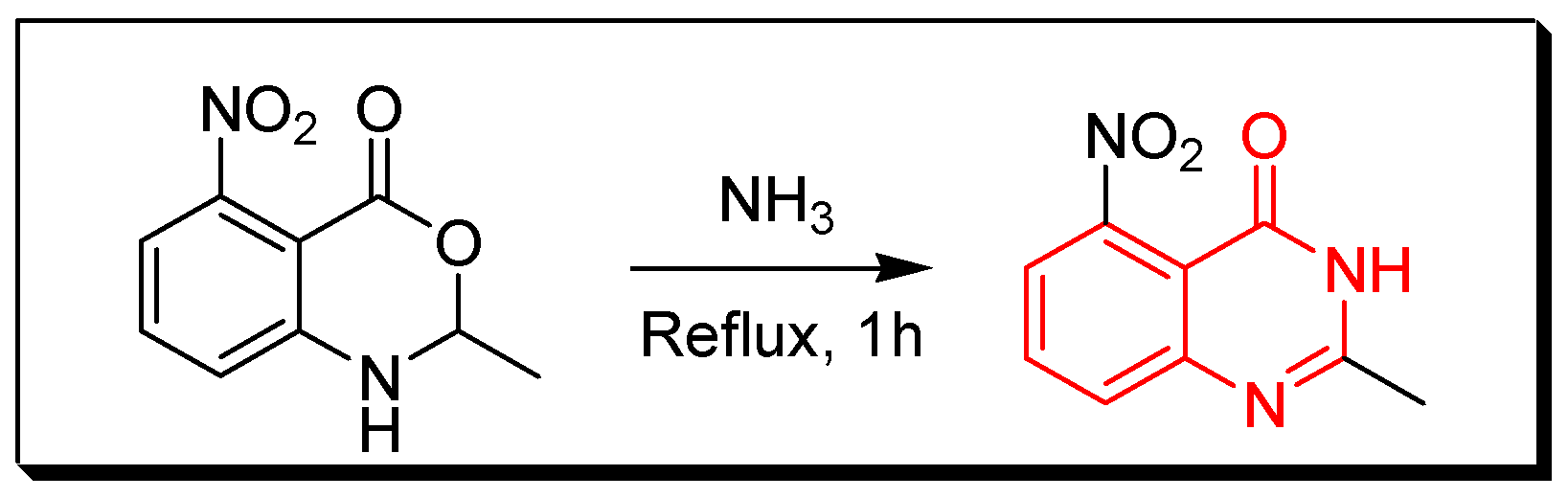{kind=link}
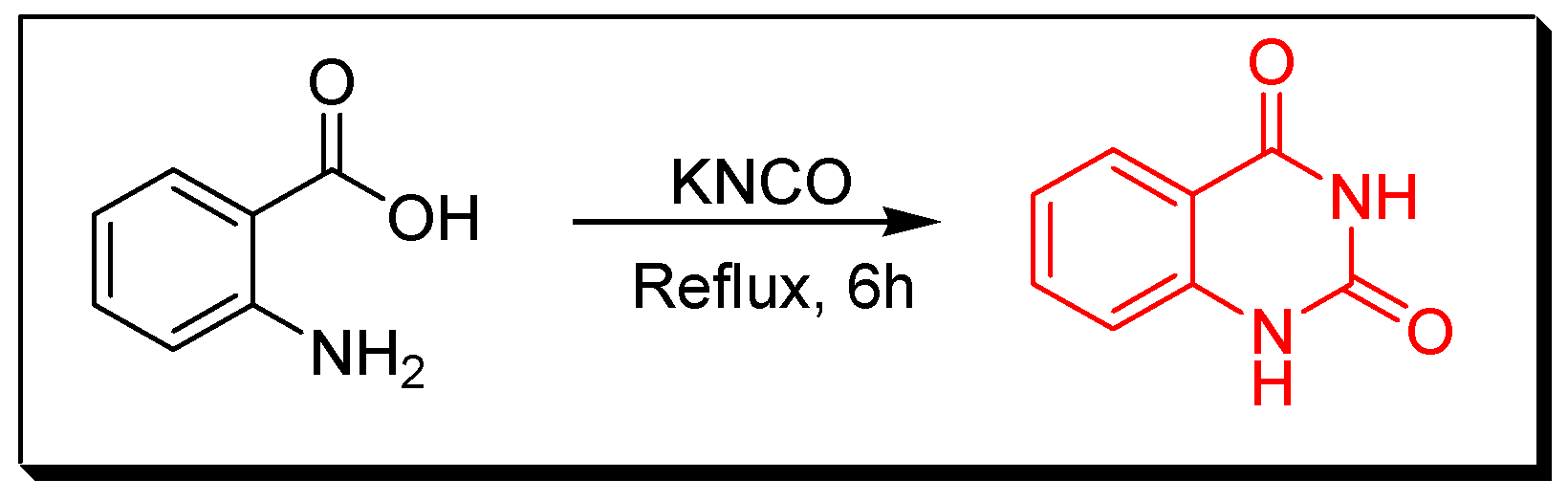{kind=link}
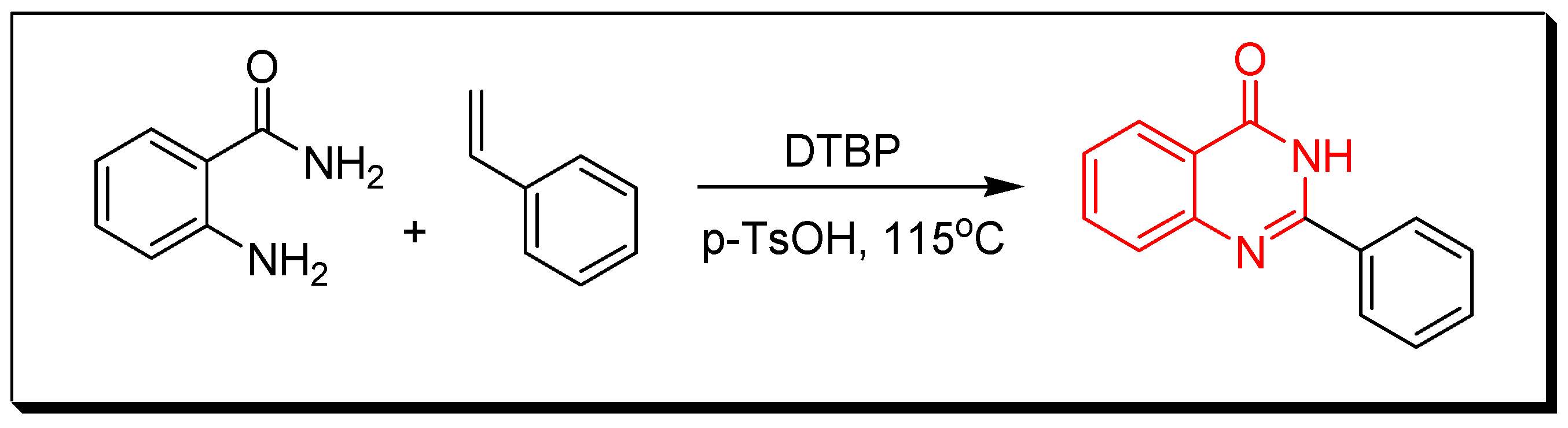{kind=link}
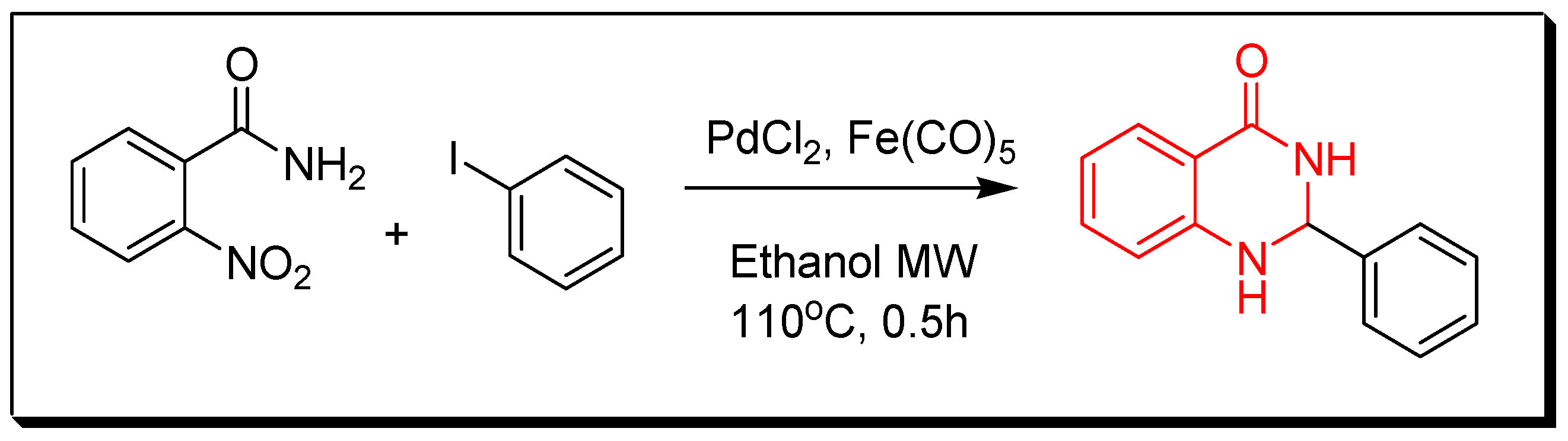{kind=link}
{kind=link}
{kind=link}
{kind=link}
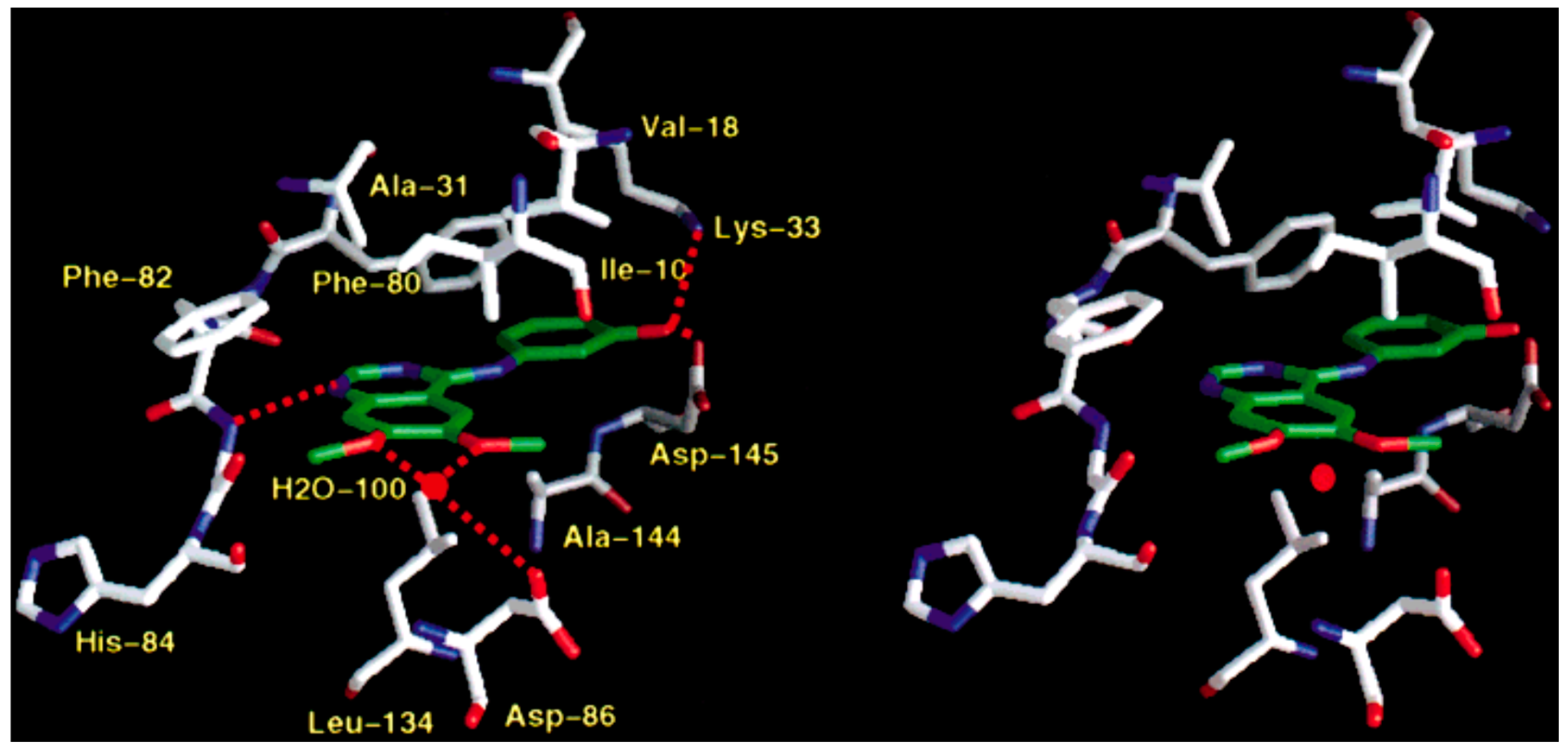{kind=link}
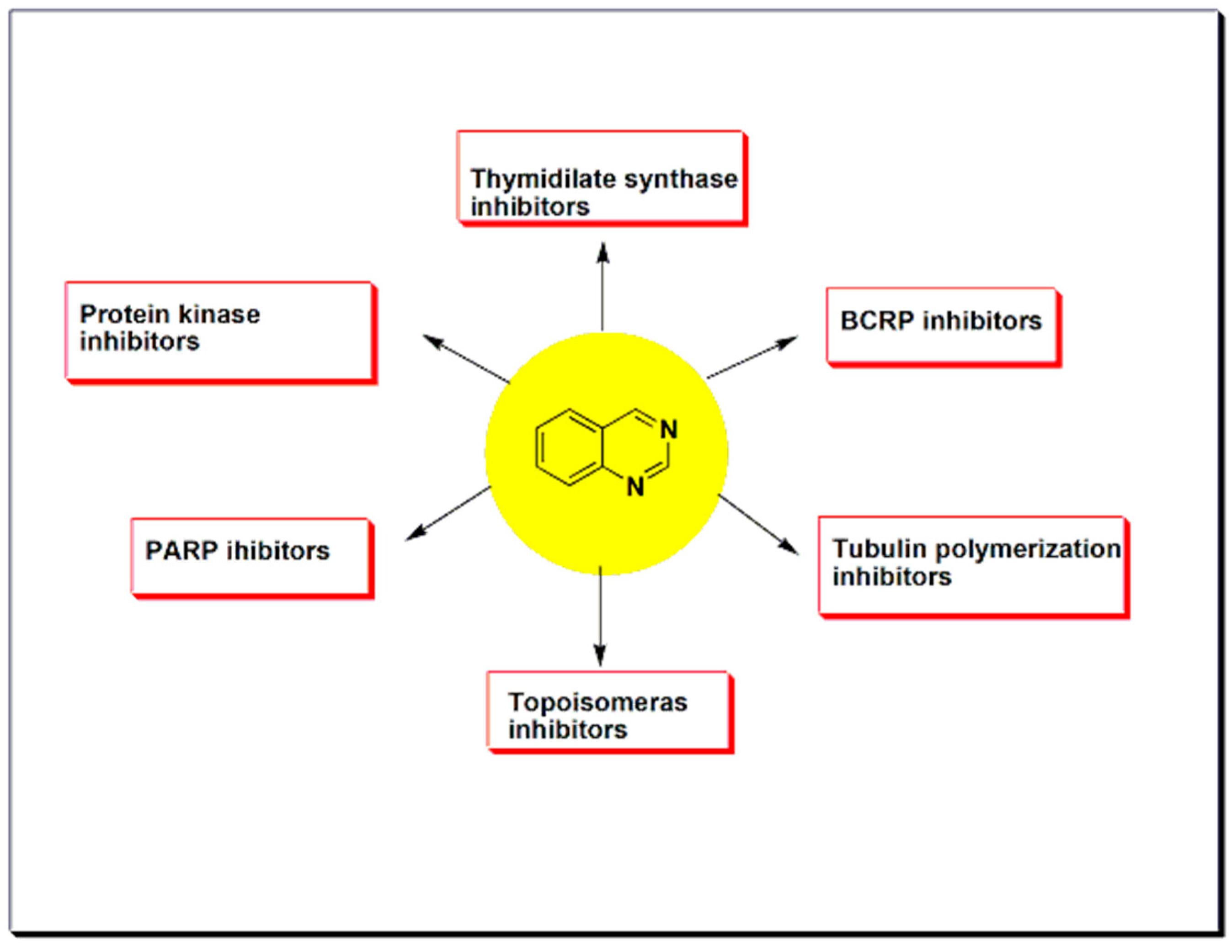{kind=link}
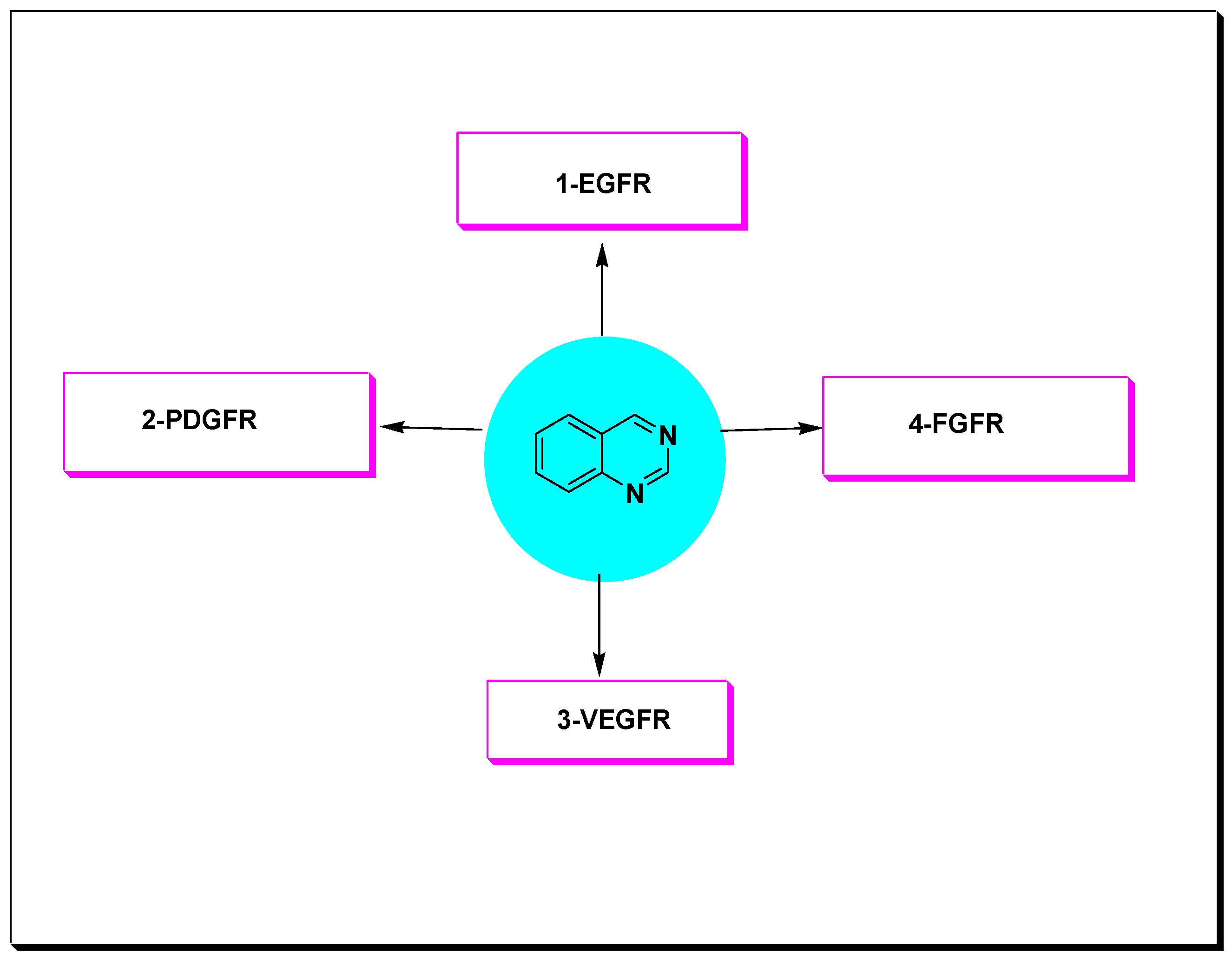{kind=link}
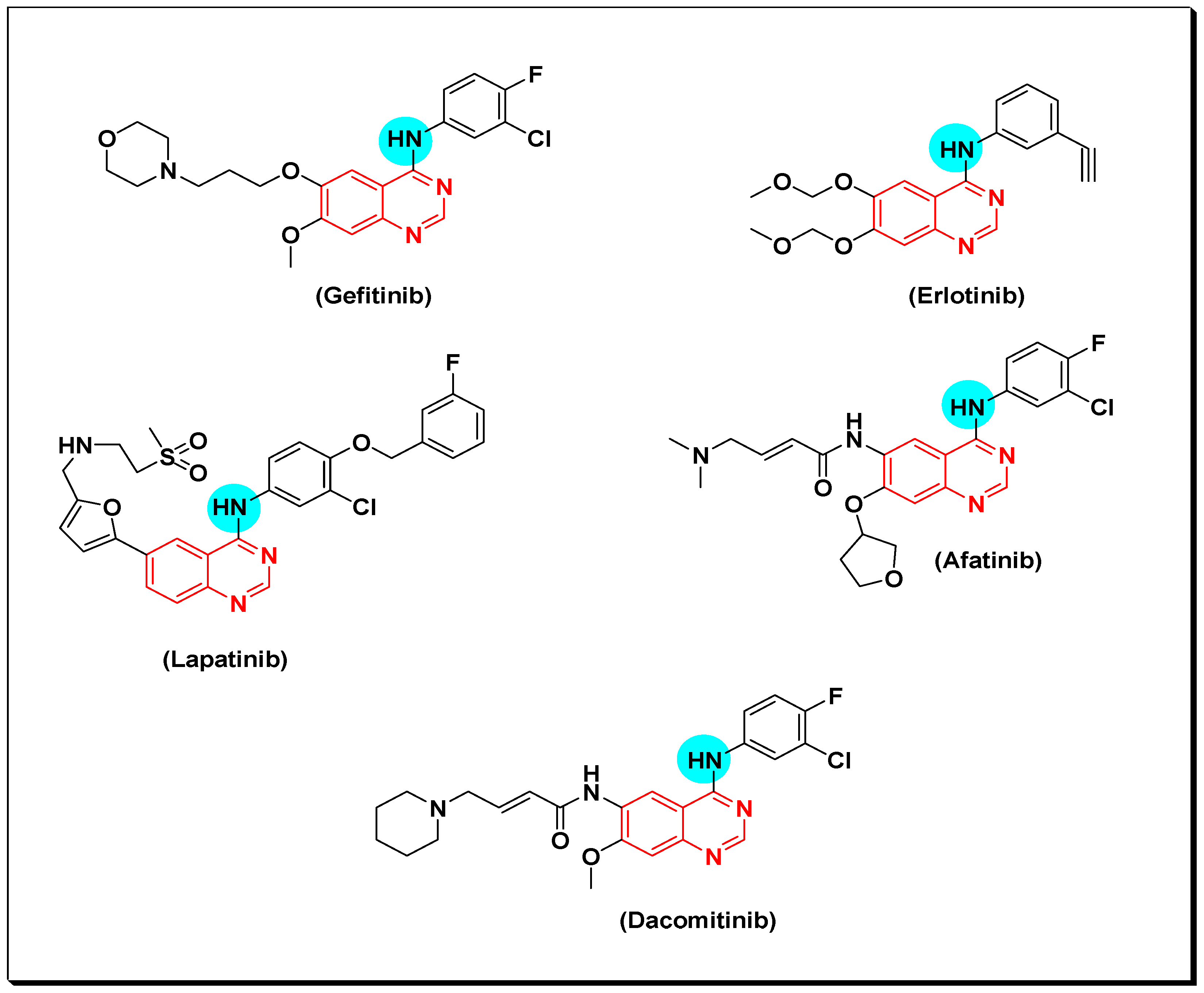{kind=link}
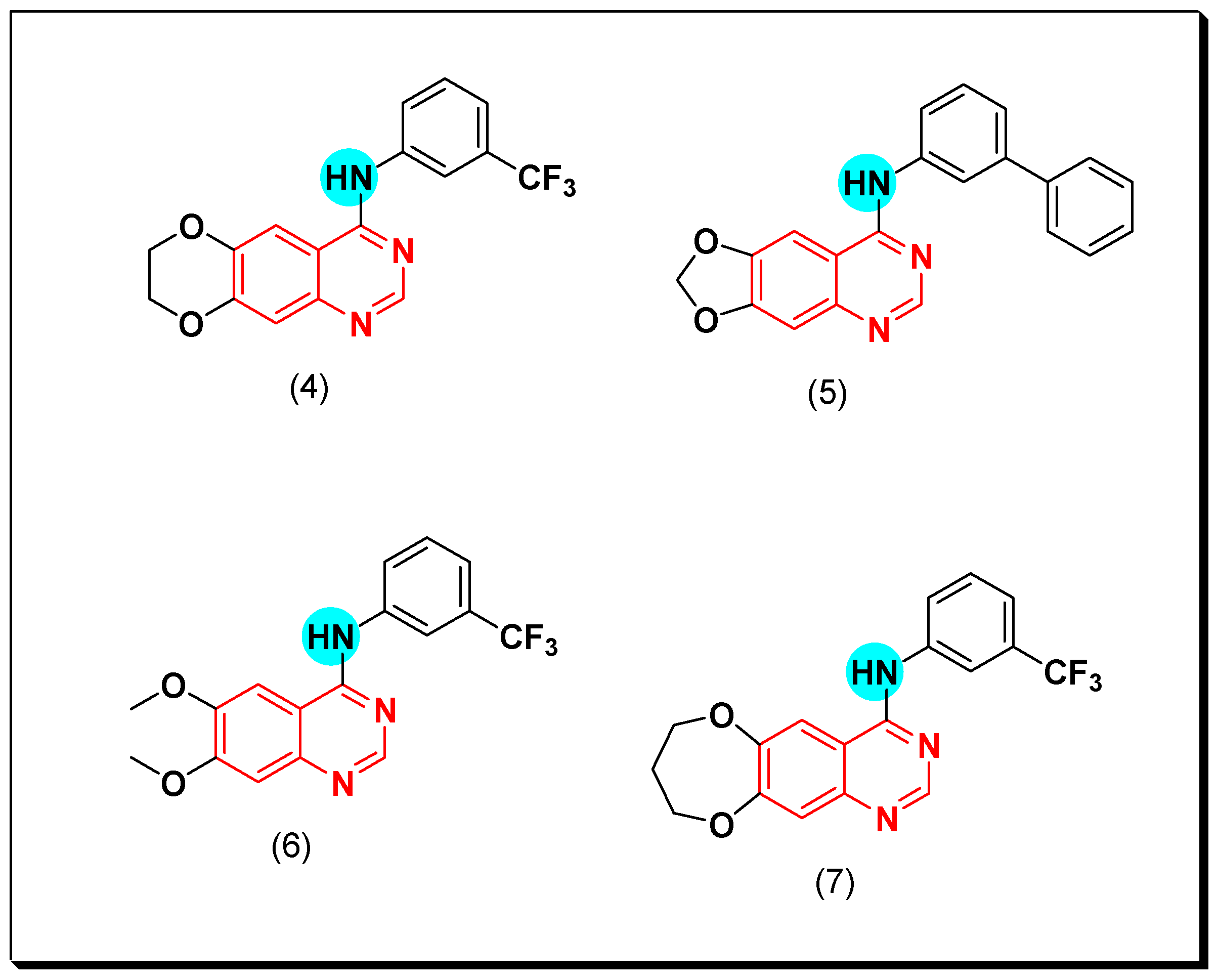{kind=link}
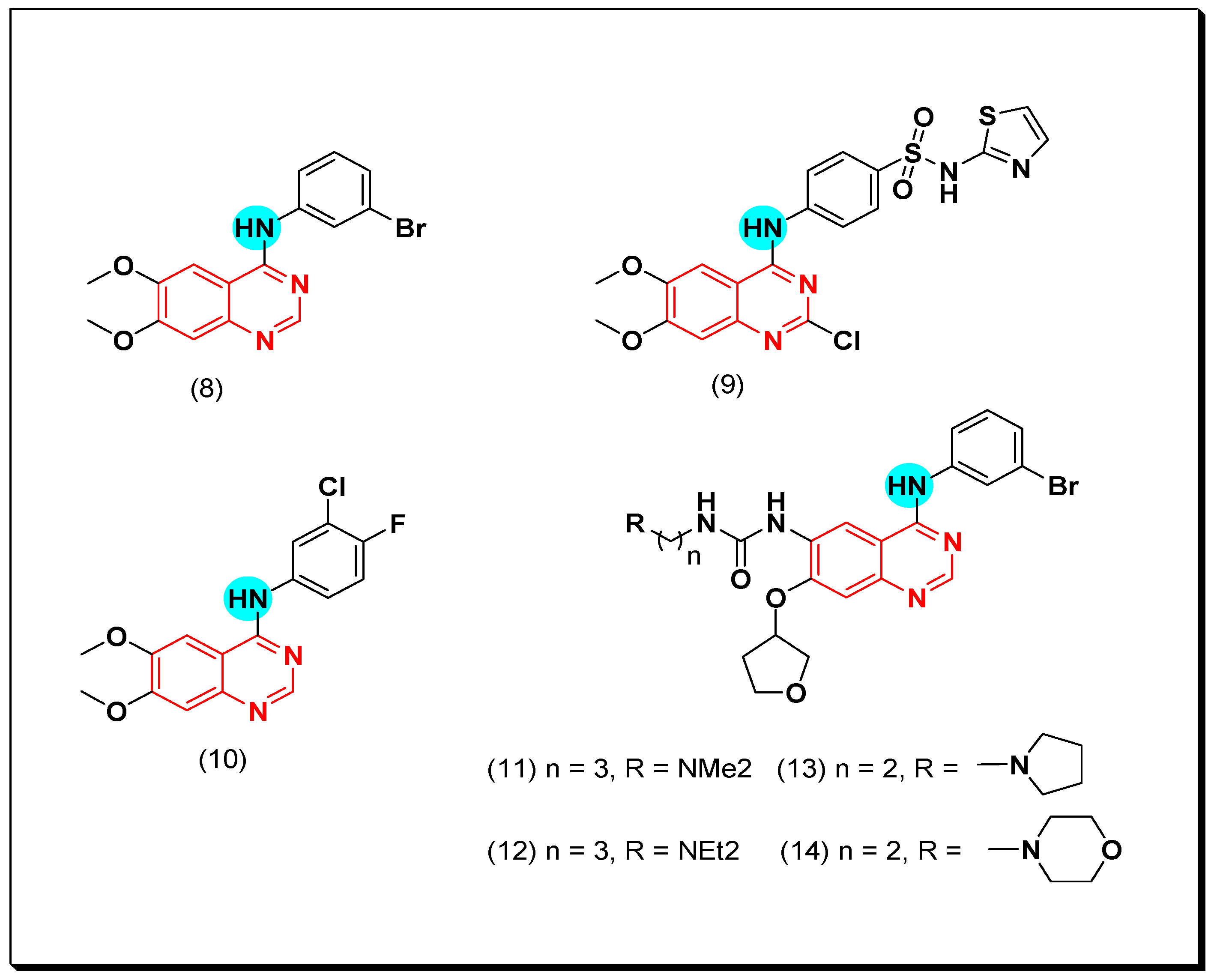{kind=link}
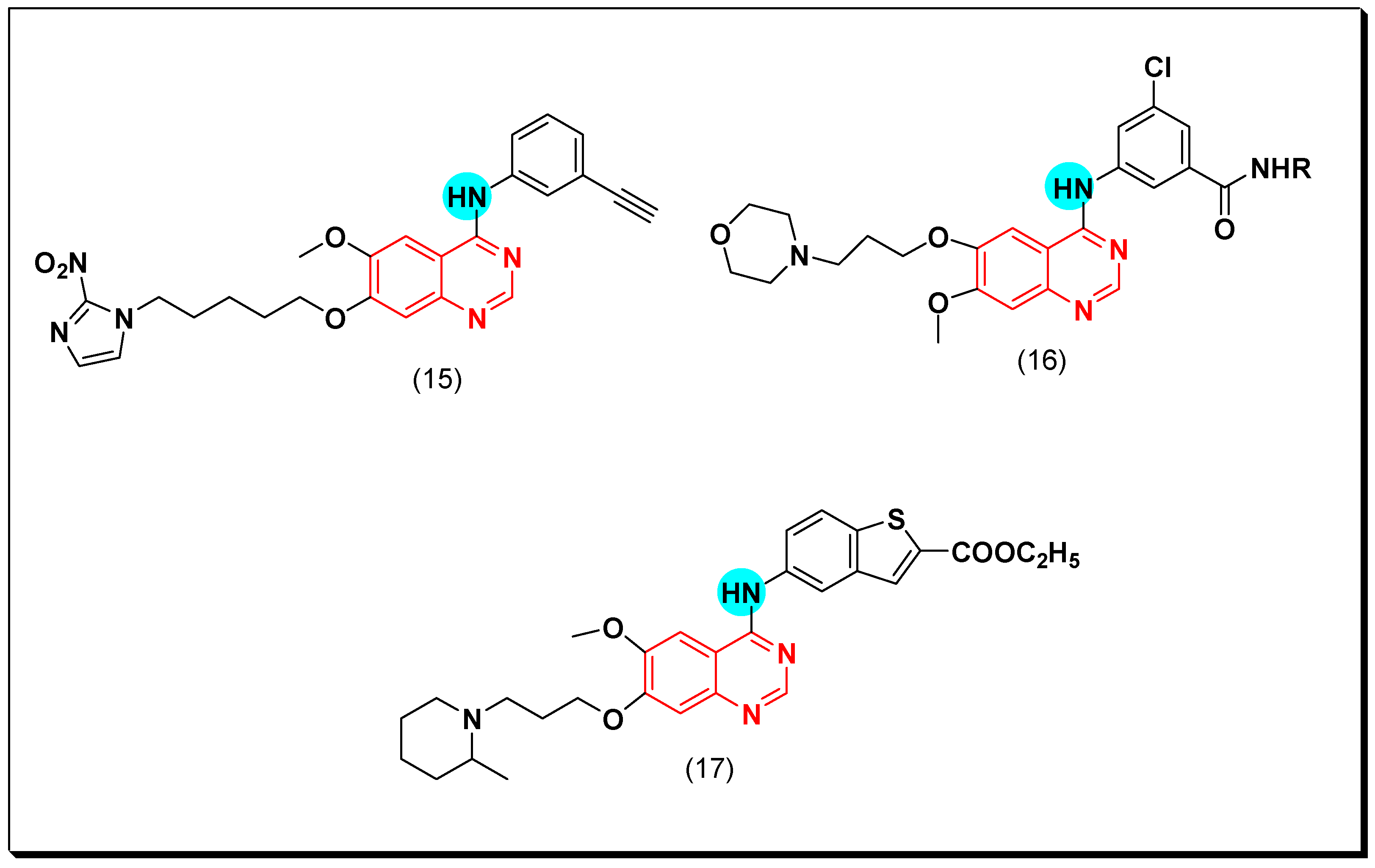{kind=link}
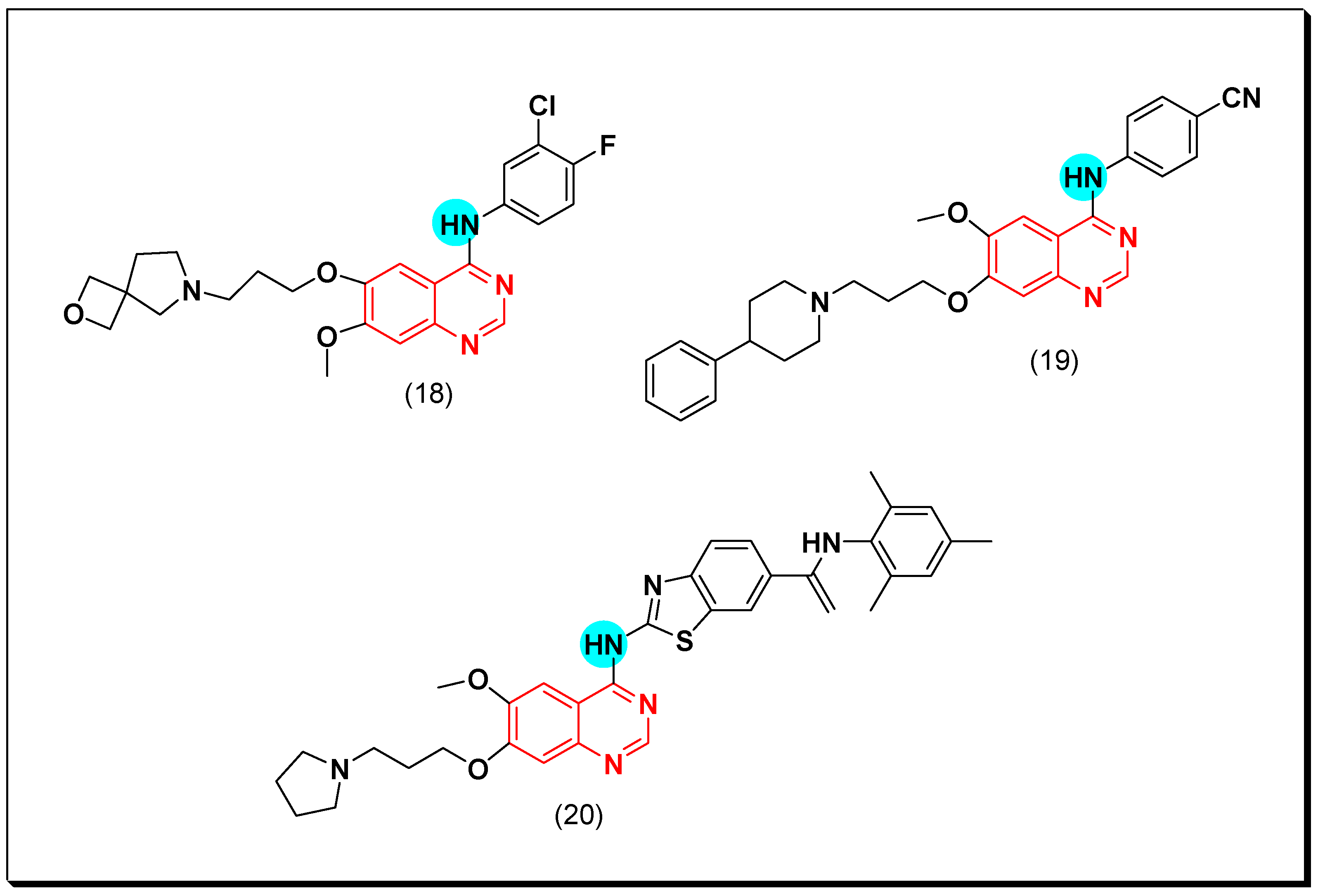{kind=link}
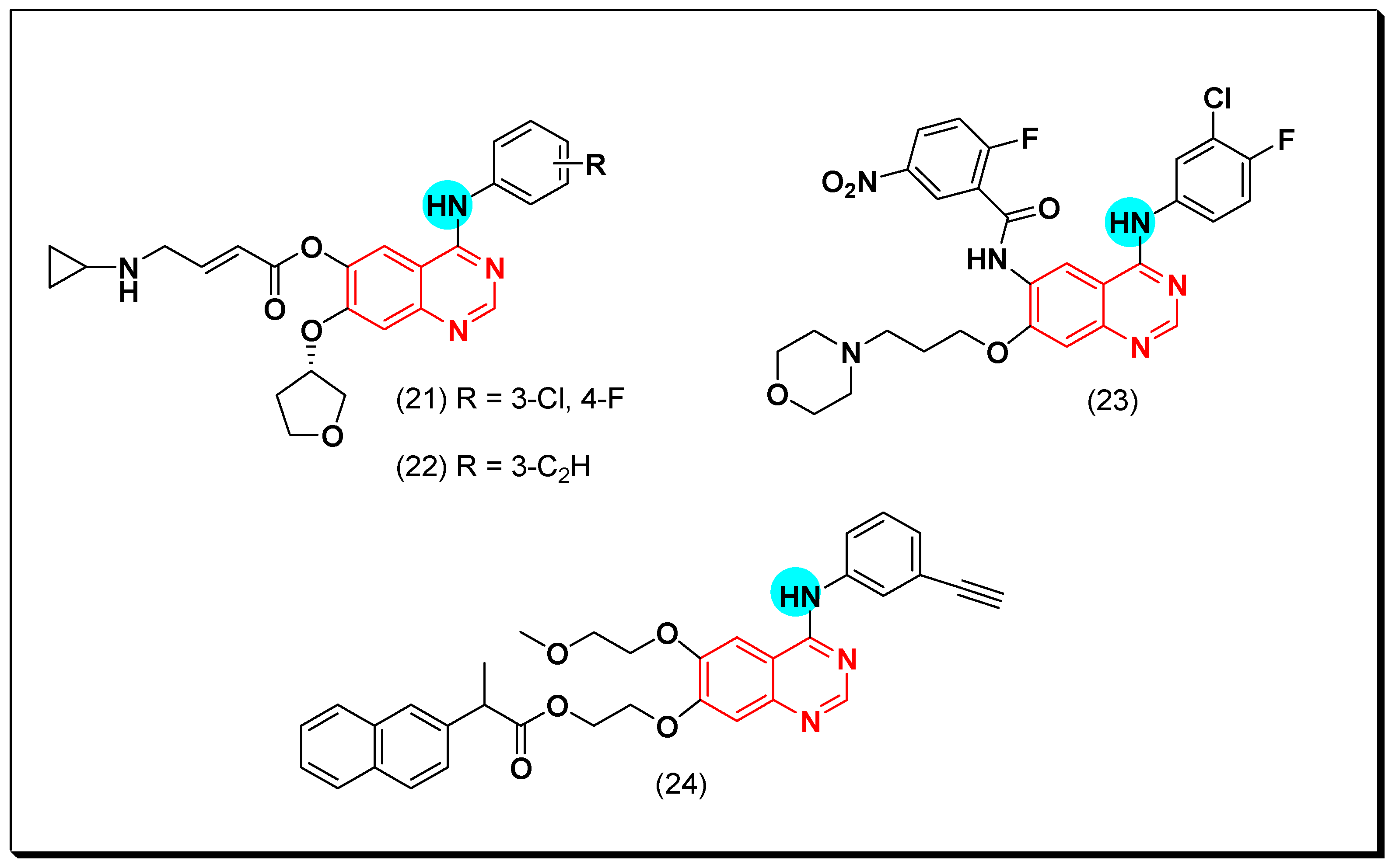{kind=link}
