
UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies

Abstract
:1. Introduction
2. UBE3A—Structure and Function
3. The 15q11–q13 Region—The Regional Association of UBE3A in ASD
4. The Underlying Molecular Mechanisms of UBE3A-Mediated ASDs

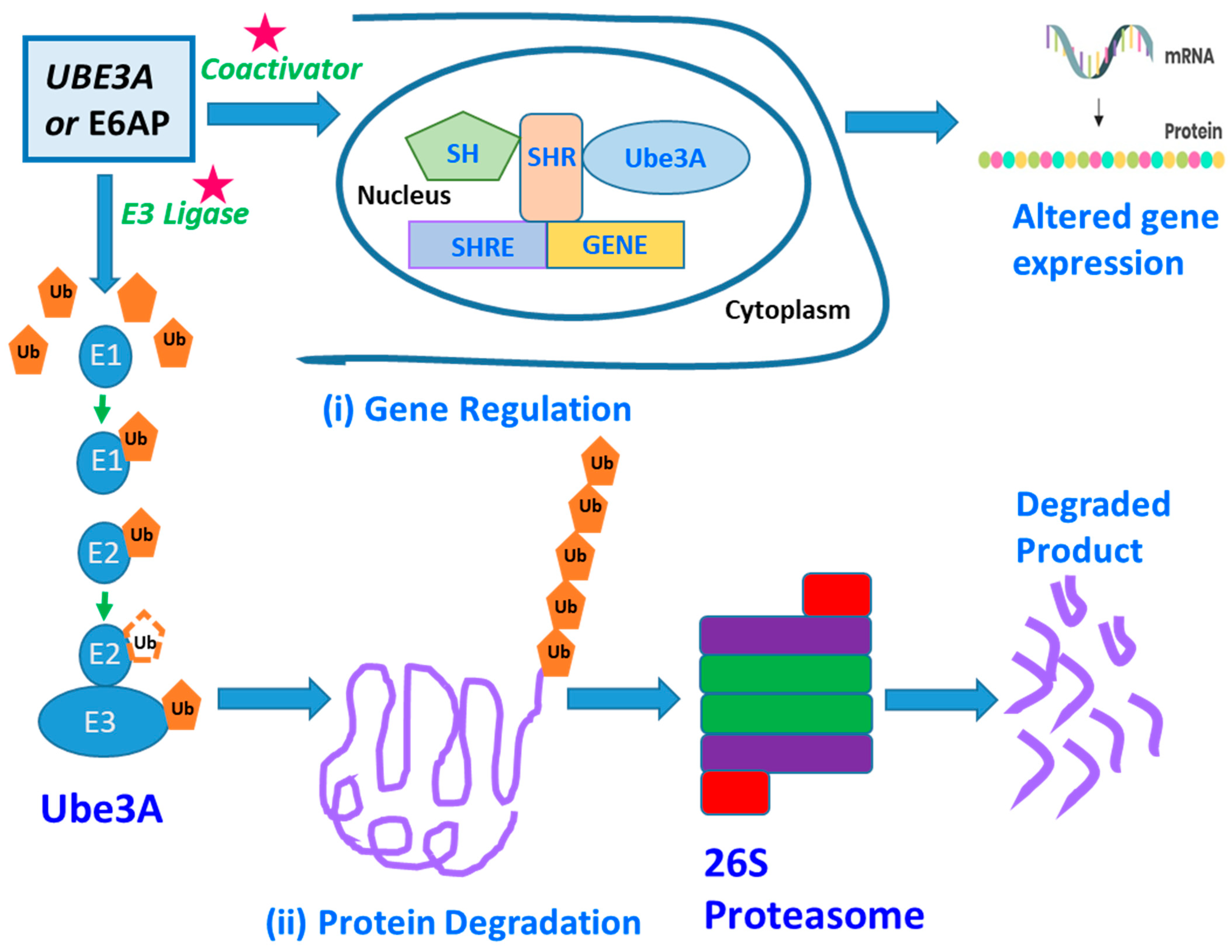{kind=link}
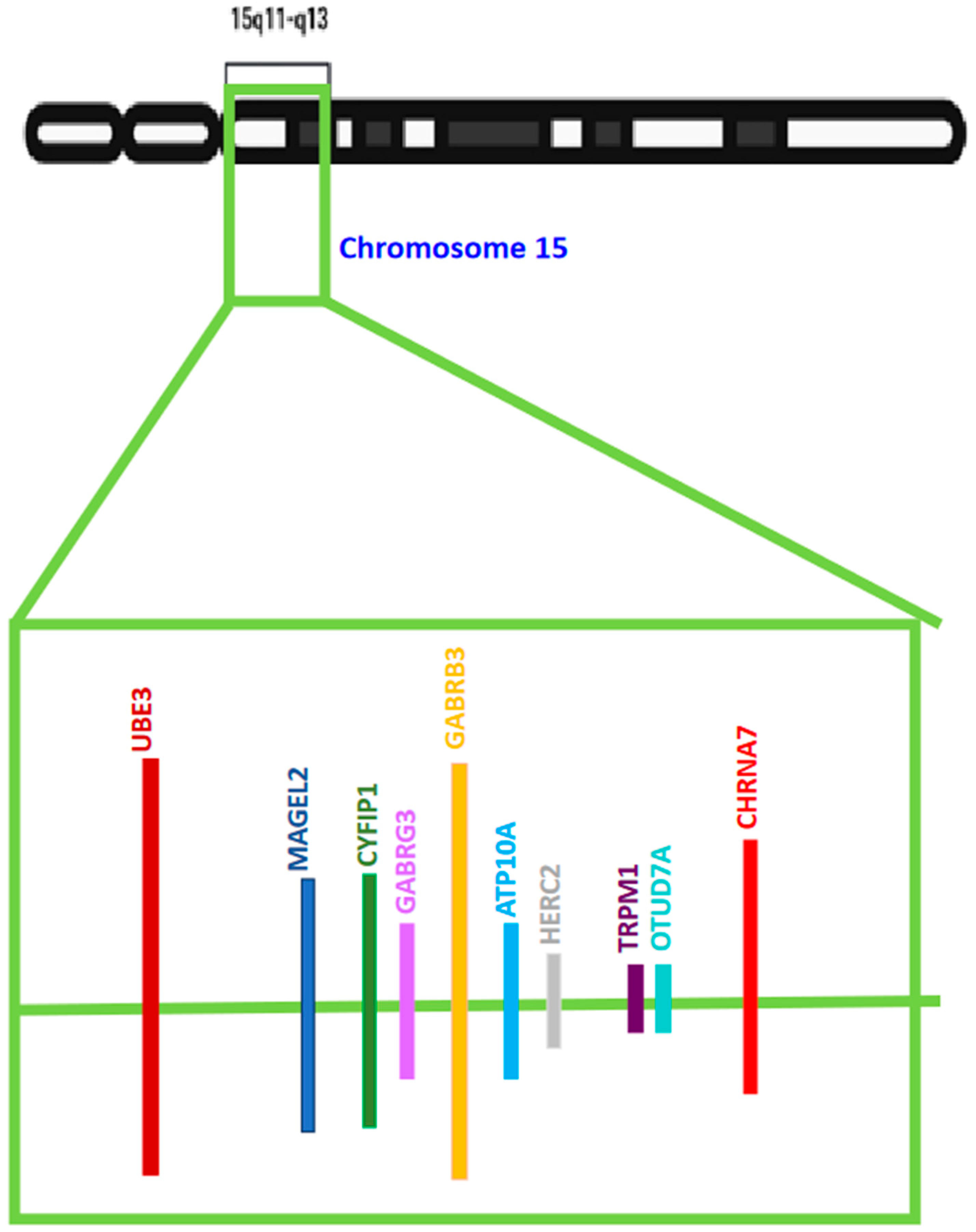{kind=link}
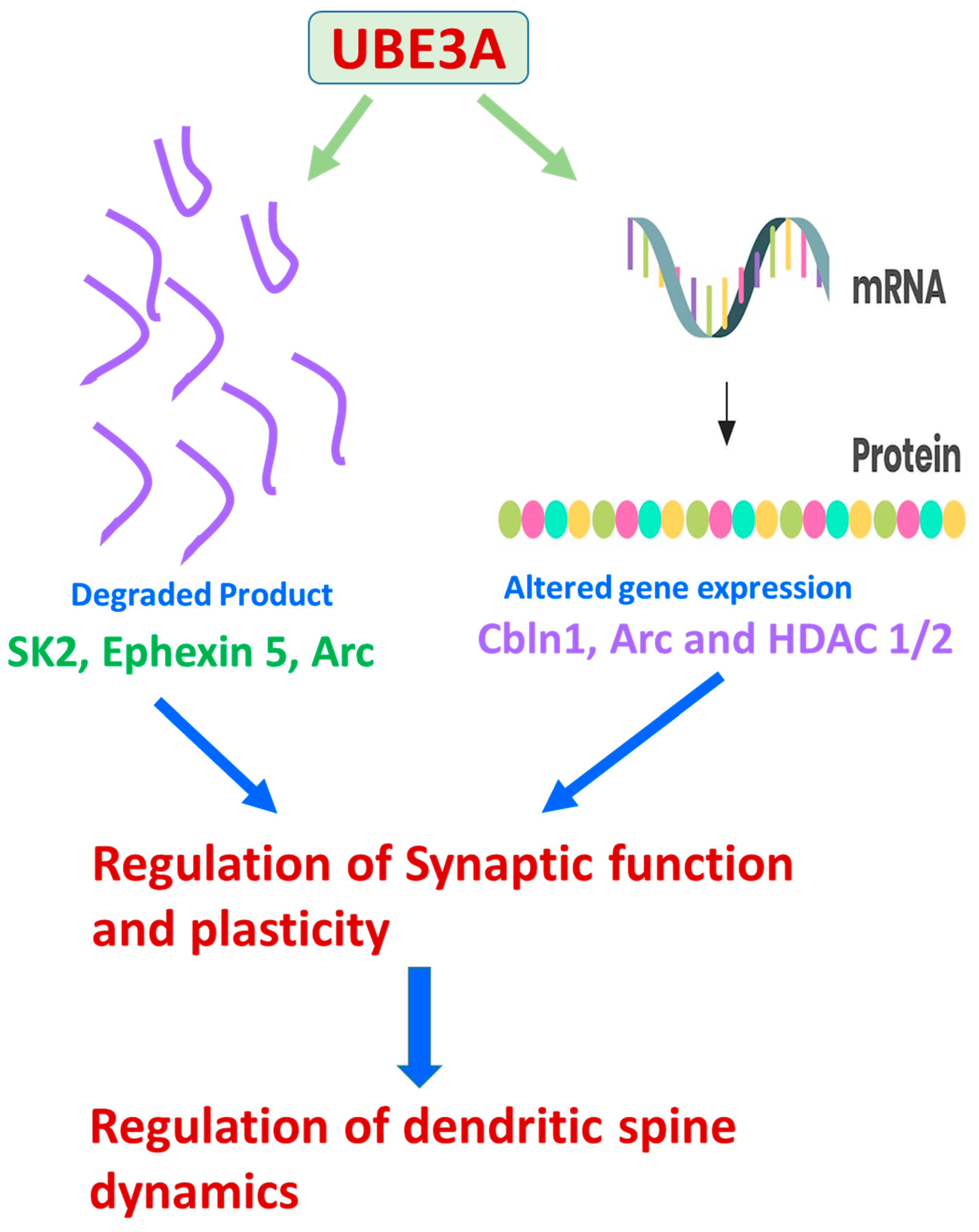{kind=link}
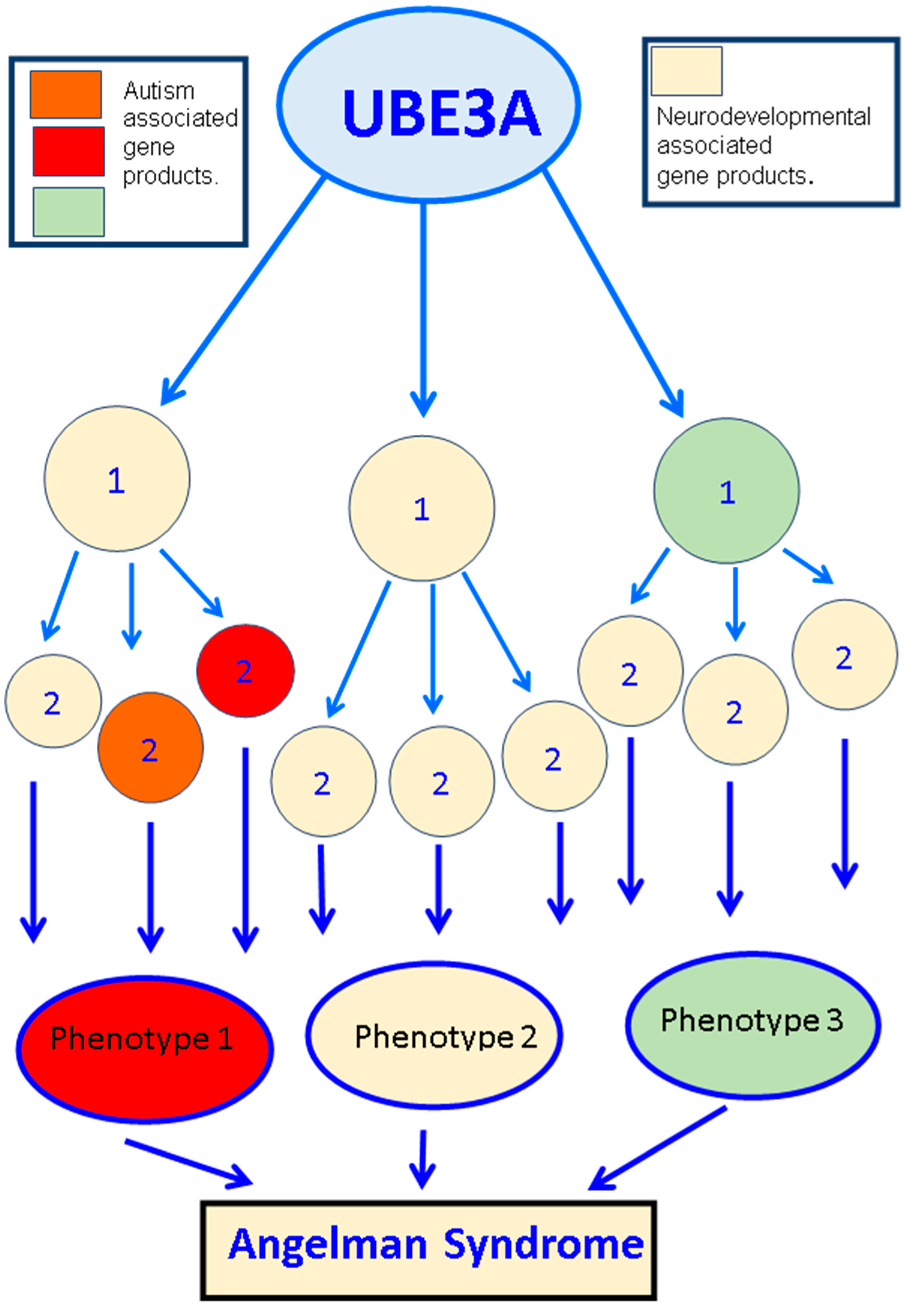{kind=link}
| Substrates/ Interactors | Role of UBE3A in Modulating the Target | Disease | References |
|---|---|---|---|
| ANXA1 | Ubiquitination | ASD | [158,159] |
| AR | Coactivator | ASD | [94,160] |
| Arc | Coactivator regulating transcription and Ubiquitination. | AS, Fragile X | [68,151,161] |
| CDKN1B | Ubiquitination | ASD | [97,162] |
| DLG1 | Ubiquitination | ASD | [163,164] |
| Ephexin 5 | Ubiquitination | Epilepsy | [69,165] |
| ESR2 | Ubiquitination | Asperger Syndrome, ASD | [141,166] |
| Herc2 | Ubiquitination | AS, ASD | [167,168,169] |
| mGluR5 | Ubiquitination | AS, Fragile X | [170,171] |
| SOD1 | Ubiquitination | ASD | [172,173] |
| TSC1 | Ubiquitination | AS, TS, ASD | [139,174] |
| TSC2 | Ubiquitination | AS, TS, ASD | [139,174,175] |
| UBE3A | Ubiquitination | AS, ASD | [176,177] |
5. Potential Biomarkers Related to UBE3A
6. Potential Therapeutic Strategies against UBE3A-Mediated ASDs: Insights from Cellular, In Vivo, and Other Pre-Clinical Models
- (i.)
