
WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: Current Status and Future Perspective
 , , , ,
, , , ,  , and
, and 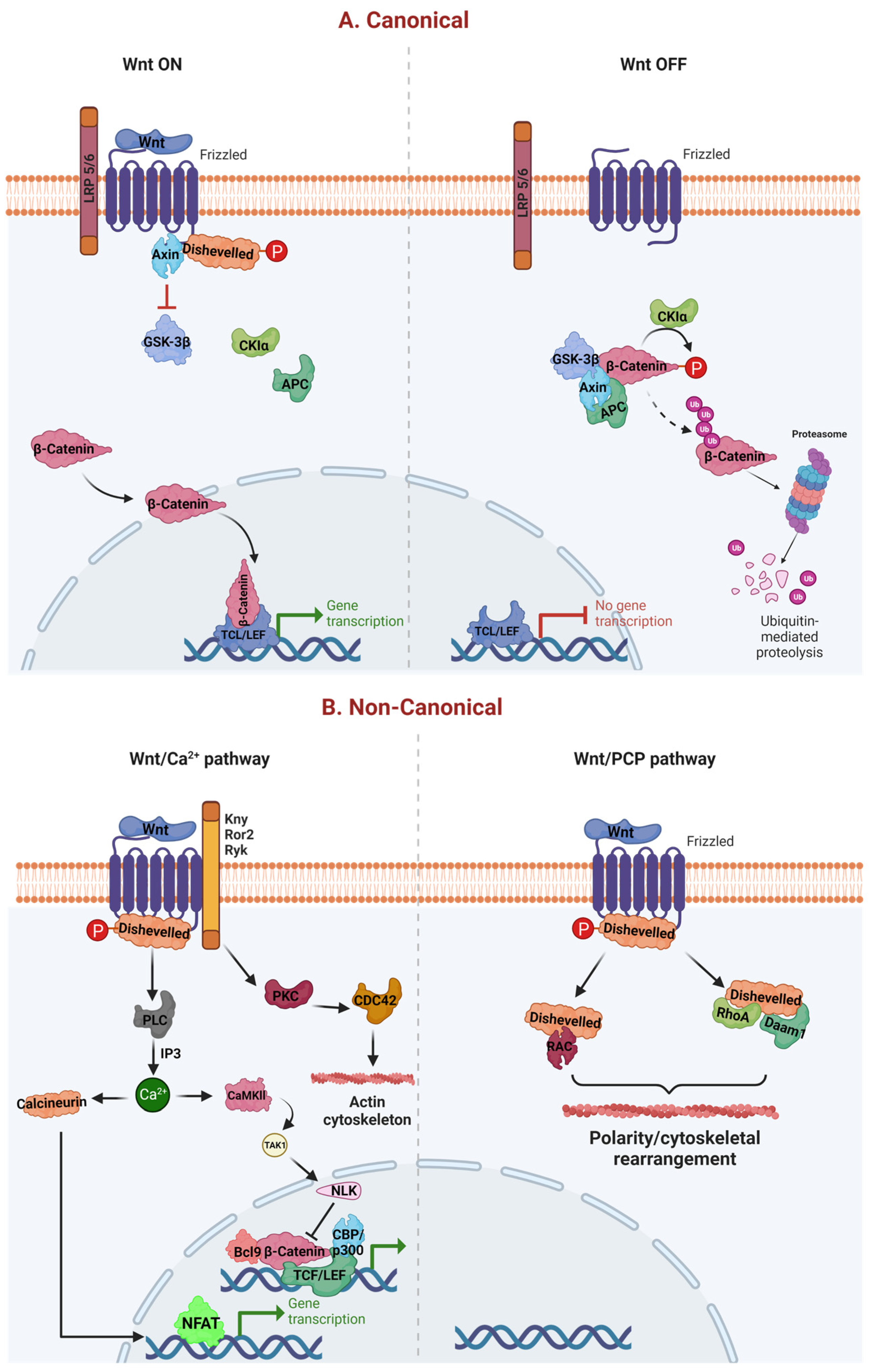{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
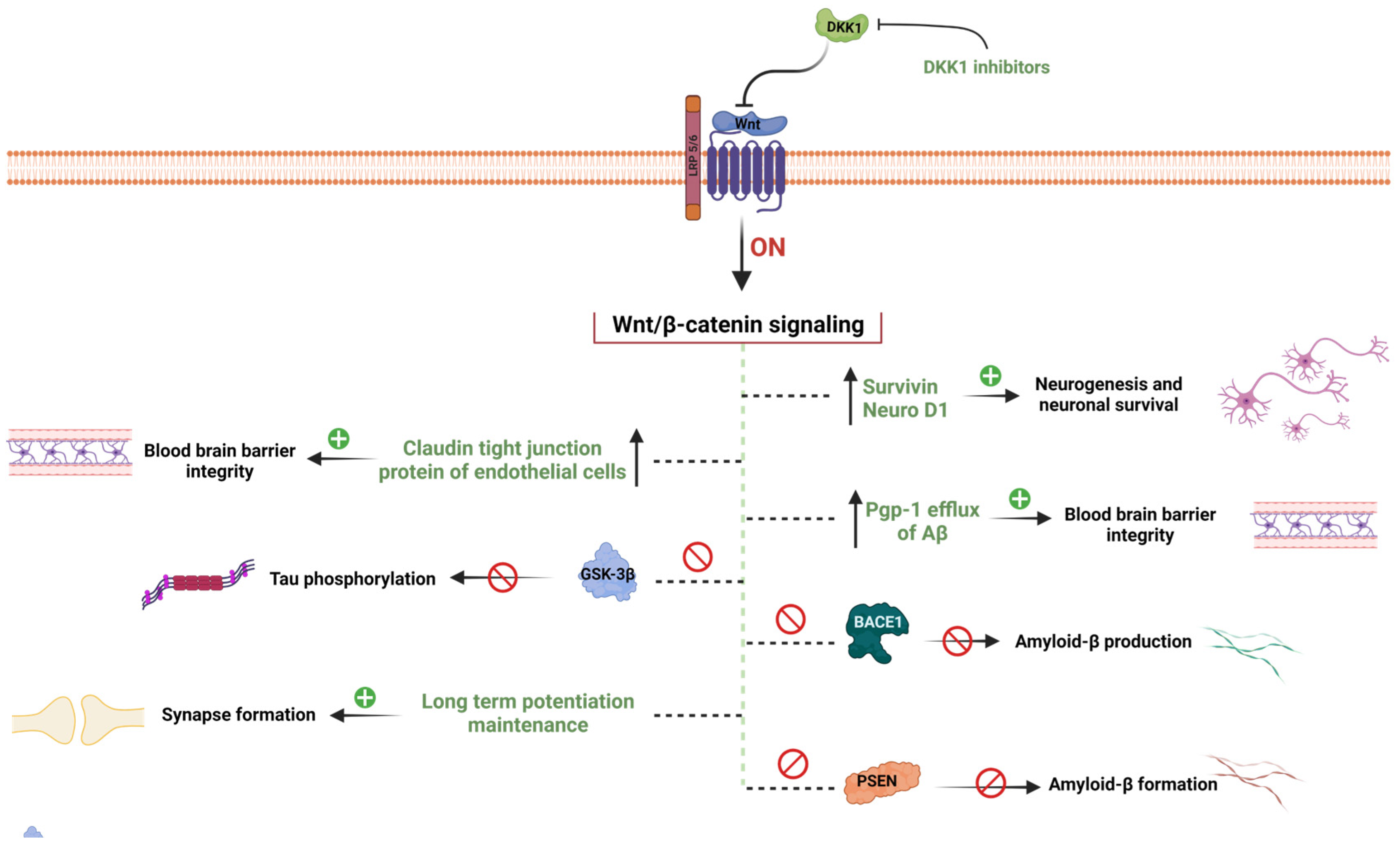
2. WβC Signaling in Alzheimer’s Disease
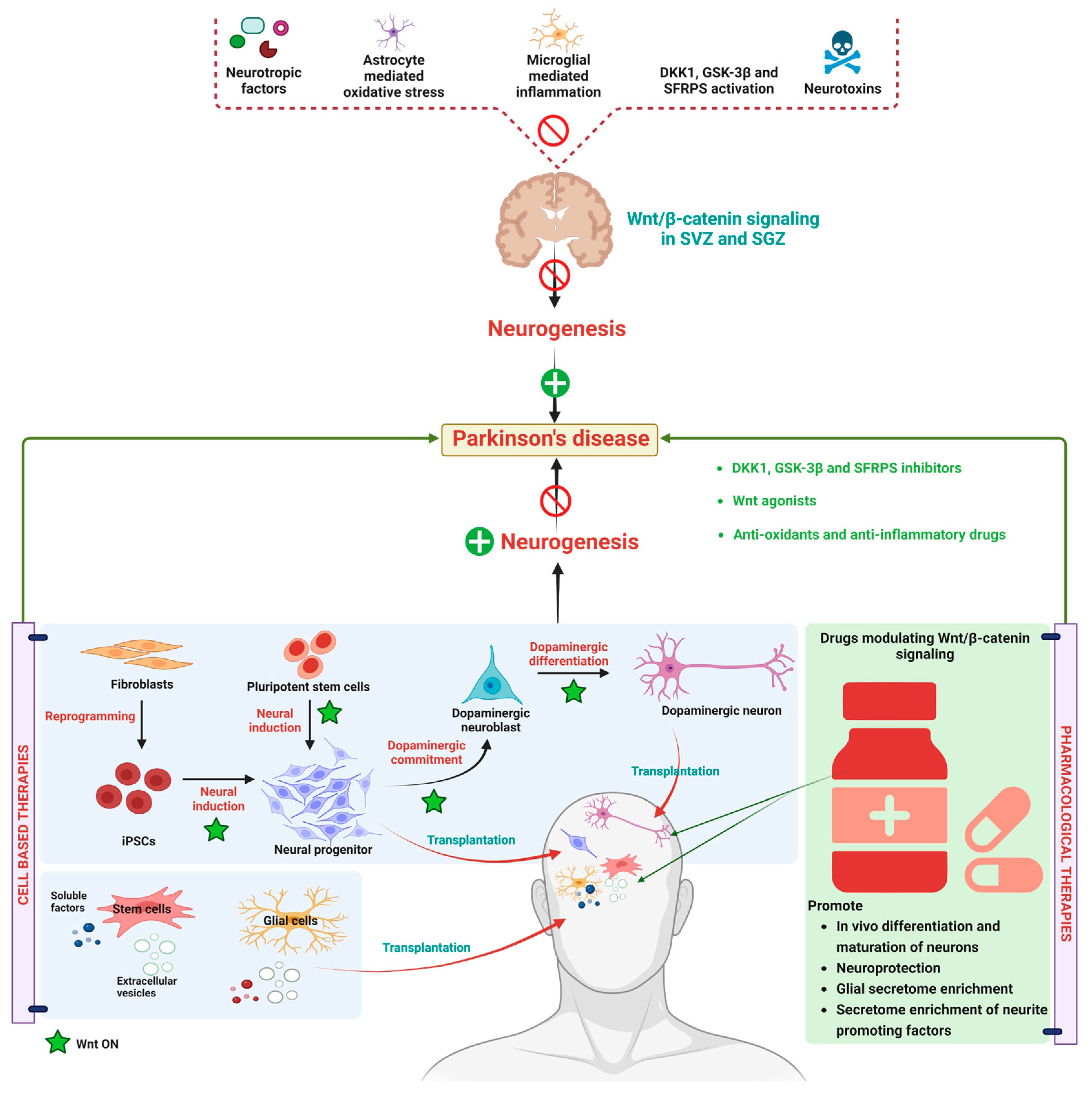
3. WβC Signaling in Parkinson’s Disease
4. WβC Signaling in Huntington’s Disease
5. WβC Signaling in Amyotrophic Lateral Sclerosis
6. WβC Signaling in Multiple Sclerosis
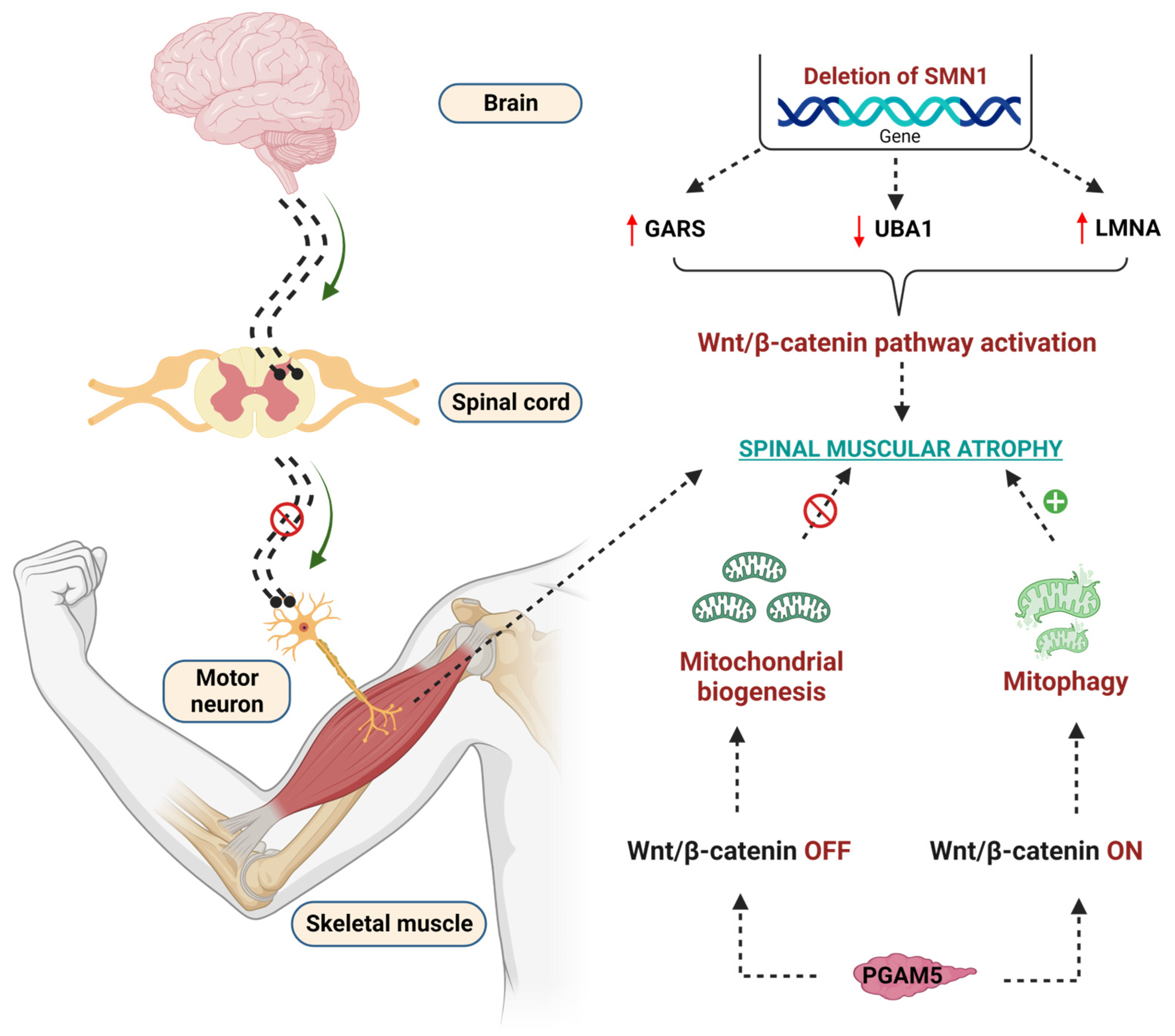
7. WβC Signaling in Spinal Muscular Atrophy
8. Summary and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchetti, B.; Tirolo, C.; L’Episcopo, F.; Caniglia, S.; Testa, N.; Smith, J.A.; Pluchino, S.; Serapide, M.F. Parkinson’s disease, aging and adult neurogenesis: Wnt/β-catenin signalling as the key to unlock the mystery of endogenous brain repair. Aging Cell 2020, 19, e13101. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Yang, S.; Zhang, J. The Wnt/β-catenin signaling pathway in the adult neurogenesis. Eur. J. Neurosci. 2011, 33, 1–8. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.N.; Lecarpentier, Y. Parkinson’s Disease: Potential Actions of Lithium by Targeting the WNT/β-Catenin Pathway, Oxidative Stress, Inflammation and Glutamatergic Pathway. Cells 2021, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Fisher, L.W.; Kilts, T.M.; Owens, R.T.; Robey, P.G.; Gutkind, J.S.; Young, M.F. Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc. Natl. Acad. Sci. USA 2011, 108, 17022–17027. [Google Scholar] [CrossRef]
- Cong, F.; Schweizer, L.; Varmus, H. Wnt signals across the plasma membrane to activate the β-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development 2004, 131, 5103–5115. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Huelsken, J.; Held, W. Canonical Wnt signalling plays essential roles. Eur. J. Immunol. 2009, 39, 3582–3583. [Google Scholar] [CrossRef]
- Kohn, A.D.; Moon, R.T. Wnt and calcium signaling: β-catenin-independent pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef]
- Ishitani, T.; Ninomiya-Tsuji, J.; Nagai, S.-i.; Nishita, M.; Meneghini, M.; Barker, N.; Waterman, M.; Bowerman, B.; Clevers, H.; Shibuya, H. The TAK1–NLK–MAPK-related pathway antagonizes signalling between β-catenin and transcription factor TCF. Nature 1999, 399, 798–802. [Google Scholar] [CrossRef]