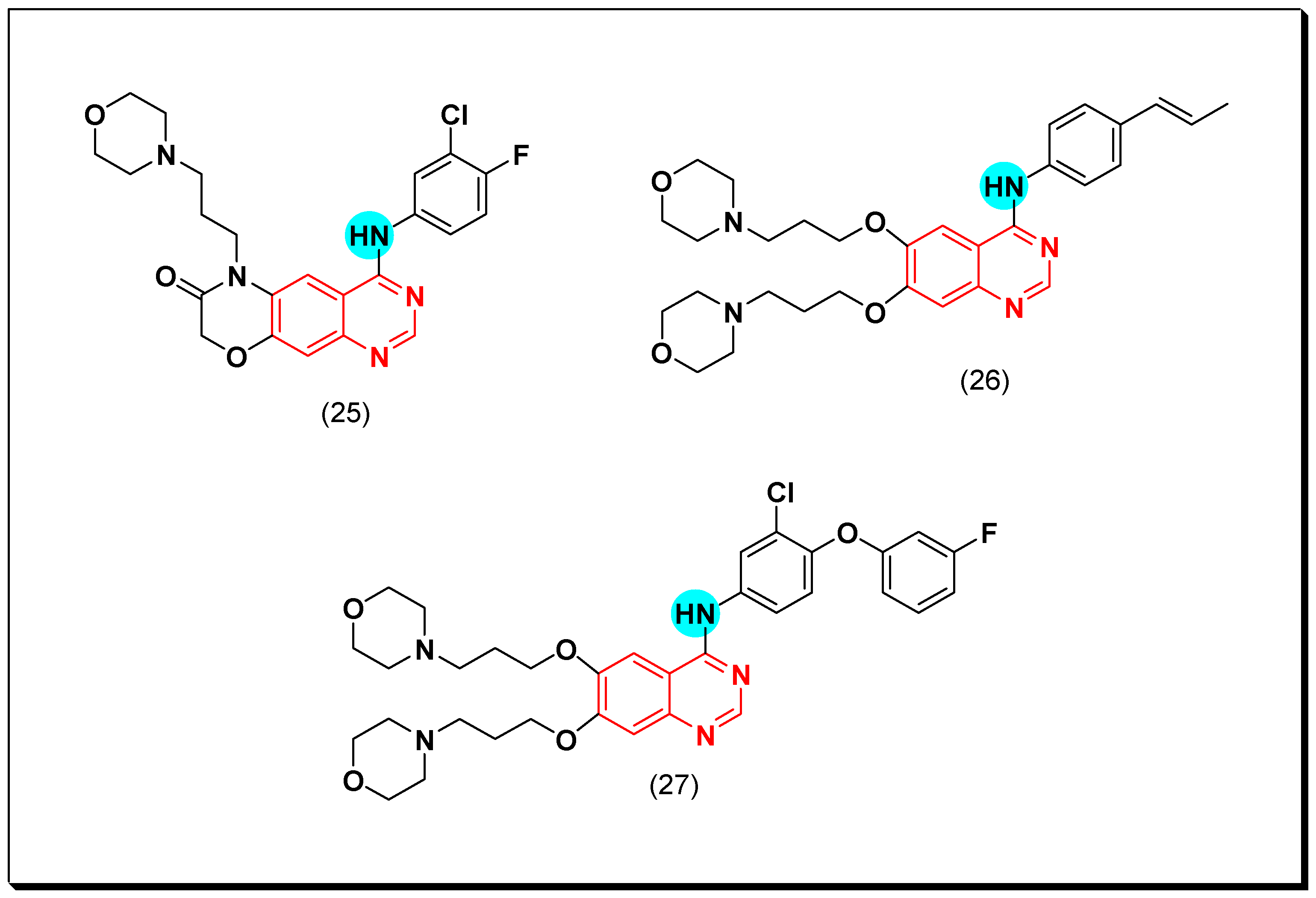{kind=link}
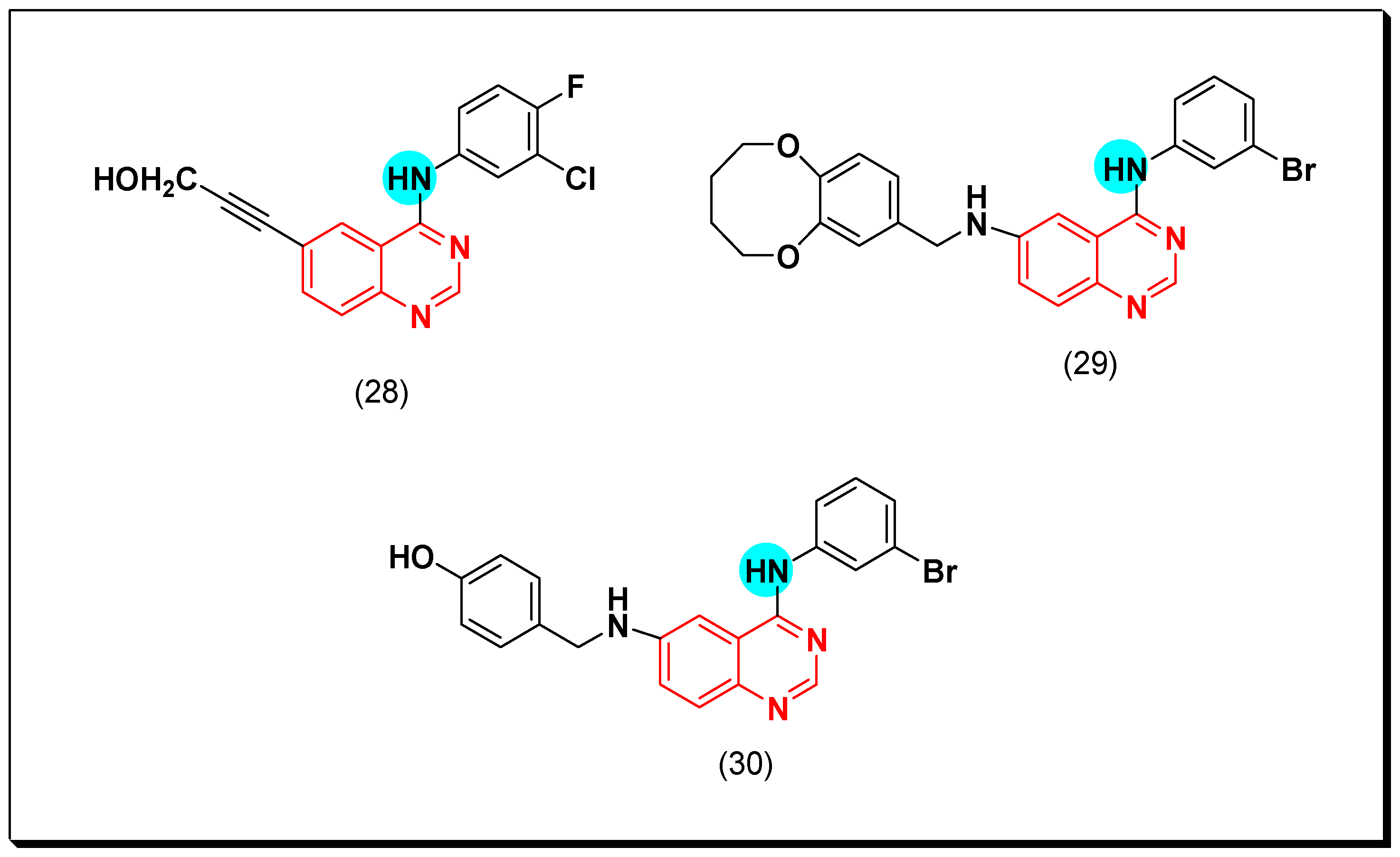{kind=link}
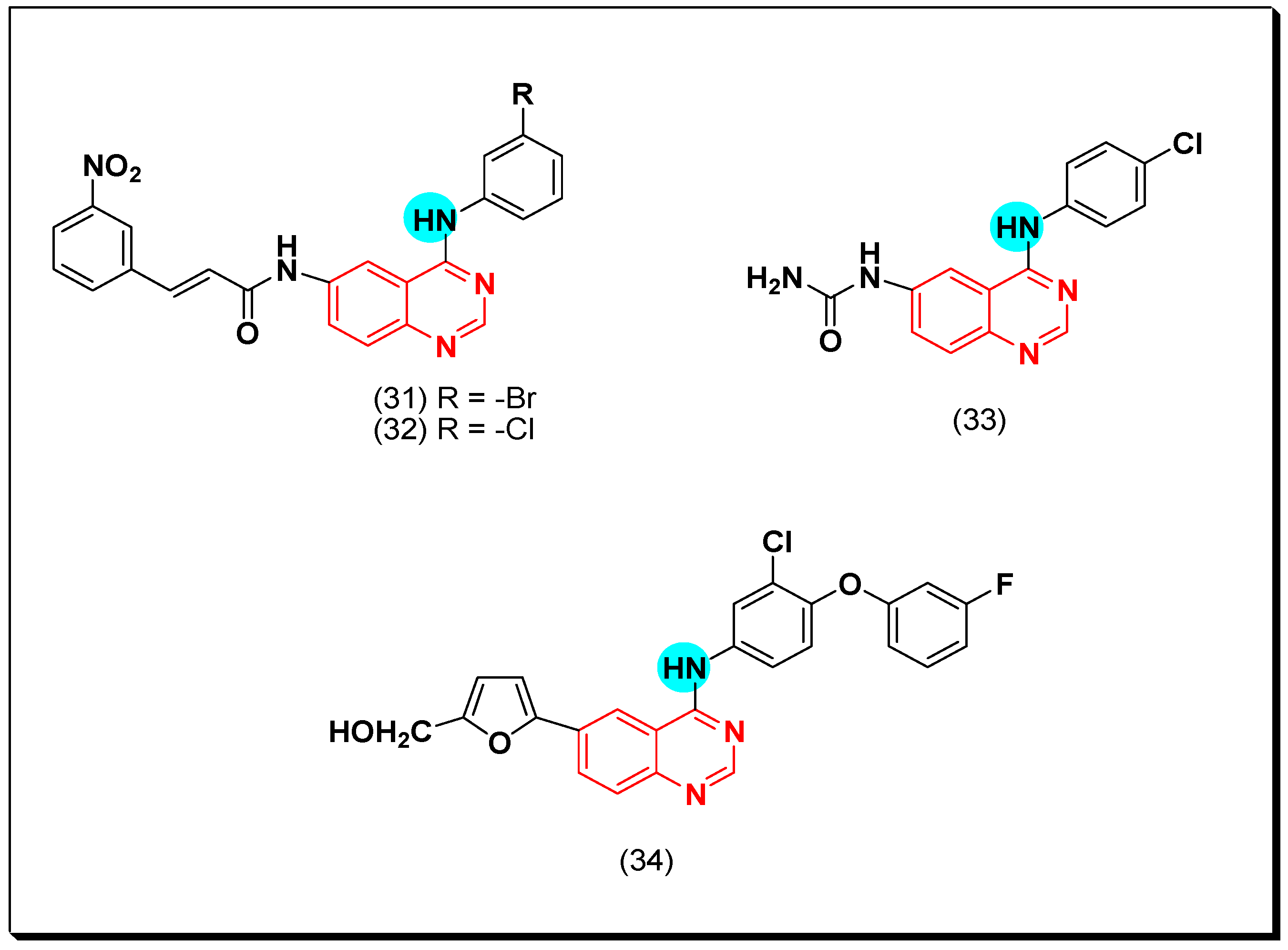{kind=link}
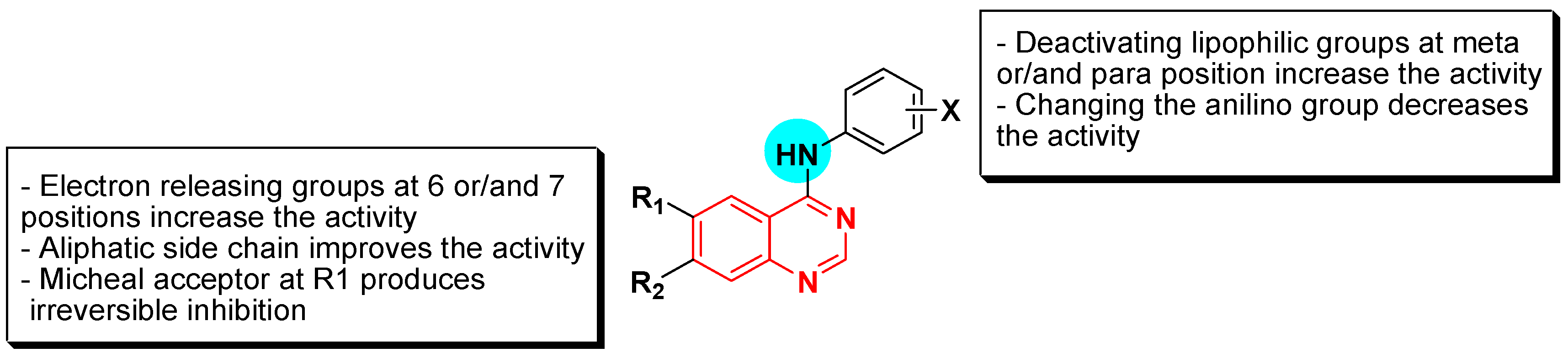{kind=link}
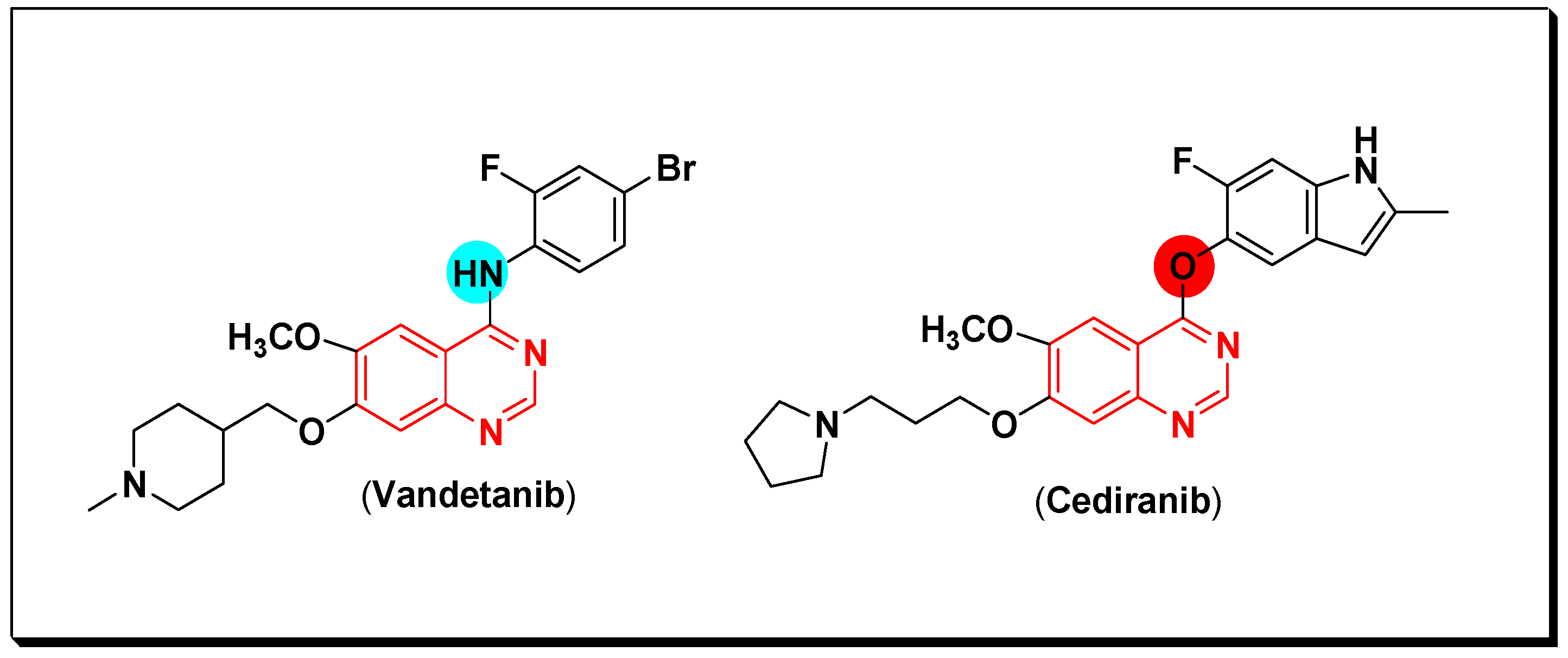{kind=link}
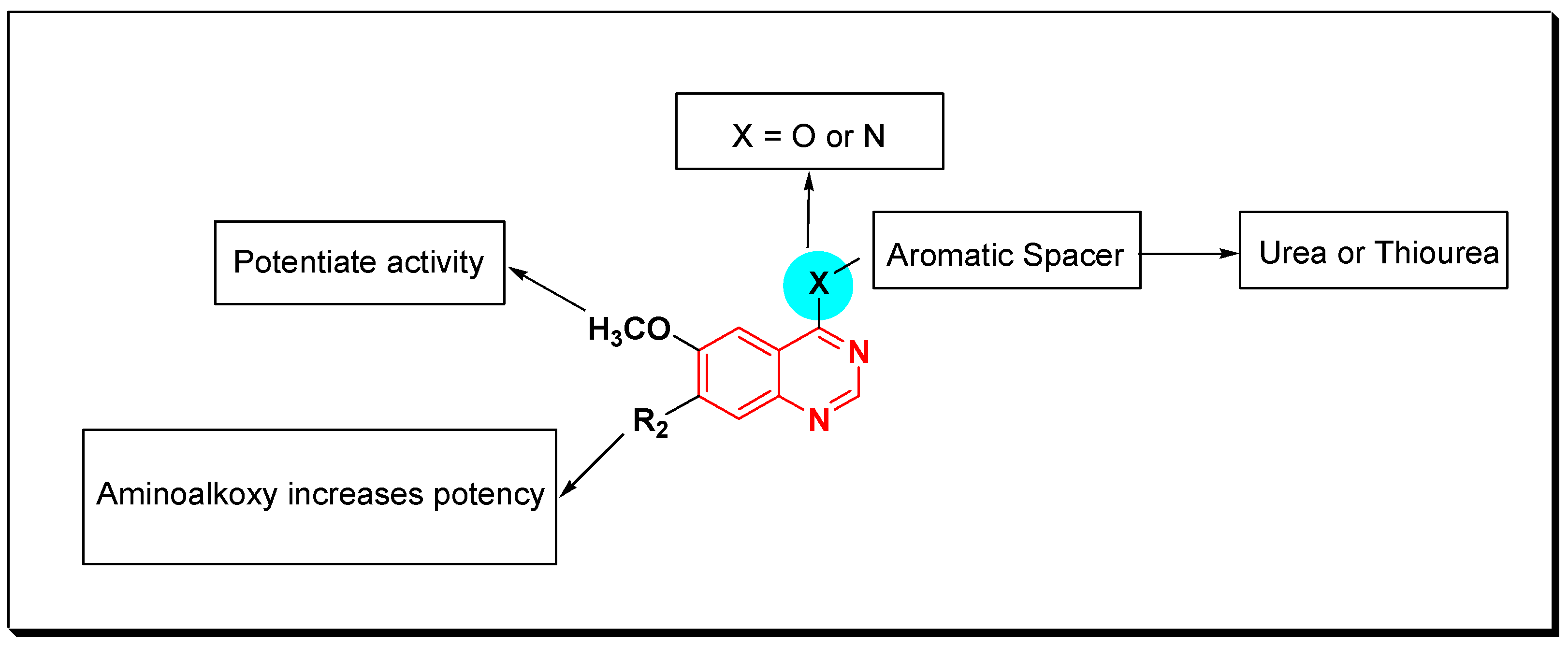{kind=link}
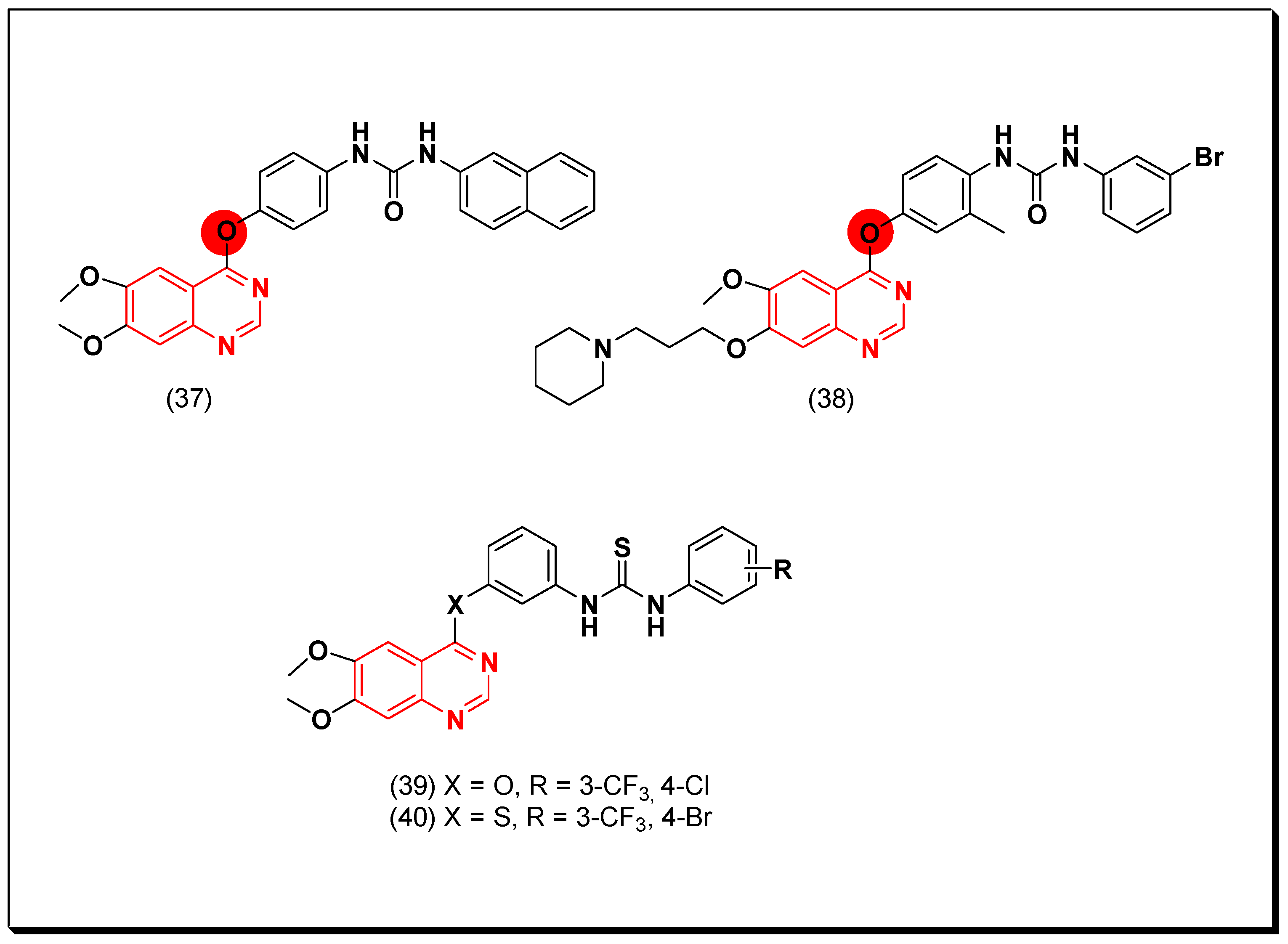{kind=link}
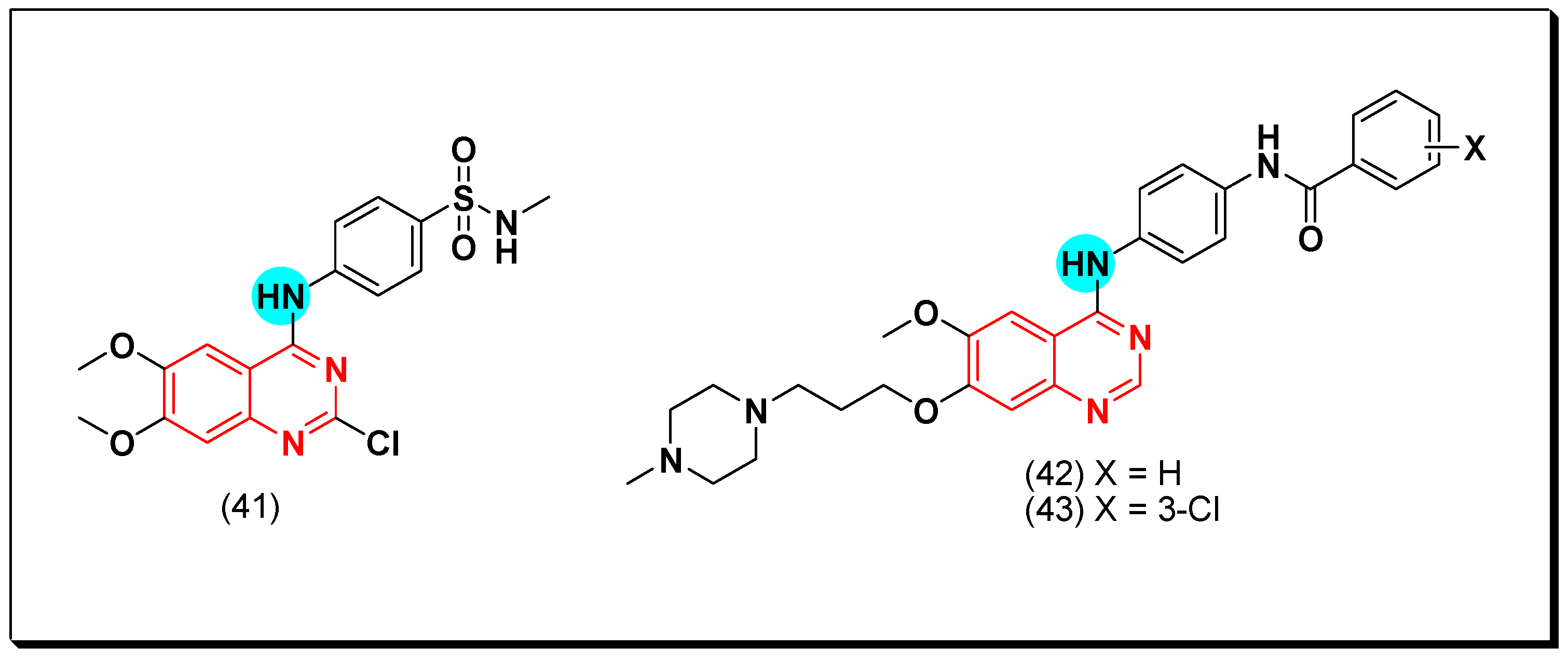{kind=link}
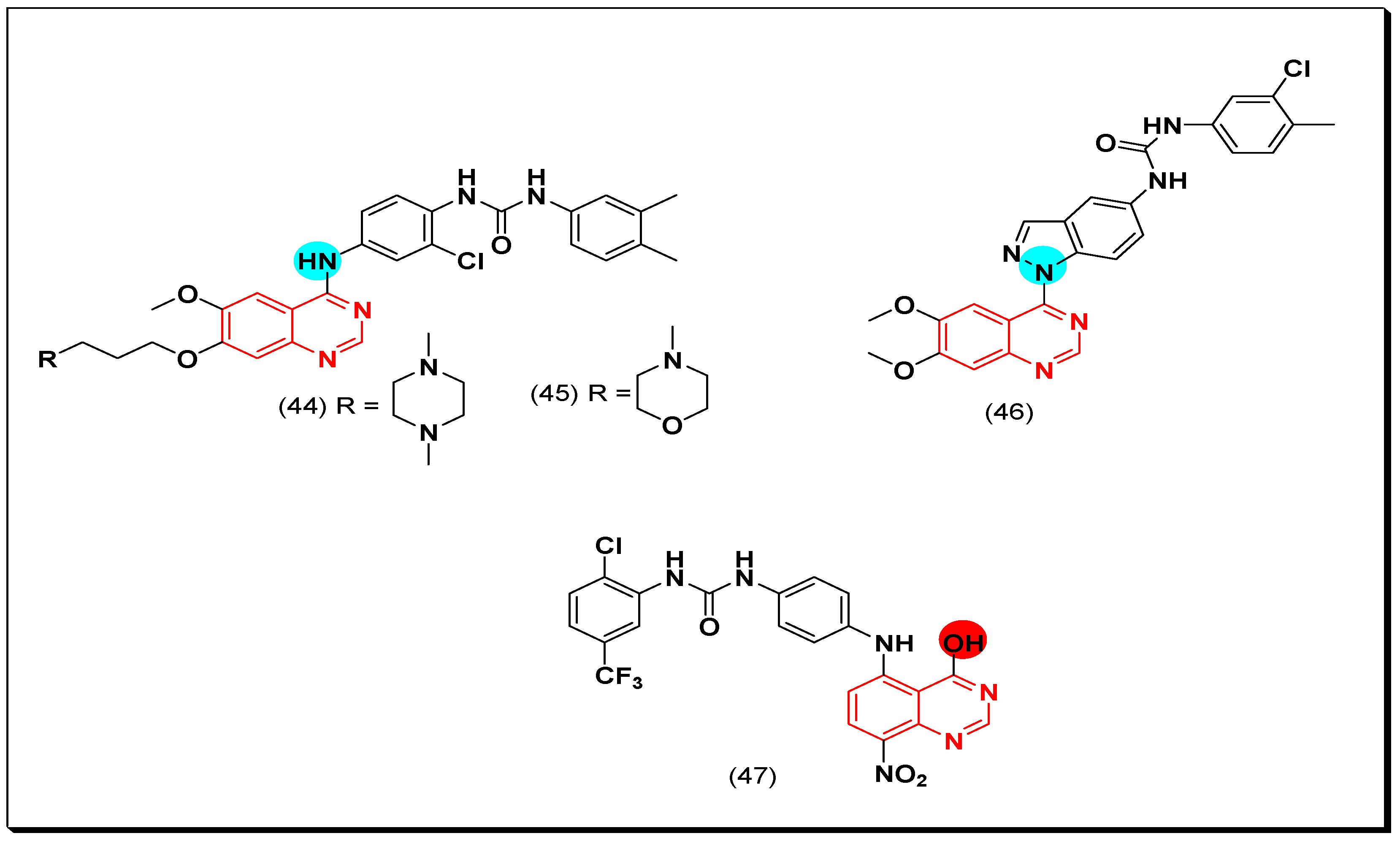{kind=link}
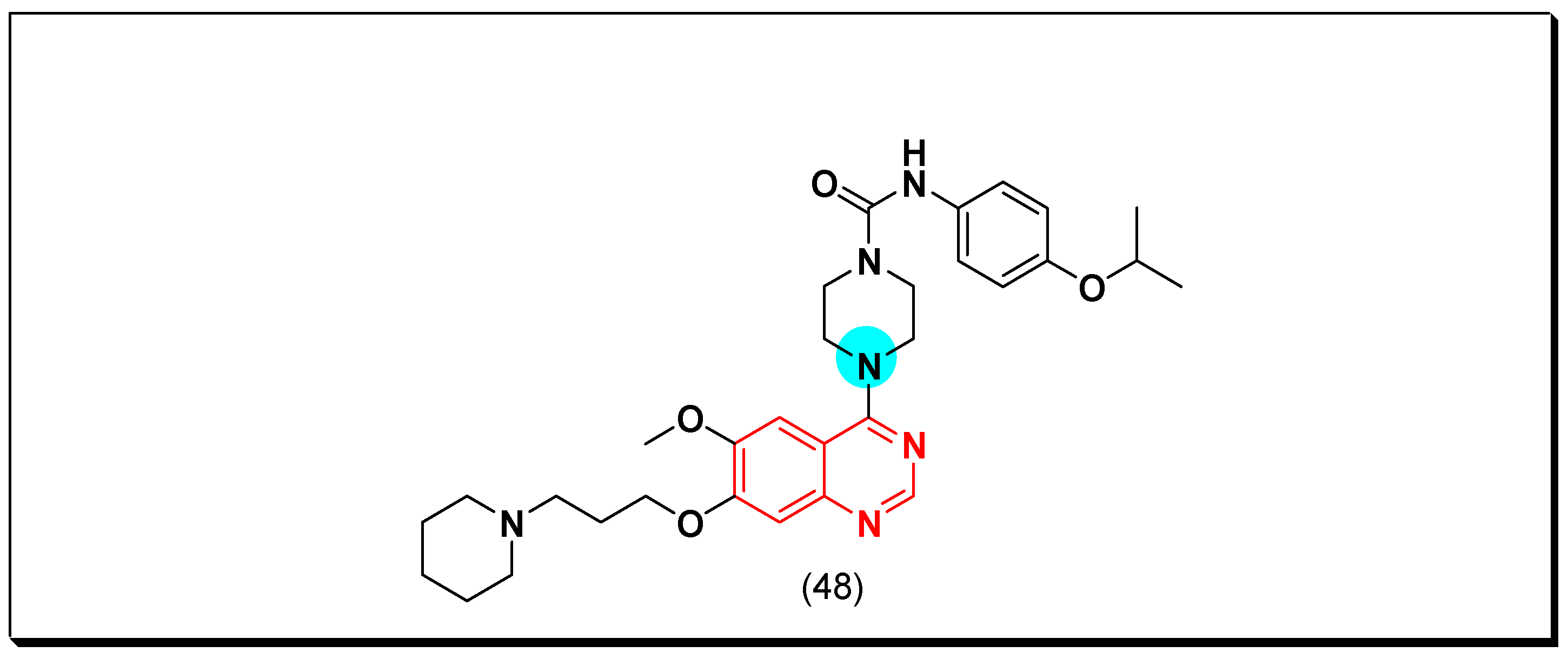{kind=link}
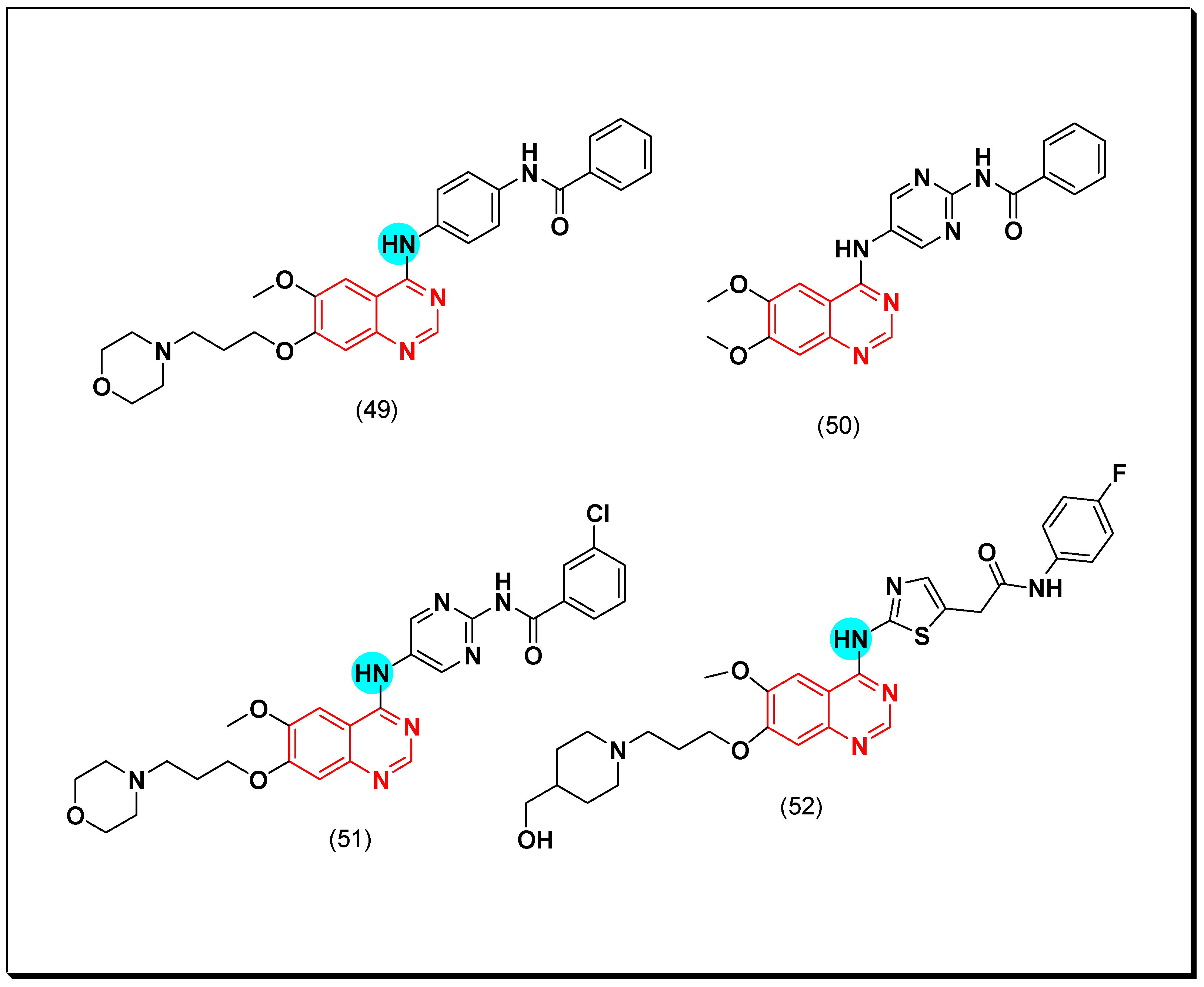{kind=link}
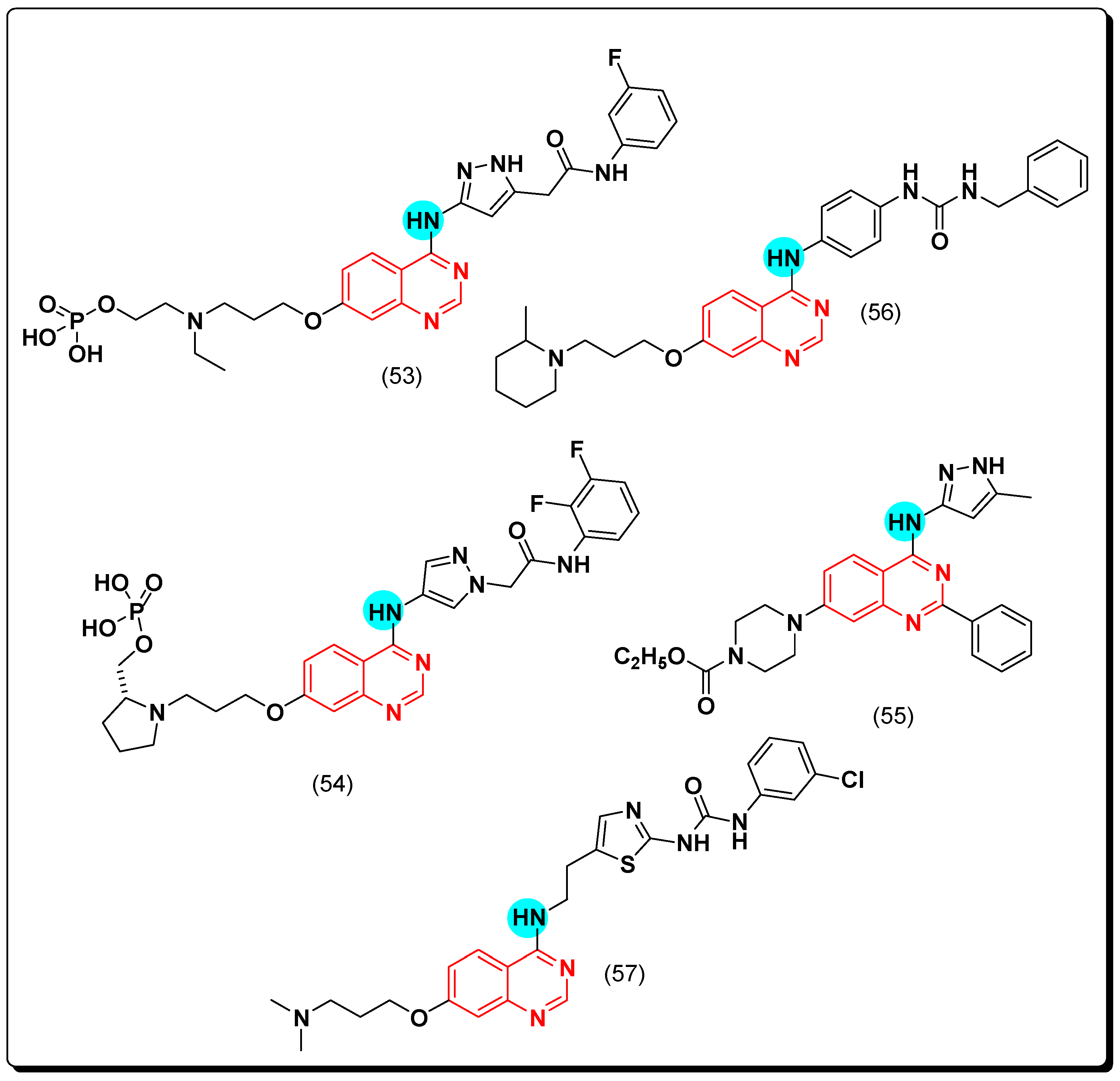{kind=link}
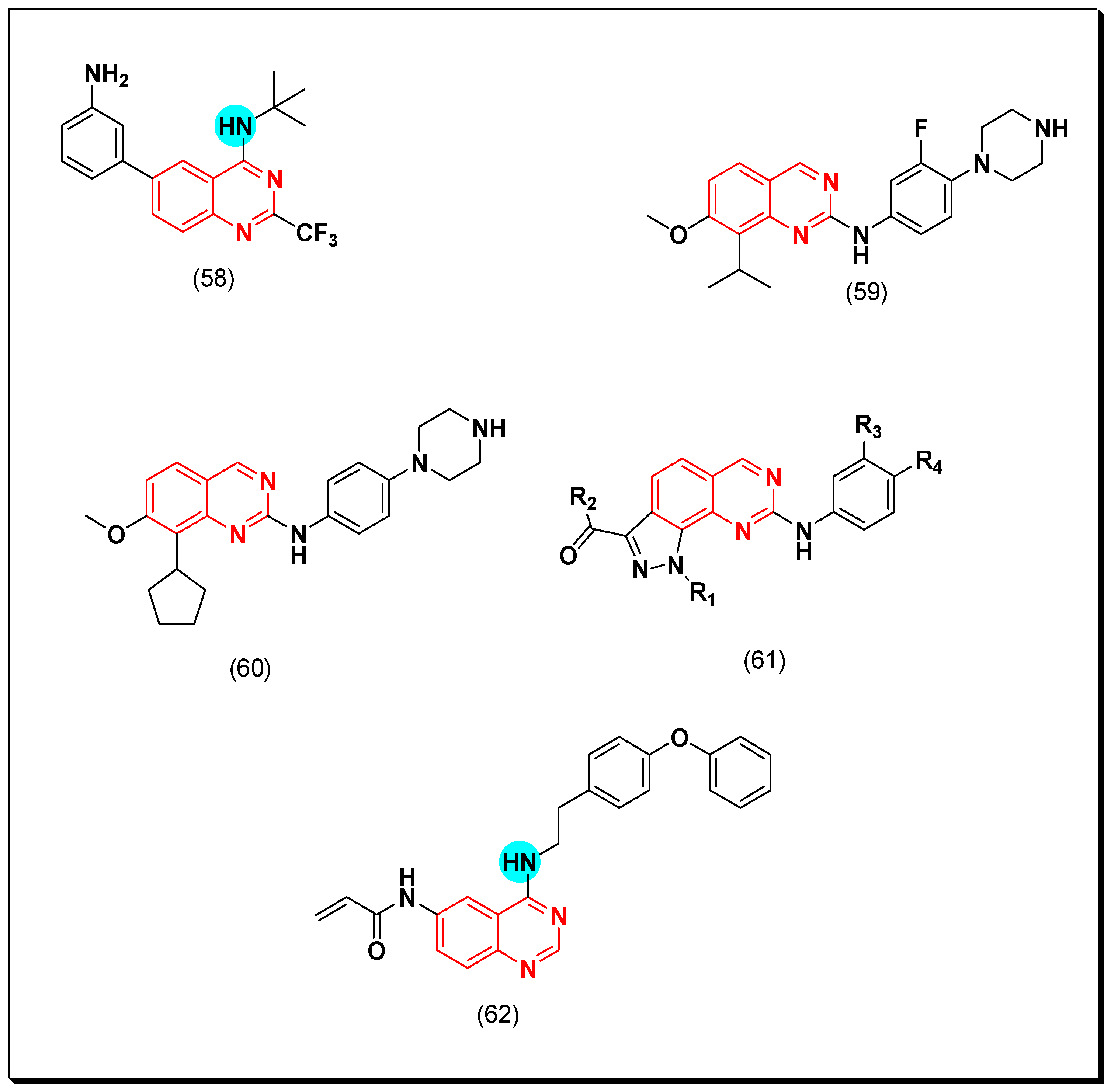{kind=link}
{kind=link}
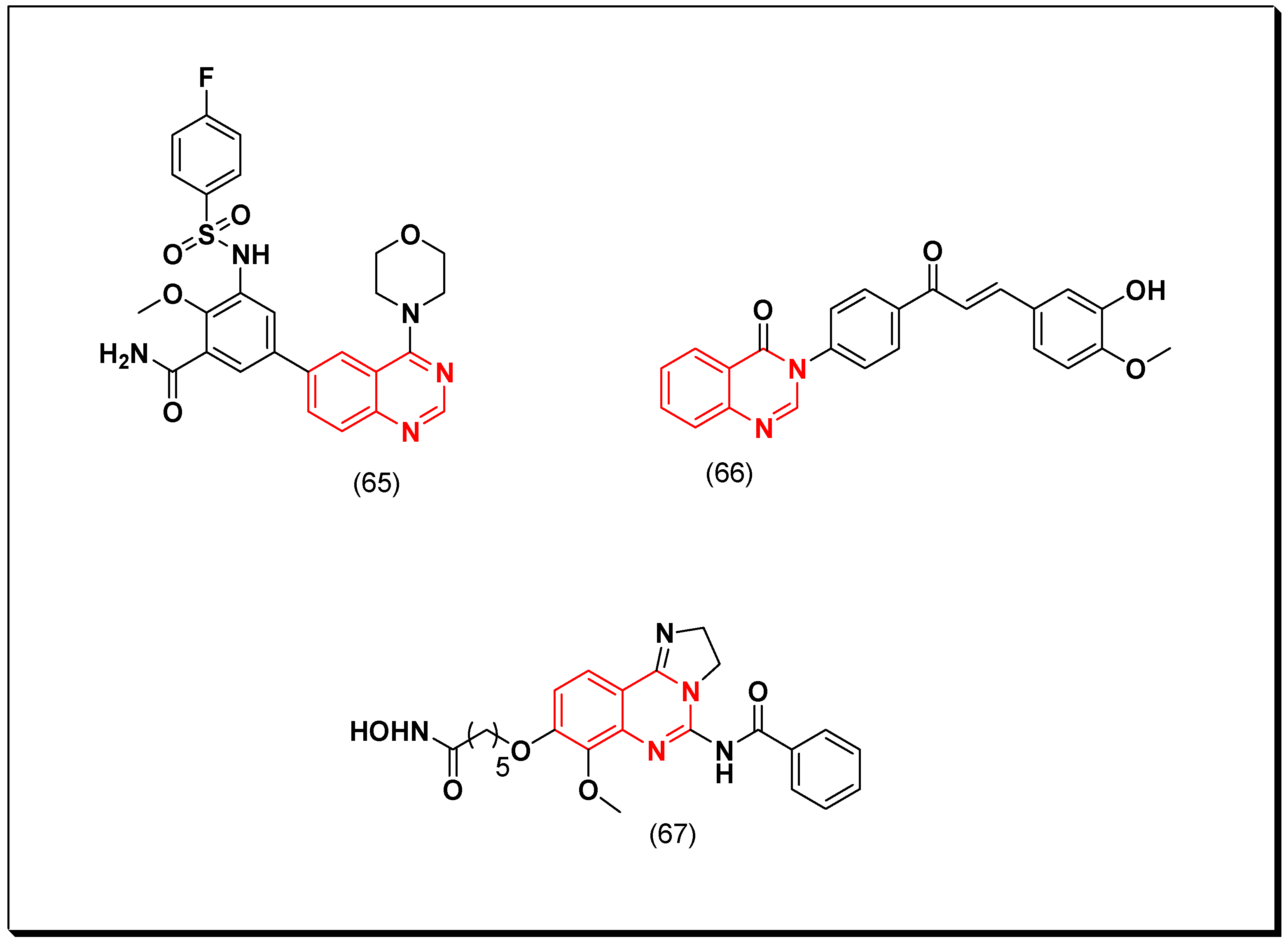{kind=link}