- Lowering ARC levels: reducing ARC levels improved seizures in UBE3A-deficient mice without any significant difference in the motor dysfunction and altered ultrasonic vocalization defects [151].
- (ii.)
- The inhibition of calmodulin-dependent protein kinase II (CAMKII) phosphorylation: Levodopa reduces CAMKII phosphorylation and decreases the seizure intensity and motor deficits and alleviates hippocampal learning behavior and synaptic plasticity in the AS-modelled mice [153]. A clinical trial for the usage of Levodopa for AS is in the pipeline and clinically significant data is still awaited for further validation (NCT01281475).
- (iii.)
- Ampakines, known to act as modulators of AMPA receptors, have been shown to enhance BDNF release. This methodology improved hippocampus-related learning paradigms and proved to be useful in alleviating LTP in AS transgenic mouse models. Obstructing SK2 channels helped in improving LTP, learning, and memory behavior, and restored activity-dependent actin polymerization. Efforts to systemically inject an SK2 channel blocker have shown promising results in restoring animal fear-conditioning paradigms in the AS model [70,205,206].
- (iv.)
- The mTOR pathway has been implicated in the development of the brain and its synaptic plasticity. The over-activation of mTORC1 and inhibition of mTORC2 in UBE3A-deficient mice leads to motor abnormalities and LTP defects, anomalies in fear conditioning, and memory processes.