- Ma, L.; Wang, H.-y. Suppression of cyclic GMP-dependent protein kinase is essential to the Wnt/cGMP/Ca2+ pathway. J. Biol. Chem. 2006, 281, 30990–31001. [Google Scholar] [CrossRef]
- Boise, L.; Petryniak, B.; Mao, X.; June, C.; Wang, C.; Lindsten, T.; Bravo, R.; Kovary, K.; Leiden, J.; Thompson, C. The NFAT-1 DNA binding complex in activated T cells contains Fra-1 and JunB. Mol. Cell. Biol. 1993, 13, 1911–1919. [Google Scholar]
- Wayman, G.A.; Impey, S.; Marks, D.; Saneyoshi, T.; Grant, W.F.; Derkach, V.; Soderling, T.R. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 2006, 50, 897–909. [Google Scholar] [CrossRef]
- Ahn, V.E.; Chu, M.L.-H.; Choi, H.-J.; Tran, D.; Abo, A.; Weis, W.I. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev. Cell 2011, 21, 862–873. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Scholz, B.; Korn, C.; Wojtarowicz, J.; Mogler, C.; Augustin, I.; Boutros, M.; Niehrs, C.; Augustin, H.G. Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca2+/NFAT signaling. Dev. Cell 2016, 36, 79–93. [Google Scholar] [CrossRef]
- Carmichael, J.; Sugars, K.L.; Bao, Y.P.; Rubinsztein, D.C. Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002, 277, 33791–33798. [Google Scholar] [CrossRef]
- Wishart, T.M.; Mutsaers, C.A.; Riessland, M.; Reimer, M.M.; Hunter, G.; Hannam, M.L.; Eaton, S.L.; Fuller, H.R.; Roche, S.L.; Somers, E. Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J. Clin. Investig. 2014, 124, 1821–1834. [Google Scholar] [CrossRef]
- Li, X.; Guan, Y.; Chen, Y.; Zhang, C.; Shi, C.; Zhou, F.; Yu, L.; Juan, J.; Wang, X. Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int. J. Clin. Exp. Pathol. 2013, 6, 1245. [Google Scholar]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Serafino, A.; Cozzolino, M. The Wnt/β-catenin signaling: A multifunctional target for neuroprotective and regenerative strategies in Parkinson’s disease. Neural Regen. Res. 2023, 18, 306. [Google Scholar] [CrossRef]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- Janssen, J.; Beck, J.; Campbell, T.; Dickinson, A.; Fox, N.; Harvey, R.; Houlden, H.; Rossor, M.; Collinge, J. Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 2003, 60, 235–239. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Weisgraber, K.H.; Goedert, M.; Saunders, A.M.; Huang, D.; Corder, E.H.; Dong, L.-M.; Jakes, R.; Alberts, M.J.; Gilbert, J.R. Hypothesis: Microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp. Neurol. 1994, 125, 163–171. [Google Scholar] [CrossRef]