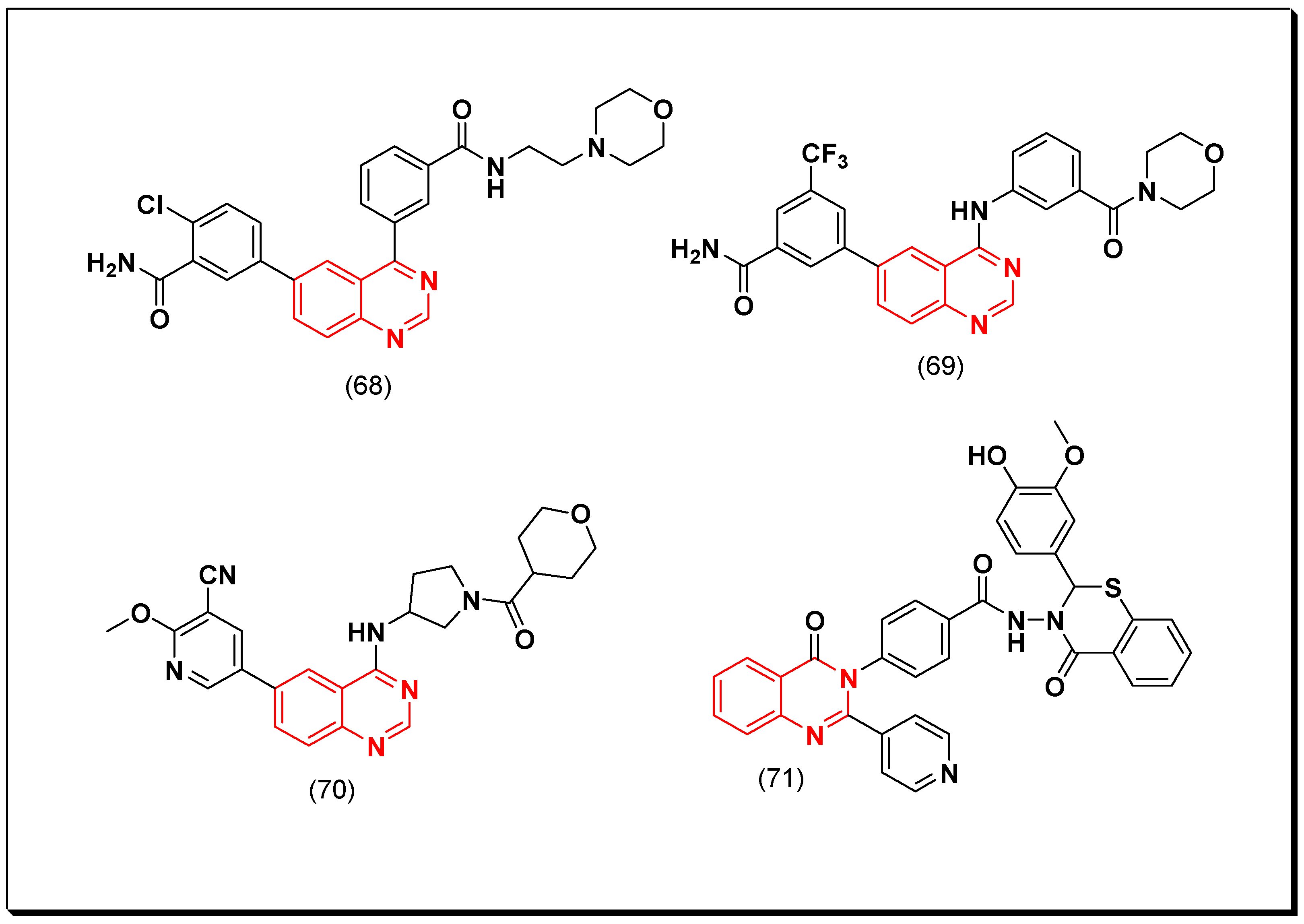{kind=link}
Table 1.
The physicochemical characters of 4-aminoarylquinazoline.
| Character | 4-Aminoquinazoline |
|---|---|
| Molecular formula | C8H9N3 |
| Molecular weight | 147.18 g/mol |
| Number of heavy atoms | 11 |
| Number of aromatic heavy atoms | 6 |
| Fraction Csp3 | 0.12 |
| Number of rotatable bonds | 0 |
| Number of H-bond acceptors | 2 |
| Number of H-bond donors | 2 |
| Molar refractivity | 51.25 |
| Tropological polar surface area | 50.41 A2 |
| Lipophilicity | 0.66 |
| Water solubility | Soluble |
| GI absorption | High |
| BBB permeation | No |
| Bioavailability score | 0.55 |
| Lipinski | Yes |
| Synthetic accessibility | Easy |
Table 2.