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, E.L.; Amin, T.; Olson, L.M.; Jacobs, M.M.; McCauley, J.L.; Lam, A.Y.; Organ, E.L.; Folstein, S.E.; Haines, J.L.; Sutcliffe, J.S. Dense linkage disequilibrium mapping in the 15q11–q13 maternal expression domain yields evidence for association in autism. Mol. Psychiatry 2003, 8, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Kato, C.; Tochigi, M.; Ohashi, J.; Koishi, S.; Kawakubo, Y.; Yamamoto, K.; Matsumoto, H.; Hashimoto, O.; Kim, S.Y.; Watanabe, K.; et al. Association study of the 15q11–q13 maternal expression domain in Japanese autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147, 1008–1012. [Google Scholar] [CrossRef]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Butler, M.G.; Hartin, S.N.; Ling, L.; Bui, M.; Francis, D.; Rogers, C.; Field, M.J.; Slee, J.; Gamage, D.; et al. Relationships between UBE3A and SNORD116 expression and features of autism in chromosome 15 imprinting disorders. Transl. Psychiatry 2020, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Leung, K.N.; Wang, N.J.; Wu, D.J.; Driscoll, J.; Vallero, R.O.; Schanen, N.C.; LaSalle, J.M. Chromosome 15q11–q13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J. Med. Genet. 2009, 46, 86–93. [Google Scholar] [CrossRef]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, H.J.; Lee, Y.M.; Park, J. Complete Penetrance but Different Phenotypes in a Korean Family with Maternal Interstitial Duplication at 15q11.2–q13.1: A Case Report. Children 2021, 8, 313. [Google Scholar] [CrossRef]
- Guffanti, G.; Strik Lievers, L.; Bonati, M.T.; Marchi, M.; Geronazzo, L.; Nardocci, N.; Estienne, M.; Larizza, L.; Macciardi, F.; Russo, S. Role of UBE3A and ATP10A genes in autism susceptibility region 15q11–q13 in an Italian population: A positive replication for UBE3A. Psychiatry Res. 2011, 185, 33–38. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Korostelev, S.A.; Zelenova, M.A.; Yurov, Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol. Cytogenet. 2015, 8, 77. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Zelenova, M.A.; Vasin, K.S.; Kurinnaia, O.S.; Korostelev, S.A.; Yurov, Y.B. Epigenomic variations manifesting as a loss of heterozygosity affecting imprinted genes represent a molecular mechanism of autism spectrum disorders and intellectual disability in children. Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2019, 119, 91–97. [Google Scholar] [CrossRef]
- Nudel, R.; Simpson, N.H.; Baird, G.; O’Hare, A.; Conti-Ramsden, G.; Bolton, P.F.; Hennessy, E.R.; Consortium, S.L.I.; Ring, S.M.; Davey Smith, G.; et al. Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment. Genes Brain Behav. 2014, 13, 418–429. [Google Scholar] [CrossRef]
- Schneider, E.; El Hajj, N.; Richter, S.; Roche-Santiago, J.; Nanda, I.; Schempp, W.; Riederer, P.; Navarro, B.; Bontrop, R.E.; Kondova, I.; et al. Widespread differences in cortex DNA methylation of the “language gene” CNTNAP2 between humans and chimpanzees. Epigenetics 2014, 9, 533–545. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef]
- Wu, D.J.; Wang, N.J.; Driscoll, J.; Dorrani, N.; Liu, D.; Sigman, M.; Schanen, N.C. Autistic disorder associated with a paternally derived unbalanced translocation leading to duplication of chromosome 15pter-q13.2: A case report. Mol. Cytogenet. 2009, 2, 27. [Google Scholar] [CrossRef]
- Fradin, D.; Cheslack-Postava, K.; Ladd-Acosta, C.; Newschaffer, C.; Chakravarti, A.; Arking, D.E.; Feinberg, A.; Fallin, M.D. Parent-of-origin effects in autism identified through genome-wide linkage analysis of 16,000 SNPs. PLoS ONE 2010, 5, e12513. [Google Scholar] [CrossRef]
- Depienne, C.; Moreno-De-Luca, D.; Heron, D.; Bouteiller, D.; Gennetier, A.; Delorme, R.; Chaste, P.; Siffroi, J.P.; Chantot-Bastaraud, S.; Benyahia, B.; et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11–q13 region in autism spectrum disorders. Biol. Psychiatry 2009, 66, 349–359. [Google Scholar] [CrossRef]
- Curran, S.; Roberts, S.; Thomas, S.; Veltman, M.; Browne, J.; Medda, E.; Pickles, A.; Sham, P.; Bolton, P.F. An association analysis of microsatellite markers across the Prader-Willi/Angelman critical region on chromosome 15 (q11–13) and autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 137, 25–28. [Google Scholar] [CrossRef]
- Ben-David, E.; Shohat, S.; Shifman, S. Allelic expression analysis in the brain suggests a role for heterogeneous insults affecting epigenetic processes in autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 4111–4124. [Google Scholar] [CrossRef] [PubMed]
- Scoles, H.A.; Urraca, N.; Chadwick, S.W.; Reiter, L.T.; Lasalle, J.M. Increased copy number for methylated maternal 15q duplications leads to changes in gene and protein expression in human cortical samples. Mol. Autism 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Dupuis, L.; Mittal, K.; Lionel, A.C.; Marshall, C.R.; Scherer, S.W.; Stockley, T.; Vincent, J.B.; Mendoza-Londono, R.; Stavropoulos, D.J. 15q11.2 Duplication Encompassing Only the UBE3A Gene Is Associated with Developmental Delay and Neuropsychiatric Phenotypes. Hum. Mutat. 2015, 36, 689–693. [Google Scholar] [CrossRef]
- Kitsiou-Tzeli, S.; Tzetis, M.; Sofocleous, C.; Vrettou, C.; Xaidara, A.; Giannikou, K.; Pampanos, A.; Mavrou, A.; Kanavakis, E. De novo interstitial duplication of the 15q11.2-q14 PWS/AS region of maternal origin: Clinical description, array CGH analysis, and review of the literature. Am. J. Med. Genet. A 2010, 152, 1925–1932. [Google Scholar] [CrossRef]
- Piard, J.; Philippe, C.; Marvier, M.; Beneteau, C.; Roth, V.; Valduga, M.; Beri, M.; Bonnet, C.; Gregoire, M.J.; Jonveaux, P.; et al. Clinical and molecular characterization of a large family with an interstitial 15q11q13 duplication. Am. J. Med. Genet. A 2010, 152, 1933–1941. [Google Scholar] [CrossRef]
- Leung, K.N.; Vallero, R.O.; DuBose, A.J.; Resnick, J.L.; LaSalle, J.M. Imprinting regulates mammalian snoRNA-encoding chromatin decondensation and neuronal nucleolar size. Hum. Mol. Genet. 2009, 18, 4227–4238. [Google Scholar] [CrossRef]
- Samaco, R.C.; Hogart, A.; LaSalle, J.M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005, 14, 483–492. [Google Scholar] [CrossRef]
- Hogart, A.; Patzel, K.A.; LaSalle, J.M. Gender influences monoallelic expression of ATP10A in human brain. Hum. Genet. 2008, 124, 235–242. [Google Scholar] [CrossRef]
- Powell, W.T.; Coulson, R.L.; Gonzales, M.L.; Crary, F.K.; Wong, S.S.; Adams, S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. USA 2013, 110, 13938–13943. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Pan, Y.; Zhu, L.; Landa, L.; Yoo, J.; Spencer, C.; Lorenzo, I.; Brilliant, M.; Noebels, J.; Beaudet, A.L. Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS ONE 2010, 5, e12278. [Google Scholar] [CrossRef]
- Chibuk, T.K.; Bischof, J.M.; Wevrick, R. A necdin/MAGE-like gene in the chromosome 15 autism susceptibility region: Expression, imprinting, and mapping of the human and mouse orthologues. BMC Genet. 2001, 2, 22. [Google Scholar] [CrossRef]
- Thatcher, K.N.; Peddada, S.; Yasui, D.H.; Lasalle, J.M. Homologous pairing of 15q11–q13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum. Mol. Genet. 2005, 14, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.J.; Dunaway, K.; Islam, M.S.; Mordaunt, C.; Vogel Ciernia, A.; Meguro-Horike, M.; Horike, S.I.; Segal, D.J.; LaSalle, J.M. UBE3A-mediated regulation of imprinted genes and epigenome-wide marks in human neurons. Epigenetics 2017, 12, 982–990. [Google Scholar] [CrossRef]
- Dunaway, K.W.; Islam, M.S.; Coulson, R.L.; Lopez, S.J.; Vogel Ciernia, A.; Chu, R.G.; Yasui, D.H.; Pessah, I.N.; Lott, P.; Mordaunt, C.; et al. Cumulative Impact of Polychlorinated Biphenyl and Large Chromosomal Duplications on DNA Methylation, Chromatin, and Expression of Autism Candidate Genes. Cell Rep. 2016, 17, 3035–3048. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef]
- Herrero, M.J.; Velmeshev, D.; Hernandez-Pineda, D.; Sethi, S.; Sorrells, S.; Banerjee, P.; Sullivan, C.; Gupta, A.R.; Kriegstein, A.R.; Corbin, J.G. Identification of amygdala-expressed genes associated with autism spectrum disorder. Mol. Autism 2020, 11, 39. [Google Scholar] [CrossRef]
- Malzac, P.; Webber, H.; Moncla, A.; Graham, J.M.; Kukolich, M.; Williams, C.; Pagon, R.A.; Ramsdell, L.A.; Kishino, T.; Wagstaff, J. Mutation analysis of UBE3A in Angelman syndrome patients. Am. J. Hum. Genet. 1998, 62, 1353–1360. [Google Scholar] [CrossRef]
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 2001, 38, 834–845. [Google Scholar] [CrossRef]
- Cook, E.H., Jr.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar]
- Schroer, R.J.; Phelan, M.C.; Michaelis, R.C.; Crawford, E.C.; Skinner, S.A.; Cuccaro, M.; Simensen, R.J.; Bishop, J.; Skinner, C.; Fender, D.; et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Med. Genet. 1998, 76, 327–336. [Google Scholar] [CrossRef]