- Tripathi, P.N.; Srivastava, P.; Sharma, P.; Tripathi, M.K.; Seth, A.; Tripathi, A.; Rai, S.N.; Singh, S.P.; Shrivastava, S.K. Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg. Chem. 2019, 85, 82–96. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Srivastava, P.; Tripathi, P.N.; Sharma, P.; Rai, S.N.; Singh, S.P.; Srivastava, R.K.; Shankar, S.; Shrivastava, S.K. Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur. J. Med. Chem. 2019, 163, 116–135. [Google Scholar] [CrossRef]
- Rai, S.N.; Chaturvedi, V.K.; Singh, B.K.; Singh, M.P. Commentary: Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42: Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. Front. Aging Neurosci. 2020, 12, 219. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.; Singh, B.K. Mitochondrial dysfunction: A potential therapeutic target to treat Alzheimer’s disease. Mol. Neurobiol. 2020, 57, 3075–3088. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Zhang, Y.-w.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer’s disease. Future Sci. OA 2015, 1, FSO11. [Google Scholar] [CrossRef]
- Metaxas, A.; Kempf, S.J. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen. Res. 2016, 11, 1579. [Google Scholar]
- Monica Moore, M.; Díaz-Santos, M.; Vossel, K. Alzheimer’s Association 2021 Facts and Figures Report; Alzheimer’s Association: Chicago, IL, USA, 2021. [Google Scholar]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sánchez-López, E.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef]
- Gutti, G.; Kakarla, R.; Kumar, D.; Beohar, M.; Ganeshpurkar, A.; Kumar, A.; Krishnamurthy, S.; Singh, S.K. Discovery of novel series of 2-substituted benzo[d]oxazol-5-amine derivatives as multi-target directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 182, 111613. [Google Scholar] [CrossRef]
- Bhanukiran, K.; Singh, R.; Gajendra, T.A.; Ramakrishna, K.; Singh, S.K.; Krishnamurthy, S.; Kumar, A.; Hemalatha, S. Vasicinone, a pyrroloquinazoline alkaloid from Adhatoda vasica Nees enhances memory and cognition by inhibiting cholinesterases in Alzheimer’s disease. Phytomed. Plus 2023, 3, 100439. [Google Scholar] [CrossRef]
- Miculas, D.C.; Negru, P.A.; Bungau, S.G.; Behl, T.; Tit, D.M. Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells 2023, 12, 131. [Google Scholar] [CrossRef]
- Huang, C.; Chu, H.; Muheremu, A.; Zuo, H. Neurorestorative strategies for Alzheimer’s disease. Neurol. India 2015, 63, 583. [Google Scholar]