Molecular structures, generic names, chemical names, and biological targets of some marketed anticancer quinazolines.
Table 2.
Molecular structures, generic names, chemical names, and biological targets of some marketed anticancer quinazolines.
| Molecular Structure | Generic Name | Chemical Name | Biological Target |
|---|---|---|---|
 | Gefitinib Iressa Irressat NSC 759856 UNII-S65743JHBS ZD 1839 CCRIS 9011 | 4-(3’-Chloro-4’-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)quinazoline | Tyrosine kinase (EGFR) IC50 = 33 nM |
 | Erlotinib HSDB 8082 UNII-J4T82NDH7E | 4-Quinazolinamine, N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)- | Tyrosine kinase (EGFR) IC50 = 2 nM |
 | Vandetanib Caprelsa HSDB 8198 UNII-YO460OQ37K Zactima ZD 6474 | 4-Quninazolinamine, N-(4-bromo-2-fluorophenyl)-6-methoxy-7-((1-methyl-4-piperidinyl)methoxy)- | Tyrosine kinase (VEGFR2) IC50 = 40 nM |
 | Dacomitinib Vizimpro UNII-5092U85G58 PF-00299804 | 2-Butenamide, N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxy-6-quinazolinyl)-4-(1-piperidinyl)-, hydrate (1:1), (2E)- | Tyrosine kinase (EGFR) IC50 = 50 nM |
 | Afatinib BIBW 2992 Tomtovok Tovok UNII-41UD74L59M | 2-Butenamide, N-(4-((3-chloro-4-fluorophenyl)amino)-7-(((3S)-tetrahydro-3-furanyl)oxy)-6-quinazolinyl)-4-(dimethylamino)-, (2E)- | Tyrosine kinase (EGFR) IC50 = 0.5 nM |
 | Canertinib UNII-C78W1K5ASF | N-(4-((3-Chloro-4-fluorophenyl)amino)-7-(3-(morpholin-4-yl)propoxy)quinazolin-6-yl)prop-2-enamide | Tyrosine kinase (EGFR) IC50 = 0.8 nM |