- Thomas, J.A.; Johnson, J.; Peterson Kraai, T.L.; Wilson, R.; Tartaglia, N.; LeRoux, J.; Beischel, L.; McGavran, L.; Hagerman, R.J. Genetic and clinical characterization of patients with an interstitial duplication 15q11–q13, emphasizing behavioral phenotype and response to treatment. Am. J. Med. Genet. A 2003, 119, 111–120. [Google Scholar] [CrossRef]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Borgatti, R.; Piccinelli, P.; Passoni, D.; Dalpra, L.; Miozzo, M.; Micheli, R.; Gagliardi, C.; Balottin, U. Relationship between clinical and genetic features in “inverted duplicated chromosome 15” patients. Pediatr. Neurol. 2001, 24, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Man, H.Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 2019, 12, 109. [Google Scholar] [CrossRef]
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008, 17, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Rougeulle, C.; Glatt, H.; Lalande, M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 1997, 17, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Joh, K.; Ohta, T.; Masuzaki, H.; Ishimaru, T.; Mukai, T.; Niikawa, N.; Ogawa, M.; Wagstaff, J.; Kishino, T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 2003, 12, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Prado, E.; Iturralde, A.L.; Simbana-Rivera, K.; Gomez-Barreno, L.; Hidalgo, I.; Rubio-Neira, M.; Espinosa, N.; Izquierdo-Condoy, J.; Arteaga-Espinosa, M.E.; Lister, A.; et al. 15q Duplication Syndrome: Report on the First Patient from Ecuador with an Unusual Clinical Presentation. Case Rep. Med. 2021, 2021, 6662054. [Google Scholar] [CrossRef]
- Urraca, N.; Hope, K.; Victor, A.K.; Belgard, T.G.; Memon, R.; Goorha, S.; Valdez, C.; Tran, Q.T.; Sanchez, S.; Ramirez, J.; et al. Significant transcriptional changes in 15q duplication but not Angelman syndrome deletion stem cell-derived neurons. Mol. Autism 2018, 9, 6. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef]
- Pelzer, C.; Kassner, I.; Matentzoglu, K.; Singh, R.K.; Wollscheid, H.P.; Scheffner, M.; Schmidtke, G.; Groettrup, M. UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 2007, 282, 23010–23014. [Google Scholar] [CrossRef]
- Shang, F.; Deng, G.; Obin, M.; Wu, C.C.; Gong, X.; Smith, D.; Laursen, R.A.; Andley, U.P.; Reddan, J.R.; Taylor, A. Ubiquitin-activating enzyme (E1) isoforms in lens epithelial cells: Origin of translation, E2 specificity and cellular localization determined with novel site-specific antibodies. Exp. Eye Res. 2001, 73, 827–836. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Hoffman, A.R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 1997, 17, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Han, J.E.; DeBruyne, J.P.; Philpot, B.D. Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci. Rep. 2016, 6, 28238. [Google Scholar] [CrossRef] [PubMed]
- Grier, M.D.; Carson, R.P.; Lagrange, A.H. Toward a Broader View of Ube3a in a Mouse Model of Angelman Syndrome: Expression in Brain, Spinal Cord, Sciatic Nerve and Glial Cells. PLoS ONE 2015, 10, e0124649. [Google Scholar] [CrossRef]
- Judson, M.C.; Sosa-Pagan, J.O.; Del Cid, W.A.; Han, J.E.; Philpot, B.D. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 2014, 522, 1874–1896. [Google Scholar] [CrossRef]
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef]
- Kalsner, L.; Chamberlain, S.J. Prader-Willi, Angelman, and 15q11–q13 Duplication Syndromes. Pediatr. Clin. N. Am. 2015, 62, 587–606. [Google Scholar] [CrossRef]
- Whittington, J.E.; Holland, A.J.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J. Med. Genet. 2001, 38, 792–798. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Sutcliffe, J.S.; Fang, P.; Galjaard, R.J.; Jiang, Y.H.; Benton, C.S.; Rommens, J.M.; Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997, 15, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Dagli, A.; Buiting, K.; Williams, C.A. Molecular and Clinical Aspects of Angelman Syndrome. Mol. Syndromol. 2012, 2, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Urraca, N.; Cleary, J.; Brewer, V.; Pivnick, E.K.; McVicar, K.; Thibert, R.L.; Schanen, N.C.; Esmer, C.; Lamport, D.; Reiter, L.T. The interstitial duplication 15q11.2–q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013, 6, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M.; Ramakers, G.J. Dendritic pathology in mental retardation: From molecular genetics to neurobiology. Genes Brain Behav. 2006, 5 (Suppl. S2), 48–60. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Reiter, L.T.; Saravanapandian, V.; DiStefano, C.; Huberty, S.; Hyde, C.; Chamberlain, S.; Bearden, C.E.; Golshani, P.; Irimia, A.; et al. Mechanisms underlying the EEG biomarker in Dup15q syndrome. Mol. Autism 2019, 10, 29. [Google Scholar] [CrossRef]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef]
- Margolis, S.S.; Salogiannis, J.; Lipton, D.M.; Mandel-Brehm, C.; Wills, Z.P.; Mardinly, A.R.; Hu, L.; Greer, P.L.; Bikoff, J.B.; Ho, H.Y.; et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 2010, 143, 442–455. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, G.; Liu, Y.; Standley, S.; Ji, A.; Tunuguntla, R.; Wang, Y.; Claus, C.; Luo, Y.; Baudry, M.; et al. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep. 2015, 12, 449–461. [Google Scholar] [CrossRef]
- Sadikovic, B.; Fernandes, P.; Zhang, V.W.; Ward, P.A.; Miloslavskaya, I.; Rhead, W.; Rosenbaum, R.; Gin, R.; Roa, B.; Fang, P. Mutation Update for UBE3A variants in Angelman syndrome. Hum. Mutat. 2014, 35, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Prabha, P.K.; Singla, R.; Kaur, G.; Sharma, A.R.; Joshi, R.; Suroy, B.; Medhi, B. Epigenetic Interface of Autism Spectrum Disorders (ASDs): Implications of Chromosome 15q11–q13 Segment. ACS Chem. Neurosci. 2022, 13, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Burette, A.C.; Judson, M.C.; Li, A.N.; Chang, E.F.; Seeley, W.W.; Philpot, B.D.; Weinberg, R.J. Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Beer-Romero, P.; Glass, S.; Rolfe, M. Antisense targeting of E6AP elevates p53 in HPV-infected cells but not in normal cells. Oncogene 1997, 14, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Dodge, A.; Willman, J.; Willman, M.; Nenninger, A.W.; Morrill, N.K.; Lamens, K.; Greene, H.; Weeber, E.J.; Nash, K.R. Identification of UBE3A Protein in CSF and Extracellular Space of the Hippocampus Suggest a Potential Novel Function in Synaptic Plasticity. Autism Res. 2021, 14, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.N.; Haynes, K.A.; Bach, S.V.; Beckelman, B.C. Local ubiquitin-proteasome-mediated proteolysis and long-term synaptic plasticity. Front. Mol. Neurosci. 2014, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.N. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010, 17, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Buiting, K.; Cassidy, S.B.; Conroy, J.M.; Driscoll, D.J.; Gabriel, J.M.; Gillessen-Kaesbach, G.; Glenn, C.C.; Greenswag, L.R.; Horsthemke, B.; et al. Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am. J. Med. Genet. 1997, 68, 195–206. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Cheon, S.; Dean, M.; Chahrour, M. The ubiquitin proteasome pathway in neuropsychiatric disorders. Neurobiol. Learn. Mem. 2019, 165, 106791. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Kishino, T.; Wagstaff, J. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics 1998, 47, 101–107. [Google Scholar] [CrossRef]
- Condon, K.H.; Ho, J.; Robinson, C.G.; Hanus, C.; Ehlers, M.D. The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J. Neurosci. 2013, 33, 3799–3814. [Google Scholar] [CrossRef]
- Krishnan, V.; Stoppel, D.C.; Nong, Y.; Johnson, M.A.; Nadler, M.J.; Ozkaynak, E.; Teng, B.L.; Nagakura, I.; Mohammad, F.; Silva, M.A.; et al. Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 2017, 543, 507–512. [Google Scholar] [CrossRef]
- Su, H.; Fan, W.; Coskun, P.E.; Vesa, J.; Gold, J.A.; Jiang, Y.H.; Potluri, P.; Procaccio, V.; Acab, A.; Weiss, J.H.; et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2011, 487, 129–133. [Google Scholar] [CrossRef]
- Llewellyn, K.J.; Nalbandian, A.; Gomez, A.; Wei, D.; Walker, N.; Kimonis, V.E. Administration of CoQ10 analogue ameliorates dysfunction of the mitochondrial respiratory chain in a mouse model of Angelman syndrome. Neurobiol. Dis. 2015, 76, 77–86. [Google Scholar] [CrossRef]
- Santini, E.; Turner, K.L.; Ramaraj, A.B.; Murphy, M.P.; Klann, E.; Kaphzan, H. Mitochondrial Superoxide Contributes to Hippocampal Synaptic Dysfunction and Memory Deficits in Angelman Syndrome Model Mice. J. Neurosci. 2015, 35, 16213–16220. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 1999, 19, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; DeVera, D.G.; Lamb, D.J.; Nawaz, Z.; Jiang, Y.H.; Beaudet, A.L.; O’Malley, B.W. Genetic ablation of the steroid receptor coactivator-ubiquitin ligase, E6-AP, results in tissue-selective steroid hormone resistance and defects in reproduction. Mol. Cell. Biol. 2002, 22, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol. 2006, 20, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Kumar, S.; Howley, P.M. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. USA 1999, 96, 9557–9562. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Jana, N.R. Regulation of turnover of tumor suppressor p53 and cell growth by E6-AP, a ubiquitin protein ligase mutated in Angelman mental retardation syndrome. Cell. Mol. Life Sci. 2008, 65, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Godavarthi, S.K.; Jana, N.R. UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol. Dis. 2009, 36, 26–34. [Google Scholar] [CrossRef]