- Dengler-Crish, C.M.; Ball, H.C.; Lin, L.; Novak, K.M.; Cooper, L.N. Evidence of Wnt/β-catenin alterations in brain and bone of a tauopathy mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 67, 148–158. [Google Scholar] [CrossRef]
- Chacón, M.A.; Varela-Nallar, L.; Inestrosa, N.C. Frizzled-1 is involved in the neuroprotective effect of Wnt3a against Aβ oligomers. J. Cell. Physiol. 2008, 217, 215–227. [Google Scholar] [CrossRef]
- Zhang, Q.-G.; Wang, R.; Khan, M.; Mahesh, V.; Brann, D.W. Role of Dickkopf-1, an antagonist of the Wnt/β-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J. Neurosci. 2008, 28, 8430–8441. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Sharma, K.; Choi, S.Y.; Zhang, Y.; Nieland, T.J.; Long, S.; Li, M.; Huganir, R.L. High-throughput genetic screen for synaptogenic factors: Identification of LRP6 as critical for excitatory synapse development. Cell Rep. 2013, 5, 1330–1341. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Chen, J.; Park, C.S.; Tang, S.J. Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 2006, 281, 11910–11916. [Google Scholar] [CrossRef]
- McLeod, F.; Bossio, A.; Marzo, A.; Ciani, L.; Sibilla, S.; Hannan, S.; Wilson, G.A.; Palomer, E.; Smart, T.G.; Gibb, A.; et al. Wnt Signaling Mediates LTP-Dependent Spine Plasticity and AMPAR Localization through Frizzled-7 Receptors. Cell Rep. 2018, 23, 1060–1071. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of Synapse Degeneration by Restoring Wnt Signaling in the Adult Hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef]
- Buechler, J.; Salinas, P.C. Deficient Wnt Signaling and Synaptic Vulnerability in Alzheimer’s Disease: Emerging Roles for the LRP6 Receptor. Front. Synaptic Neurosci. 2018, 10, 38. [Google Scholar] [CrossRef]
- Liu, C.-C.; Tsai, C.-W.; Deak, F.; Rogers, J.; Penuliar, M.; Sung, Y.M.; Maher, J.N.; Fu, Y.; Li, X.; Xu, H. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron 2014, 84, 63–77. [Google Scholar] [CrossRef]
- Salta, E.; Lazarov, O.; Fitzsimons, C.P.; Tanzi, R.; Lucassen, P.J.; Choi, S.H. Adult hippocampal neurogenesis in Alzheimer’s disease: A roadmap to clinical relevance. Cell Stem Cell 2023, 30, 120–136. [Google Scholar] [CrossRef]
- Fortress, A.M.; Schram, S.L.; Tuscher, J.J.; Frick, K.M. Canonical Wnt signaling is necessary for object recognition memory consolidation. J. Neurosci. 2013, 33, 12619–12626. [Google Scholar] [CrossRef]