 | Trimetrexate Trimetrexatum HSDB 6545 JB 11 NSC 249008 TMQ UNII-UPN4ITI8T4 | 2,4-Quinazolinediamine, 5-methyl-6-(((3,4,5-trimethoxyphenyl)amino)methyl) | Dihydrofolate reductase (DHFR) IC50 = 0.04 nM |
 | Lapatinib GSK 572016 HSDB 8209 Tykerb UNII-0VUA21238F | 4-Quinazolinamine, N-(3-chloro-4-((3-fluorophenyl)methoxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)-2-furanyl) | Tyrosine Kinase (EGFR) IC50 = 10.8 nM; (HER) IC50 = 29.2 nM |
 | Raltitrexed D 1694 UNII-FCB9EGG971 ZD1694 | L-Glutamic acid, N-((5-(((1,4-dihydro-2-methyl-4-oxo-6-quinazolinyl)methyl)methylamino)-2-thienyl)carbonyl) | Thymidylate synthase inhibitor IC50 = 9 nM |
 | Cediranib USAN AZD-2171 Recentin UNII-NQU9IPY4K9 | Quinazoline, 4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxy-7-(3-(1-pyrrolidinyl)propoxy) | Tyrosine Kinase (VEGFR) IC50 = 0.4 nM |
 | Tandutinib CT 53518 MLN 518 NSC 759851 NII-E1IO3ICJ9A | 1-Piperazinecarboxamide, 4-(6-methoxy-7-(3-(1-piperidinyl)propoxy)-4-quinazolinyl)-N-(4-(1-methylethoxy)phenyl) | Tyrosine Kinase (PDGFR) IC50 = 0.20 μM |
 | Barasertib AZD 1152 UNII-16XC2U7W8N | 1H-Pyrazole-3-acetamide, 5-((7-(3-(ethyl(2- (phosphonooxy)ethyl)amino)propoxy)-4-quinazolinyl)amino)-N-(3-fluorophenyl) | Tyrosine Kinase (Aurora B) IC50 = 0.37 nM |
 | Idelalisib GS 1101 UNII-YG57I8T5M0 Zydelig HSDB 8408 CAL-101 | 5-Fluoro-3-phenyl-2-((S)-1-(9H-purin-6-ylamino)-propyl)-3H-quinazolin-4-one | Tyrosine Kinase (PI3Kδ) IC50 = 2.5 nM |
 | Copanlisib BAY 80-6946 UNII-WI6V529FZ9 Aliqopa | 2-amino-N-(7-methoxy-8-(3-morpholinopropoxy)-2,3-dihydroimidazo(1,2-c)quinazolin-4-yl)pyrimidine-5-carboxamide | Tyrosine Kinase (PI3Kα) (PI3Kϒ) IC50 = 19 nM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zayed, M.F. Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Sci. Pharm. 2023, 91, 18. https://doi.org/10.3390/scipharm91020018
AMA Style
Zayed MF. Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases. Scientia Pharmaceutica. 2023; 91(2):18. https://doi.org/10.3390/scipharm91020018
Chicago/Turabian StyleZayed, Mohamed F. 2023. "Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases" Scientia Pharmaceutica 91, no. 2: 18. https://doi.org/10.3390/scipharm91020018