- Reiter, L.T.; Seagroves, T.N.; Bowers, M.; Bier, E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum. Mol. Genet. 2006, 15, 2825–2835. [Google Scholar] [CrossRef]
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. 2011, 31, 17637–17648. [Google Scholar] [CrossRef]
- Kaphzan, H.; Hernandez, P.; Jung, J.I.; Cowansage, K.K.; Deinhardt, K.; Chao, M.V.; Abel, T.; Klann, E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol. Psychiatry 2012, 72, 182–190. [Google Scholar] [CrossRef]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Dikshit, P.; Purkayastha, S.; Sharma, J.; Nukina, N.; Jana, N.R. E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 2008, 283, 7648–7656. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Godavarthi, S.K.; Maheshwari, M.; Goswami, A.; Jana, N.R. The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 2009, 284, 10537–10545. [Google Scholar] [CrossRef] [PubMed]
- Mulherkar, S.A.; Sharma, J.; Jana, N.R. The ubiquitin ligase E6-AP promotes degradation of alpha-synuclein. J. Neurochem. 2009, 110, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Valluy, J.; Bicker, S.; Aksoy-Aksel, A.; Lackinger, M.; Sumer, S.; Fiore, R.; Wust, T.; Seffer, D.; Metge, F.; Dieterich, C.; et al. A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci. 2015, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R. Glial cells. Int. J. Biochem. Cell Biol. 2004, 36, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–q13 duplication seen in autism. Cell 2009, 137, 1235–1246. [Google Scholar] [CrossRef]
- Smith, S.E.; Zhou, Y.D.; Zhang, G.; Jin, Z.; Stoppel, D.C.; Anderson, M.P. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 2011, 3, 103ra197. [Google Scholar] [CrossRef]
- Kuhnle, S.; Mothes, B.; Matentzoglu, K.; Scheffner, M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. USA 2013, 110, 8888–8893. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Heck, D.H.; Zhao, Y.; Roy, S.; LeDoux, M.S.; Reiter, L.T. Analysis of cerebellar function in Ube3a-deficient mice reveals novel genotype-specific behaviors. Hum. Mol. Genet. 2008, 17, 2181–2189. [Google Scholar] [CrossRef]
- Godavarthi, S.K.; Dey, P.; Maheshwari, M.; Jana, N.R. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum. Mol. Genet. 2012, 21, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.Q.; Bichell, T.J.; Ihrie, R.A.; Johnson, C.H. Ube3a imprinting impairs circadian robustness in Angelman syndrome models. Curr. Biol. 2015, 25, 537–545. [Google Scholar] [CrossRef]
- Weeber, E.J.; Jiang, Y.H.; Elgersma, Y.; Varga, A.W.; Carrasquillo, Y.; Brown, S.E.; Christian, J.M.; Mirnikjoo, B.; Silva, A.; Beaudet, A.L.; et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 2003, 23, 2634–2644. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Riday, T.T.; Condon, K.H.; Roberts, A.C.; Bernardo, D.R.; Prakash, R.; Weinberg, R.J.; Ehlers, M.D.; Philpot, B.D. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 2009, 12, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Stryker, M.P. Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. USA 2010, 107, 5611–5616. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.L.; Burette, A.C.; Weinberg, R.J.; Philpot, B.D. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 2012, 74, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Ling, L.; Gamage, D.; Baker, E.K.; Bui, M.; Field, M.J.; Rogers, C.; Butler, M.G.; Murgia, A.; Leonardi, E.; et al. Feasibility of Screening for Chromosome 15 Imprinting Disorders in 16579 Newborns by Using a Novel Genomic Workflow. JAMA Netw. Open 2022, 5, e2141911. [Google Scholar] [CrossRef]
- Carli, D.; Riberi, E.; Ferrero, G.B.; Mussa, A. Syndromic Disorders Caused by Disturbed Human Imprinting. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Flashner, B.M.; Russo, M.E.; Boileau, J.E.; Leong, D.W.; Gallicano, G.I. Epigenetic factors and autism spectrum disorders. Neuromolecular. Med. 2013, 15, 339–350. [Google Scholar] [CrossRef]
- Li, J.; Lin, X.; Wang, M.; Hu, Y.; Xue, K.; Gu, S.; Lv, L.; Huang, S.; Xie, W. Potential role of genomic imprinted genes and brain developmental related genes in autism. BMC Med. Genom. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Lalande, M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11–q13. Neurobiol. Dis. 2010, 39, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A. The inv dup(15) or idic(15) syndrome: A clinically recognisable neurogenetic disorder. Brain Dev. 2005, 27, 365–369. [Google Scholar] [CrossRef]
- Kumar, S.; Reynolds, K.; Ji, Y.; Gu, R.; Rai, S.; Zhou, C.J. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Sell, G.L.; Xin, W.; Cook, E.K.; Zbinden, M.A.; Schaffer, T.B.; O’Meally, R.N.; Cole, R.N.; Margolis, S.S. Deleting a UBE3A substrate rescues impaired hippocampal physiology and learning in Angelman syndrome mice. Sci. Rep. 2021, 11, 19414. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Chen, R.; Ye, J.; Tan, G.H.; Li, S.; Zhang, J.; Jiang, Y.H.; Xiong, Z.Q. The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci. 2013, 33, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Gilbert, J.P.; Huo, Y.; Sharaflari, R.; Nee, M.; Qiao, H.; Man, H.Y. The Autism Protein Ube3A/E6AP Remodels Neuronal Dendritic Arborization via Caspase-Dependent Microtubule Destabilization. J. Neurosci. 2018, 38, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Fiumara, A.; Pittala, A.; Cocuzza, M.; Sorge, G. Epilepsy in patients with Angelman syndrome. Ital. J. Pediatr. 2010, 36, 31. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef]
- Egawa, K.; Kitagawa, K.; Inoue, K.; Takayama, M.; Takayama, C.; Saitoh, S.; Kishino, T.; Kitagawa, M.; Fukuda, A. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci. Transl. Med. 2012, 4, 163ra157. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Carstens, K.E.; Judson, M.C.; Dalton, K.A.; Rougie, M.; Clark, E.P.; Dudek, S.M.; Philpot, B.D. Ube3a reinstatement mitigates epileptogenesis in Angelman syndrome model mice. J. Clin. Investig. 2019, 129, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Pelc, K.; Cheron, G.; Boyd, S.G.; Dan, B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008, 9, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Gossan, N.C.; Zhang, F.; Guo, B.; Jin, D.; Yoshitane, H.; Yao, A.; Glossop, N.; Zhang, Y.Q.; Fukada, Y.; Meng, Q.J. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014, 42, 5765–5775. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Paranjape, S.R.; Walker, M.P.; Choudhury, R.; Wolter, J.M.; Fragola, G.; Emanuele, M.J.; Major, M.B.; Zylka, M.J. The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/beta-catenin pathway by inhibiting the proteasome. J. Biol. Chem. 2017, 292, 12503–12515. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; O’Roak, B.J.; Shendure, J.; Eichler, E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014, 37, 95–105. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Moreno, S.; Baudry, M.; Bi, X. Imbalanced mechanistic target of rapamycin C1 and C2 activity in the cerebellum of Angelman syndrome mice impairs motor function. J. Neurosci. 2015, 35, 4706–4718. [Google Scholar] [CrossRef]
- Zaaroor-Regev, D.; de Bie, P.; Scheffner, M.; Noy, T.; Shemer, R.; Heled, M.; Stein, I.; Pikarsky, E.; Ciechanover, A. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6788–6793. [Google Scholar] [CrossRef]
- Picard, N.; Charbonneau, C.; Sanchez, M.; Licznar, A.; Busson, M.; Lazennec, G.; Tremblay, A. Phosphorylation of activation function-1 regulates proteasome-dependent nuclear mobility and E6-associated protein ubiquitin ligase recruitment to the estrogen receptor beta. Mol. Endocrinol. 2008, 22, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Fan, J.; Zhao, Y.; Bi, S.; Si, L.; Liu, Q. Estrogen receptor beta treats Alzheimer’s disease. Neural Regen. Res. 2013, 8, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; He, P.; Shen, Y.; Li, R. New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: Deficiency of estrogen receptor-beta. J. Alzheimers Dis. 2012, 30, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Sohrabji, F.; Miranda, R.C.; Toran-Allerand, C.D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11110–11114. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; Jayaraman, A.; Pike, C.; Baudry, M. Progesterone inhibits estrogen-mediated neuroprotection against excitotoxicity by down-regulating estrogen receptor-beta. J. Neurochem. 2010, 115, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.D.; MacFadden, A.; Wu, Z.; Peng, J.; Liu, C.W. Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell 2014, 25, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Avagliano Trezza, R.; Sonzogni, M.; Bossuyt, S.N.V.; Zampeta, F.I.; Punt, A.M.; van den Berg, M.; Rotaru, D.C.; Koene, L.M.C.; Munshi, S.T.; Stedehouder, J.; et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019, 22, 1235–1247. [Google Scholar] [CrossRef]
- Xu, X.; Li, C.; Gao, X.; Xia, K.; Guo, H.; Li, Y.; Hao, Z.; Zhang, L.; Gao, D.; Xu, C.; et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018, 28, 48–68. [Google Scholar] [CrossRef]