- Parr, C.; Mirzaei, N.; Christian, M.; Sastre, M. Activation of the Wnt/β-catenin pathway represses the transcription of the β-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J. 2015, 29, 623–635. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Burgos, P.V.; Inestrosa, N.C. Inhibition of Wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of Amyloid-β (Aβ) 42 peptides. J. Neurochem. 2016, 139, 1175–1191. [Google Scholar] [CrossRef]
- Noll, E.; Medina, M.; Hartley, D.; Zhou, J.; Perrimon, N.; Kosik, K.S. Presenilin affects Arm/β-catenin localization and function in Drosophila. Dev. Biol. 2000, 227, 450–464. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.; Hardy, J.; Hutton, M.; Kukull, W. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Lee, S.H.; Lutz, D.; Mossalam, M.; Bolshakov, V.Y.; Frotscher, M.; Shen, J. Presenilins regulate synaptic plasticity and mitochondrial calcium homeostasis in the hippocampal mossy fiber pathway. Mol. Neurodegener. 2017, 12, 48. [Google Scholar] [CrossRef]
- Soriano, S.; Kang, D.E.; Fu, M.; Pestell, R.; Chevallier, N.; Zheng, H.; Koo, E.H. Presenilin 1 negatively regulates β-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of β-amyloid precursor protein and notch processing. J. Cell Biol. 2001, 152, 785–794. [Google Scholar] [CrossRef]
- Zhang, Z.; Hartmann, H.; Minh Do, V.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; van de Wetering, M.; Clevers, H.; Saftig, P. Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395, 698–702. [Google Scholar] [CrossRef]
- Qu, Q.; Sun, G.; Murai, K.; Ye, P.; Li, W.; Asuelime, G.; Cheung, Y.-T.; Shi, Y. Wnt7a regulates multiple steps of neurogenesis. Mol. Cell. Biol. 2013, 33, 2551–2559. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- García-Velázquez, L.; Arias, C. The emerging role of Wnt signaling dysregulation in the understanding and modification of age-associated diseases. Ageing Res. Rev. 2017, 37, 135–145. [Google Scholar] [CrossRef]
- Hofmann, J.W.; McBryan, T.; Adams, P.D.; Sedivy, J.M. The effects of aging on the expression of Wnt pathway genes in mouse tissues. Age 2014, 36, 1033–1040. [Google Scholar] [CrossRef]
- Caruso, A.; Motolese, M.; Iacovelli, L.; Caraci, F.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; Gaviraghi, G.; Caricasole, A. Inhibition of the canonical Wnt signaling pathway by apolipoprotein E4 in PC12 cells. J. Neurochem. 2006, 98, 364–371. [Google Scholar] [CrossRef]