- Wang, J.; Lou, S.S.; Wang, T.; Wu, R.J.; Li, G.; Zhao, M.; Lu, B.; Li, Y.Y.; Zhang, J.; Cheng, X.; et al. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc. Natl. Acad. Sci. USA 2019, 116, 12500–12505. [Google Scholar] [CrossRef]
- Margolis, S.S.; Sell, G.L.; Zbinden, M.A.; Bird, L.M. Angelman Syndrome. Neurotherapeutics 2015, 12, 641–650. [Google Scholar] [CrossRef]
- Mandel-Brehm, C.; Salogiannis, J.; Dhamne, S.C.; Rotenberg, A.; Greenberg, M.E. Seizure-like activity in a juvenile Angelman syndrome mouse model is attenuated by reducing Arc expression. Proc. Natl. Acad. Sci. USA 2015, 112, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Mabb, A.M.; Je, H.S.; Wall, M.J.; Robinson, C.G.; Larsen, R.S.; Qiang, Y.; Correa, S.A.; Ehlers, M.D. Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron 2014, 82, 1299–1316. [Google Scholar] [CrossRef] [PubMed]
- van Woerden, G.M.; Harris, K.D.; Hojjati, M.R.; Gustin, R.M.; Qiu, S.; de Avila Freire, R.; Jiang, Y.H.; Elgersma, Y.; Weeber, E.J. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat. Neurosci. 2007, 10, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Kaphzan, H.; Buffington, S.A.; Ramaraj, A.B.; Lingrel, J.B.; Rasband, M.N.; Santini, E.; Klann, E. Genetic reduction of the alpha1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013, 4, 405–412. [Google Scholar] [CrossRef]
- Jensen, L.; Farook, M.F.; Reiter, L.T. Proteomic profiling in Drosophila reveals potential Dube3a regulation of the actin cytoskeleton and neuronal homeostasis. PLoS ONE 2013, 8, e61952. [Google Scholar] [CrossRef] [PubMed]
- Sell, G.L.; Margolis, S.S. From UBE3A to Angelman syndrome: A substrate perspective. Front. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Jasper, H.H. Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; Oxford University Press: Bethesda, MD, USA, 2012. [Google Scholar]
- Shimoji, T.; Murakami, K.; Sugiyama, Y.; Matsuda, M.; Inubushi, S.; Nasu, J.; Shirakura, M.; Suzuki, T.; Wakita, T.; Kishino, T.; et al. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J. Cell. Biochem. 2009, 106, 1123–1135. [Google Scholar] [CrossRef]
- Correia, C.T.; Conceicao, I.C.; Oliveira, B.; Coelho, J.; Sousa, I.; Sequeira, A.F.; Almeida, J.; Cafe, C.; Duque, F.; Mouga, S.; et al. Recurrent duplications of the annexin A1 gene (ANXA1) in autism spectrum disorders. Mol. Autism 2014, 5, 28. [Google Scholar] [CrossRef]
- Henningsson, S.; Jonsson, L.; Ljunggren, E.; Westberg, L.; Gillberg, C.; Rastam, M.; Anckarsater, H.; Nygren, G.; Landen, M.; Thuresson, K.; et al. Possible association between the androgen receptor gene and autism spectrum disorder. Psychoneuroendocrinology 2009, 34, 752–761. [Google Scholar] [CrossRef]
- Park, S.; Park, J.M.; Kim, S.; Kim, J.A.; Shepherd, J.D.; Smith-Hicks, C.L.; Chowdhury, S.; Kaufmann, W.; Kuhl, D.; Ryazanov, A.G.; et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 2008, 59, 70–83. [Google Scholar] [CrossRef]
- Grey, W.; Izatt, L.; Sahraoui, W.; Ng, Y.M.; Ogilvie, C.; Hulse, A.; Tse, E.; Holic, R.; Yu, V. Deficiency of the cyclin-dependent kinase inhibitor, CDKN1B, results in overgrowth and neurodevelopmental delay. Hum. Mutat. 2013, 34, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Nakagawa, S.; Yano, T.; Takizawa, S.; Nagasaka, K.; Nakagawa, K.; Minaguchi, T.; Wada, O.; Ooishi, H.; Matsumoto, K.; et al. Involvement of a cellular ubiquitin-protein ligase E6AP in the ubiquitin-mediated degradation of extensive substrates of high-risk human papillomavirus E6. J. Med. Virol. 2006, 78, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, M.; Ma, Z.; Zhao, S.; Euskirchen, G.; Ziskin, J.; Urban, A.; Hallmayer, J.; Snyder, M. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 2014, 10, 774. [Google Scholar] [CrossRef]
- Veeramah, K.R.; Johnstone, L.; Karafet, T.M.; Wolf, D.; Sprissler, R.; Salogiannis, J.; Barth-Maron, A.; Greenberg, M.E.; Stuhlmann, T.; Weinert, S.; et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia 2013, 54, 1270–1281. [Google Scholar] [CrossRef]
- Chakrabarti, B.; Dudbridge, F.; Kent, L.; Wheelwright, S.; Hill-Cawthorne, G.; Allison, C.; Banerjee-Basu, S.; Baron-Cohen, S. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009, 2, 157–177. [Google Scholar] [CrossRef]
- Kuhnle, S.; Kogel, U.; Glockzin, S.; Marquardt, A.; Ciechanover, A.; Matentzoglu, K.; Scheffner, M. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 2011, 286, 19410–19416. [Google Scholar] [CrossRef] [PubMed]
- Puffenberger, E.G.; Jinks, R.N.; Wang, H.; Xin, B.; Fiorentini, C.; Sherman, E.A.; Degrazio, D.; Shaw, C.; Sougnez, C.; Cibulskis, K.; et al. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum. Mutat. 2012, 33, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Harlalka, G.V.; Baple, E.L.; Cross, H.; Kuhnle, S.; Cubillos-Rojas, M.; Matentzoglu, K.; Patton, M.A.; Wagner, K.; Coblentz, R.; Ford, D.L.; et al. Mutation of HERC2 causes developmental delay with Angelman-like features. J. Med. Genet. 2013, 50, 65–73. [Google Scholar] [CrossRef]
- Dolen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef]
- Pignatelli, M.; Piccinin, S.; Molinaro, G.; Di Menna, L.; Riozzi, B.; Cannella, M.; Motolese, M.; Vetere, G.; Catania, M.V.; Battaglia, G.; et al. Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling angelman syndrome. J. Neurosci. 2014, 34, 4558–4566. [Google Scholar] [CrossRef]
- Mishra, A.; Maheshwari, M.; Chhangani, D.; Fujimori-Tonou, N.; Endo, F.; Joshi, A.P.; Jana, N.R.; Yamanaka, K. E6-AP association promotes SOD1 aggresomes degradation and suppresses toxicity. Neurobiol. Aging 2013, 34, 1310.e11–1310.e23. [Google Scholar] [CrossRef] [PubMed]
- Kovac, J.; Macedoni Luksic, M.; Trebusak Podkrajsek, K.; Klancar, G.; Battelino, T. Rare single nucleotide polymorphisms in the regulatory regions of the superoxide dismutase genes in autism spectrum disorder. Autism Res. 2014, 7, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Smalley, S.L. Autism and tuberous sclerosis. J. Autism Dev. Disord. 1998, 28, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.E.; Rosa, J.L.; Scheffner, M. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998, 273, 12148–12154. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, E.L.; Bradford, Y.; Chen, Y.; Hall, J.; Arnone, B.; Gardiner, M.B.; Hutcheson, H.B.; Gilbert, J.R.; Pericak-Vance, M.A.; Copeland-Yates, S.A.; et al. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 2001, 77, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Al Ageeli, E.; Drunat, S.; Delanoe, C.; Perrin, L.; Baumann, C.; Capri, Y.; Fabre-Teste, J.; Aboura, A.; Dupont, C.; Auvin, S.; et al. Duplication of the 15q11–q13 region: Clinical and genetic study of 30 new cases. Eur. J. Med. Genet. 2014, 57, 5–14. [Google Scholar] [CrossRef]
- Patzold, L.M.; Richdale, A.L.; Tonge, B.J. An investigation into sleep characteristics of children with autism and Asperger’s Disorder. J. Paediatr. Child Health 1998, 34, 528–533. [Google Scholar] [CrossRef]
- Wiggs, L.; Stores, G. Sleep patterns and sleep disorders in children with autistic spectrum disorders: Insights using parent report and actigraphy. Dev. Med. Child Neurol. 2004, 46, 372–380. [Google Scholar] [CrossRef]
- Liu, X.; Hubbard, J.A.; Fabes, R.A.; Adam, J.B. Sleep disturbances and correlates of children with autism spectrum disorders. Child Psychiatry Hum. Dev. 2006, 37, 179–191. [Google Scholar] [CrossRef]
- Souders, M.C.; Mason, T.B.; Valladares, O.; Bucan, M.; Levy, S.E.; Mandell, D.S.; Weaver, T.E.; Pinto-Martin, J. Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep 2009, 32, 1566–1578. [Google Scholar] [CrossRef]
- Devnani, P.A.; Hegde, A.U. Autism and sleep disorders. J. Pediatr. Neurosci. 2015, 10, 304–307. [Google Scholar] [CrossRef]
- Cohen, S.; Conduit, R.; Lockley, S.W.; Rajaratnam, S.M.; Cornish, K.M. The relationship between sleep and behavior in autism spectrum disorder (ASD): A review. J. Neurodev. Disord. 2014, 6, 44. [Google Scholar] [CrossRef]
- Ferrarelli, F.; Tononi, G. Reduced sleep spindle activity point to a TRN-MD thalamus-PFC circuit dysfunction in schizophrenia. Schizophr. Res. 2017, 180, 36–43. [Google Scholar] [CrossRef]
- Wamsley, E.J.; Tucker, M.A.; Shinn, A.K.; Ono, K.E.; McKinley, S.K.; Ely, A.V.; Goff, D.C.; Stickgold, R.; Manoach, D.S. Reduced sleep spindles and spindle coherence in schizophrenia: Mechanisms of impaired memory consolidation? Biol. Psychiatry 2012, 71, 154–161. [Google Scholar] [CrossRef]
- Limoges, E.; Mottron, L.; Bolduc, C.; Berthiaume, C.; Godbout, R. Atypical sleep architecture and the autism phenotype. Brain 2005, 128 Pt 5, 1049–1061. [Google Scholar] [CrossRef]
- Tessier, S.; Lambert, A.; Chicoine, M.; Scherzer, P.; Soulieres, I.; Godbout, R. Intelligence measures and stage 2 sleep in typically-developing and autistic children. Int. J. Psychophysiol. 2015, 97, 58–65. [Google Scholar] [CrossRef]
- Christensen, J.A.; Kempfner, J.; Zoetmulder, M.; Leonthin, H.L.; Arvastson, L.; Christensen, S.R.; Sorensen, H.B.; Jennum, P. Decreased sleep spindle density in patients with idiopathic REM sleep behavior disorder and patients with Parkinson’s disease. Clin. Neurophysiol. 2014, 125, 512–519. [Google Scholar] [CrossRef]
- Fernandez, L.M.J.; Luthi, A. Sleep Spindles: Mechanisms and Functions. Physiol. Rev. 2020, 100, 805–868. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Ammanuel, S.; O’Driscoll, C.; Wozniak, A.; Naidu, S.; Kadam, S.D. Twenty-four hour quantitative-EEG and in-vivo glutamate biosensor detects activity and circadian rhythm dependent biomarkers of pathogenesis in Mecp2 null mice. Front. Syst. Neurosci. 2014, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Ammanuel, S.; Chan, W.C.; Adler, D.A.; Lakshamanan, B.M.; Gupta, S.S.; Ewen, J.B.; Johnston, M.V.; Marcus, C.L.; Naidu, S.; Kadam, S.D. Heightened Delta Power during Slow-Wave-Sleep in Patients with Rett Syndrome Associated with Poor Sleep Efficiency. PLoS ONE 2015, 10, e0138113. [Google Scholar] [CrossRef]
- Arazi, A.; Meiri, G.; Danan, D.; Michaelovski, A.; Flusser, H.; Menashe, I.; Tarasiuk, A.; Dinstein, I. Reduced sleep pressure in young children with autism. Sleep 2020, 43, zsz309. [Google Scholar] [CrossRef] [PubMed]
- Saravanapandian, V.; Nadkarni, D.; Hsu, S.H.; Hussain, S.A.; Maski, K.; Golshani, P.; Colwell, C.S.; Balasubramanian, S.; Dixon, A.; Geschwind, D.H.; et al. Abnormal sleep physiology in children with 15q11.2–q13.1 duplication (Dup15q) syndrome. Mol. Autism 2021, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Sun, J.; Ji, A.X.; Baudry, M. Potential therapeutic approaches for Angelman syndrome. Expert Opin. Ther. Targets 2016, 20, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Daily, J.L.; Nash, K.; Jinwal, U.; Golde, T.; Rogers, J.; Peters, M.M.; Burdine, R.D.; Dickey, C.; Banko, J.L.; Weeber, E.J. Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE 2011, 6, e27221. [Google Scholar] [CrossRef] [PubMed]
- Schanen, N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006, 15, R138–R150. [Google Scholar] [CrossRef]
- Owais, A.; Mishra, R.K.; Kiyokawa, H. The HECT E3 Ligase E6AP/UBE3A as a Therapeutic Target in Cancer and Neurological Disorders. Cancers 2020, 12, 2108. [Google Scholar] [CrossRef]
- Vatsa, N.; Jana, N.R. UBE3A and Its Link with Autism. Front. Mol. Neurosci. 2018, 11, 448. [Google Scholar] [CrossRef]
- Huang, H.S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W., Jr.; Lee, H.M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef]
- Peters, S.U.; Bird, L.M.; Kimonis, V.; Glaze, D.G.; Shinawi, L.M.; Bichell, T.J.; Barbieri-Welge, R.; Nespeca, M.; Anselm, I.; Waisbren, S.; et al. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid. Am. J. Med. Genet. A 2010, 152, 1994–2001. [Google Scholar] [CrossRef]
- Bird, L.M.; Tan, W.H.; Bacino, C.A.; Peters, S.U.; Skinner, S.A.; Anselm, I.; Barbieri-Welge, R.; Bauer-Carlin, A.; Gentile, J.K.; Glaze, D.G.; et al. A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome. Am. J. Med. Genet. A 2011, 155, 2956–2963. [Google Scholar] [CrossRef]
- Bailus, B.J.; Pyles, B.; McAlister, M.M.; O’Geen, H.; Lockwood, S.H.; Adams, A.N.; Nguyen, J.T.; Yu, A.; Berman, R.F.; Segal, D.J. Protein Delivery of an Artificial Transcription Factor Restores Widespread Ube3a Expression in an Angelman Syndrome Mouse Brain. Mol. Ther. 2016, 24, 548–555. [Google Scholar] [CrossRef]
- Lizarraga, S.B.; Morrow, E.M. Uncovering a Role for SK2 in Angelman Syndrome. Cell Rep. 2015, 12, 359–360. [Google Scholar] [CrossRef]
- Ngo-Anh, T.J.; Bloodgood, B.L.; Lin, M.; Sabatini, B.L.; Maylie, J.; Adelman, J.P. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 2005, 8, 642–649. [Google Scholar] [CrossRef]
- Sun, J.; Baudry, M.; Bi, X. Novel neurobiological roles of UBE3A. Oncotarget 2017, 8, 12548–12549. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Jia, Y.; Hao, X.; Lin, W.J.; Tran, J.; Lynch, G.; Baudry, M.; Bi, X. UBE3A-mediated p18/LAMTOR1 ubiquitination and degradation regulate mTORC1 activity and synaptic plasticity. Elife 2018, 7, e37993. [Google Scholar] [CrossRef]
- Sato, A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef]
- Burket, J.A.; Benson, A.D.; Tang, A.H.; Deutsch, S.I. Rapamycin improves sociability in the BTBR T(+)Itpr3(tf)/J mouse model of autism spectrum disorders. Brain Res. Bull. 2014, 100, 70–75. [Google Scholar] [CrossRef]
- Robak, T.; Huang, H.; Jin, J.; Zhu, J.; Liu, T.; Samoilova, O.; Pylypenko, H.; Verhoef, G.; Siritanaratkul, N.; Osmanov, E.; et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N. Engl. J. Med. 2015, 372, 944–953. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Landgren, O.; Sonneveld, P.; Jakubowiak, A.; Mohty, M.; Iskander, K.S.; Mezzi, K.; Siegel, D.S. Carfilzomib with immunomodulatory drugs for the treatment of newly diagnosed multiple myeloma. Leukemia 2019, 33, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Thomas, S.M.; Chan, E.T.; Kirk, C.J.; Freilino, M.L.; DeLancey, H.M.; Grandis, J.R.; Li, C.; Johnson, D.E. Carfilzomib and ONX 0912 inhibit cell survival and tumor growth of head and neck cancer and their activities are enhanced by suppression of Mcl-1 or autophagy. Clin. Cancer Res. 2012, 18, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Lee, B.H. Chemically Induced Cellular Proteolysis: An Emerging Therapeutic Strategy for Undruggable Targets. Mol. Cells 2018, 41, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Okuhira, K.; Ohoka, N.; Sai, K.; Nishimaki-Mogami, T.; Itoh, Y.; Ishikawa, M.; Hashimoto, Y.; Naito, M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 2011, 585, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef]
- Grieco, J.C.; Ciarlone, S.L.; Gieron-Korthals, M.; Schoenberg, M.R.; Smith, A.G.; Philpot, R.M.; Heussler, H.S.; Banko, J.L.; Weeber, E.J. An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurol. 2014, 14, 232. [Google Scholar] [CrossRef]
- Germain, N.D.; Chen, P.F.; Plocik, A.M.; Glatt-Deeley, H.; Brown, J.; Fink, J.J.; Bolduc, K.A.; Robinson, T.M.; Levine, E.S.; Reiter, L.T.; et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11–q13.1. Mol. Autism 2014, 5, 44. [Google Scholar] [CrossRef]
- Yi, J.J.; Berrios, J.; Newbern, J.M.; Snider, W.D.; Philpot, B.D.; Hahn, K.M.; Zylka, M.J. An Autism-Linked Mutation Disables Phosphorylation Control of UBE3A. Cell 2015, 162, 795–807. [Google Scholar] [CrossRef]
- Roy, B.; Lee, E.; Li, T.; Rampersaud, M. Role of miRNAs in Neurodegeneration: From Disease Cause to Tools of Biomarker Discovery and Therapeutics. Genes 2022, 13, 425. [Google Scholar] [CrossRef]
- Cruz, E.; Descalzi, G.; Steinmetz, A.; Scharfman, H.E.; Katzman, A.; Alberini, C.M. CIM6P/IGF-2 Receptor Ligands Reverse Deficits in Angelman Syndrome Model Mice. Autism Res. 2021, 14, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Person, R.E.; Huang, W.; Zhu, P.J.; Costa-Mattioli, M.; Beaudet, A.L. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013, 9, e1004039. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, S.; van Woerden, G.M.; Bruinsma, C.F.; Mientjes, E.; Jolfaei, M.A.; Distel, B.; Kushner, S.A.; Elgersma, Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Investig. 2015, 125, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Sonzogni, M.; Zhai, P.; Mientjes, E.J.; van Woerden, G.M.; Elgersma, Y. Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol. Autism 2020, 11, 70. [Google Scholar] [CrossRef]
- Roy, B.; Han, J.; Hope, K.A.; Peters, T.L.; Palmer, G.; Reiter, L.T. An Unbiased Drug Screen for Seizure Suppressors in Duplication 15q Syndrome Reveals 5-HT1A and Dopamine Pathway Activation as Potential Therapies. Biol. Psychiatry 2020, 88, 698–709. [Google Scholar] [CrossRef]




| (A) | |||||||
| S.No. | Case Study Group | Author Details | Analytical Methodology | Gene Location | Genes Modulated | Type of Mutation | Reference |
| 1 | 100 families, of which 42 affected sibling-pair families and 58 trios were tested. | Nurmi et al., 2003. | Genotyping of SNPs was performed either by using allele-specific oligonucleotide hybridization (ASO), pyrosequencing analysis (PSQ), or fluorescent polarization-template, directed-dye terminator incorporation assay [FP-TD1]. | The maternal expression domain of 15q11–q13 and ICRs. | Two SNPs are located within the gene ATP10C. | SNPs | [2] |
| 2 | 166 ASD patients and 416 healthy individuals. | Kato et al., 2008 | SNP analysis based on sequencing technology. | Maternal expression 15q11– q13 domain. | Marker haplotype located in ATP10C gene. | SNPs | [3] |
| 3 | 20 ASD patients and controls. | Gregory et al., 2009 | Control patients and 20 ASD patients. Defects in oxytocin function were found in the ASD patients. | 15q11.2 | Receptor for Oxytocin [OXTR] | Hypermethylation or excessive methylation of the gene promoter, with a reduced mRNA expression. | [4] |
| 4 | 58 participants (affected with a C15-imprinting disorder), 20 normal control patients. | Baker et al., 2020 | Reverse transcription ddPCR (droplet digital polymerase chain reaction) was used for gene expression assay | 15q11–q13 | UBE3A, SNORD116. | Duplication | [5] |
| 5 | 2 postmortem brain tissue samples. | Hogart et al., 2009 | RT-PCR followed by Southern blot, bisulfite sequencing, and fluorescence in situ hybridization. | 15q11–q13 | Paternally expressed transcripts of SNRPN, NDN, HBII85, and HBII52 showed deficiencies. Increased DNA methylation of the 15q11–q13-imprinted control region (ICR) was observed. | Duplication | [6] |