- Elliott, C.; Rojo, A.I.; Ribe, E.; Broadstock, M.; Xia, W.; Morin, P.; Semenov, M.; Baillie, G.; Cuadrado, A.; Al-Shawi, R. A role for APP in Wnt signalling links synapse loss with β-amyloid production. Transl. Psychiatry 2018, 8, 179. [Google Scholar] [CrossRef]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The secreted Wnt antagonist Dickkopf-1 is required for amyloid β-mediated synaptic loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Hussain, B.; Fang, C.; Chang, J. Blood–brain barrier breakdown: An emerging biomarker of cognitive impairment in normal aging and dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Xiao, M.; Xiao, Z.J.; Yang, B.; Lan, Z.; Fang, F. Blood-brain barrier: More contributor to disruption of central nervous system homeostasis than victim in neurological disorders. Front. Neurosci. 2020, 14, 764. [Google Scholar] [CrossRef]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A role for P-glycoprotein in clearance of Alzheimer amyloid β-peptide from the brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef]
- Zhou, Y.; Nathans, J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev. Cell 2014, 31, 248–256. [Google Scholar] [CrossRef]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M. Wnt/β-catenin signaling controls development of the blood–brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B. Activation of β-catenin signalling by GSK-3 inhibition increases p-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.-E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.-w. TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT signaling in activated microglia is proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt signaling in the prefrontal cortex of Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 873–891. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3β-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef]
- Sun, X.; Sato, S.; Murayama, O.; Murayama, M.; Park, J.-M.; Yamaguchi, H.; Takashima, A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 2002, 321, 61–64. [Google Scholar] [CrossRef]
- Li, B.; Ryder, J.; Su, Y.; Zhou, Y.; Liu, F.; Ni, B. FRAT1 peptide decreases Aβ production in swAPP751 cells. FEBS Lett. 2003, 553, 347–350. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Lee, K.; Becker, K.G.; Zhang, Y.; Maudsley, S.; Martin, B.; Cutler, R.G.; Mattson, M.P. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol. Aging 2010, 31, 1937–1949. [Google Scholar] [CrossRef]