| 6 | 146 subjects with autism/ASD. | Burnside et al., 2001 | Cytogenomic microarray analytical tools were implemented. | Proximal 15q11.2. | Deletion or duplication of the BP1-BP2 region on proximal 15q. | [7] | |
| 7 | 15-year-old female case report study. | Han, 2021 | Array-comparative genomic hybridization (aCGH) of chromosome 15q11.2–q13.1 was performed. | Chromosome 15q11.2–q13.1. | Maternally expressed gene product in the critical region of PWS/AS. | Interstitial (int) dup15q | [8] |
| 8 | 146 parents, 79 pro-bands, and seven healthy siblings amongst a total of 232 subjects. | Guffanti et al., 2011 | Low-density genotyping followed by secondary analysis using a densely spaced set of tag-SNPs spanning the target area. | 15q11–q13 | UBE3A and ATP10A. | An association of autistic phenotypes with an SNP located in the intergenic region between UBE3A and ATP10A was observed. | [9] |
| 9 | 267 patients showing clinical manifestations like intellectual disability, autism, epilepsy, and congenital defects. | Iourov et al., 2015 | SNP chromosomal microarray. | Long continuous stretches of homozygous (LCSH) regions of 7q21.3, 7q31.2, 11p15.5, and 15p11.2. | UBE3A | Single nucleotide polymorph-hism (SNP). | [10] |
| 10 | 504 children with ASD and intellectual disability defects. | Iourov et al., 2019 | SNP-based chromosomal microarray. | Long continuous stretches of homozygous in situ (LCSH) regions of 7q21.3, 7q31.2, 11p15.5, and 15p11.2. | UBE3A, MAGEL2. | Single nucleotide polymorphism (SNP). | [11] |
| 11 | 297 affected children with language impairment). | Nudel et al., 2014 | GWAS [Genome wide association] analysis, using a genotyping array. | Chromosome 14q12, Chromosome 5p13. | Several genes including the NOP9 gene in the paternal region; chromosome 5 regions fall between the PTGER4 and DAB2 genes | Paternal parent-of-origin effects on chromosome 14q12; suggestive maternal parent-of-origin effects on chromosome 5p13. | [12] |
| 12 | 6 chimpanzees and 25 human brain tissue samples were analyzed. | Schneider et al., 2014 | Methylated DNA immunoprecipitation (MeDIP) combined with DNA methylation arrays. | CNTNAP2 | Protein-coding splice variant CNTNAP2-201 is 1.6 times upregulated in the human cortex, with widespread cortical DNA methylation changes in CNTNAP2. | [13] | |
| 13 | 43 ASD patients and 38 controls (a total of 223 post-mortem tissue samples isolated from three brain regions [prefrontal cortex, the temporal cortex, and the cerebellum]). | Wong et al., 2019 | Genome-wide DNA methylation profiling. | Chromosome 15q (dup15q). | UBE3A, ATP10A. | Duplication and DNA methylation. | [14] |
| 14 | A single male patient case report study. | Wu et al., 2009 | Array comparative genome hybridization (aCGH). | Chromosome 15. | Chromosome 15 duplication arising from a 3:1 segregation error of a paternally derived translocation between chromosome 15q13.2 and chromosome 9q34.12, which led to trisomy of chromosome 15p-q13.2 and 9q34.12-q. | [15] | |
| 15 | 993 families with 896 sibling pairs from the AGRE (Autism Genetic Resource Exchange) and 223 families (174 affected sibling pairs) from the NIMH (National Institute of Mental Health) Autism Genetics initiative were included. | Fradin et al., 2010 | SNP genotyping, followed by a genome-wide linkage scan using parametric and non-parametric linkage analysis. | Chromosome 15q. | CLOCK gene. | SNPs | [16] |
| 16 | 522 patients. | Depien-ne et al., 2009. | Multiplex ligation-dependent probe amplification (MLPA). | Chromosome 15q11–q13 region. | UBE3A, GABRB3. | Deletions, duplications, and methylation abnormalities. | [17] |
| 17 | 82 patients with autism. | Curran et al., 2005. | Extended transmission disequilibrium test (ETDT). | Chromosome 15 (q11–q13). | GABRB3 | Microsatellite markers, hemizygous deletion. | [18] |
| 18 | 18 patients with ASD and 15 controls; post mortem tissue samples were analyzed. | Ben-David et al., 2014. | Genome-wide survey of allele expression imbalance (AEI). | Chromosome 15q11–q13. | UBE3A | SNPs | [19] |
| 19 | Postmortem human brain tissue (8 from dup15q, 10 patients with idiopathic autism, and 21 controls) | Scoles et al., 2011 | Quantitative RT-PCR and Western blot analyses. | Chromosome 15q11–q13. | UBE3A | Duplication | [20] |
| 20 | A single female patient case study. | Noor et al., 2015. | Array comparative genomic hybridization (aCGH) analysis. | 15q11.2 duplication encompassing only the UBE3A gene. | UBE3A | Maternally inherited 129 Kb duplication in chromosome region 15q11.2 encompassing only the UBE3A gene. | [21] |
| 21 | 10 autistic patients. | Veenst-ra et al., 1999 | Mutation screening of the UBE3A/E6-AP gene in autistic disorder. | 15q11–q13. | UBE3A | Coding region and a putative promoter region. | [16] |
| 22 | 6-month-old girl case report study. | Kitsiou-Tzeli et al., 2010 | Array comparative genomic hybridization (aCGH) analysis. | 15q11–q13. | Duplication of maternal origin of the 15q11.2–q14 PWS/AS region. | [22] | |
| 23 | 12 family members, in a family, tested positive for the 15q11q13 duplication. | Piard et al., 2010 | Fluorescence in in situ hybridization (FISH), PCR analysis of microsatellite markers, array-comparative genomic hybridization analysis (Array-CGH) and semi-quantitative methylation-sensitive PCR. | Chromosome 15q11–q13. | CYFIP1, MAGEL2, SNRPN, UBE3A, and GABRB3. | Interstitial duplication of chromosome 15q11–q13. | [23] |
| (B) | |||||||
| S.No. | Details of the Authors | Model Organism Being Investigated | Position on the Chromosome | Genes Involved | Type of Mutation | Role of the Gene in the Cellular Process | Reference in Pubmed |
| 1 | Leung and coworkers, 2009 | C57BL/6 mice | 15q11–q13 snoRNA HBII-85 locus. | snoRNA | Locus of UBE3A gene with the mutation for transcriptionally regulated chromatin decondensation or unfolding. | Gene transcription, neurodevelopmental processes and long-term memory. | [24] |
| 2 | Samaco and coworkers, 2005 | Mouse null for Mecp2. Brain region was investigated. | Maternal 15q11–q13 or UBE3A deficiency. | Mecp2, UBE3A and Gabrb3. | Mutation in Mecp2. | Synaptic excitatory or inhibitory homeostasis maintenance. | [25] |
| 3 | Hogart and coworkers, 2008 | Mouse | 15q11–q13 | Atp10a/ATP10A. | Imprinting of the maternally expressed gene Atp10a/ATP10A. | [26] | |
| 4 | Powell and coworkers, 2013 | Mice | 15q11–q13 | UBE3A | Mice with Snord116 deletion exhibit increased UBE3A-ATS levels. | [27] | |
| 5 | Jiang and coworkers, 2010 | C57BL/6 mouse | UBE3A Gabrb3 deletions of 15q11–q13 | UBE3A, Gabrb3, Atp10a | Maternal deletion from the UBE3A to the Gabrb3 gene region. | [28] | |
| 6 | Chibuk and coworkers, 2001 | C57BL/6JEi and M. spretus SPRET/Ei DNA | Chromo-some 15q. | NDNL2 | [29] | ||
| (C) | |||||||
| S.No. | Author Details | Investigation Cellular Models | Position | Genes Involved | Mutation Type | Reference | |
| 1 | Thatcher et al., 2005 | SH-SY5Y neuroblastoma cells. | 15q11–q13 | SNURF/SNRPN | Imprinting of 15q11–q13. | [30] | |
| 2 | Lopez et al., 2017 | Human neuroblastoma cells, including parental SH-SY5Y and the SH(15M) cellular model. | 15q11–q13 | UBE3A | Duplication of the 15q11–q13 locus. | [31] | |
| 3 | Dunaway et al., 2016 | Dup15q cell model (SH15M) and parental SH-SY5Y (SH) cell lines. | 15q11.2–q13.3 | UBE3A | 15q11.2–q13.3 maternal duplication. | [32] | |
| S.No. | GENES | No. of Times Genes Detected in Various Clinical, In Vitro and In Vivo Studies. [Total 31 Studies] |
|---|---|---|
| 1 | ATP10C | 2 |
| 2 | OXTR | 1 |
| 3 | UBE3A | 16 |
| 4 | SNORD116 | 1 |
| 5 | SNRPN | 1 |
| 6 | NDN | 1 |
| 7 | HB1185 | 1 |
| 8 | HB1152 | 1 |
| 9 | ATP10A | 4 |
| 10 | MAGEL2 | 2 |
| 11 | NOP9 | 1 |
| 12 | PTGER4 | 1 |
| 13 | DAB2 | 1 |
| 14 | CNTNAP2 | 1 |
| 15 | CLOCK | 1 |
| 16 | GABRB3 | 5 |
| 17 | CYF1P1 | 1 |
| 18 | SNRPN/SNURF | 1 |
| 19 | SnoRNA | 1 |
| 20 | Mecp2 | 1 |
| 21 | NDNL2 | 1 |
| Biomarker | Methodology | Reference |
|---|---|---|
| Beta EEG | Spontaneous EEG recordings. | [4] |
| Awake electroencephalography (EEG) | Spindle density, beta power, and slow-wave sleep (SWS) percentage in sleep EEG recordings as quantitative parameters for measurements of clinical manifestation of ASD symptoms. | [78] |
| UBE3A and SNORD116 mRNA levels | Semi-quantitative techniques like reverse transcription droplet digital polymerase chain reaction (PCR) were used for UBE3A and SNORD116mRNA analysis from peripheral blood mononuclear cells (PBMCs). The data were normalized to a panel of internal control genes using the geNorm approach. | [79] |
| UBE3A protein | Micro-dialysis of CSF fluid located in the rat hippocampus. | [5] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, B.; Amemasor, E.; Hussain, S.; Castro, K. UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies. Diseases 2024, 12, 7. https://doi.org/10.3390/diseases12010007
Roy B, Amemasor E, Hussain S, Castro K. UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies. Diseases. 2024; 12(1):7. https://doi.org/10.3390/diseases12010007
Chicago/Turabian StyleRoy, Bidisha, Enyonam Amemasor, Suhail Hussain, and Kimberly Castro. 2024. "UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies" Diseases 12, no. 1: 7. https://doi.org/10.3390/diseases12010007