- Bayod, S.; Menella, I.; Sanchez-Roige, S.; Lalanza, J.; Escorihuela, R.; Camins, A.; Pallàs, M.; Canudas, A. Wnt pathway regulation by long-term moderate exercise in rat hippocampus. Brain Res. 2014, 1543, 38–48. [Google Scholar] [CrossRef]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R.; Diaz Brinton, R.; Mosconi, L. Female sex and Alzheimer’s risk: The menopause connection. J. Prev. Alzheimer’s Dis. 2018, 5, 225–230. [Google Scholar] [CrossRef]
- Scott, E.L.; Zhang, Q.-g.; Han, D.; Desai, B.N.; Brann, D.W. Long-term estrogen deprivation leads to elevation of Dickkopf-1 and dysregulation of Wnt/β-Catenin signaling in hippocampal CA1 neurons. Steroids 2013, 78, 624–632. [Google Scholar] [CrossRef]
- Li, X.; Shan, J.; Chang, W.; Kim, I.; Bao, J.; Lee, H.-J.; Zhang, X.; Samuel, V.T.; Shulman, G.I.; Liu, D. Chemical and genetic evidence for the involvement of Wnt antagonist Dickkopf2 in regulation of glucose metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 11402–11407. [Google Scholar] [CrossRef]
- Thysiadis, S.; Katsamakas, S.; Mpousis, S.; Avramidis, N.; Efthimiopoulos, S.; Sarli, V. Design and synthesis of gallocyanine inhibitors of dkk1/lrp6 interactions for treatment of alzheimer’s disease. Bioorg. Chem. 2018, 80, 230–244. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Ahumada, J.; Arrázola, M.S.; Fuenzalida, M.; Inestrosa, N.C. WASP-1, a canonical Wnt signaling potentiator, rescues hippocampal synaptic impairments induced by Aβ oligomers. Exp. Neurol. 2015, 264, 14–25. [Google Scholar] [CrossRef]
- Sanei, M.; Saberi-Demneh, A. Effect of curcumin on memory impairment: A systematic review. Phytomedicine 2019, 52, 98–106. [Google Scholar] [CrossRef]
- Robin, N.C.; Agoston, Z.; Biechele, T.L.; James, R.G.; Berndt, J.D.; Moon, R.T. Simvastatin promotes adult hippocampal neurogenesis by enhancing Wnt/β-catenin signaling. Stem Cell Rep. 2014, 2, 9–17. [Google Scholar] [CrossRef]
- Salins, P.; Shawesh, S.; He, Y.; Dibrov, A.; Kashour, T.; Arthur, G.; Amara, F. Lovastatin protects human neurons against Abeta-induced toxicity and causes activation of beta-catenin-TCF/LEF signaling. Neurosci. Lett. 2007, 412, 211–216. [Google Scholar] [CrossRef]
- Miranda, C.J.; Braun, L.; Jiang, Y.; Hester, M.E.; Zhang, L.; Riolo, M.; Wang, H.; Rao, M.; Altura, R.A.; Kaspar, B.K. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 2012, 11, 542–552. [Google Scholar] [CrossRef]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef]
- Rai, S.N.; Tiwari, N.; Singh, P.; Singh, A.K.; Mishra, D.; Imran, M.; Singh, S.; Hooshmandi, E.; Vamanu, E.; Singh, S.K. Exploring the paradox of COVID-19 in neurological complications with emphasis on Parkinson’s and Alzheimer’s disease. Oxidative Med. Cell. Longev. 2022, 2022, 3012778. [Google Scholar] [CrossRef]
- Rai, S.N.; Tiwari, N.; Singh, P.; Mishra, D.; Singh, A.K.; Hooshmandi, E.; Vamanu, E.; Singh, M.P. Therapeutic potential of vital transcription factors in Alzheimer’s and Parkinson’s disease with particular emphasis on transcription factor EB mediated autophagy. Front. Neurosci. 2021, 15, 777347. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis; Exon Publications: Brisbane, QLD, Australia, 2018; pp. 3–26. [Google Scholar]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective effect of chlorogenic acid on mitochondrial dysfunction-mediated apoptotic death of DA neurons in a Parkinsonian mouse model. Oxidative Med. Cell. Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Front. Pharmacol. 2018, 9, 757. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Prakash, J.; Chouhan, S.; Yadav, S.K.; Westfall, S.; Rai, S.N.; Singh, S.P. Withania somnifera alleviates parkinsonian phenotypes by inhibiting apoptotic pathways in dopaminergic neurons. Neurochem. Res. 2014, 39, 2527–2536. [Google Scholar] [CrossRef]
- Rai, S.N.; Birla, H.; Singh, S.S.; Zahra, W.; Patil, R.R.; Jadhav, J.P.; Gedda, M.R.; Singh, S.P. Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Front. Aging Neurosci. 2017, 9, 421. [Google Scholar] [CrossRef]
- Yadav, S.K.; Rai, S.N.; Singh, S.P. Mucuna pruriens reduces inducible nitric oxide synthase expression in Parkinsonian mice model. J. Chem. Neuroanat. 2017, 80, 1–10. [Google Scholar] [CrossRef]
- Rai, S.N.; Yadav, S.K.; Singh, D.; Singh, S.P. Ursolic acid attenuates oxidative stress in nigrostriatal tissue and improves neurobehavioral activity in MPTP-induced Parkinsonian mouse model. J. Chem. Neuroanat. 2016, 71, 41–49. [Google Scholar] [CrossRef]
- Rai, S.N.; Chaturvedi, V.K.; Singh, P.; Singh, B.K.; Singh, M. Mucuna pruriens in Parkinson’s and in some other diseases: Recent advancement and future prospective. 3 Biotech 2020, 10, 522. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, P. Advancement in the modelling and therapeutics of Parkinson’s disease. J. Chem. Neuroanat. 2020, 104, 101752. [Google Scholar] [CrossRef]
- Zahra, W.; Rai, S.N.; Birla, H.; Singh, S.S.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Keswani, C.; Singh, R.K.; Singh, S.P. Neuroprotection of rotenone-induced parkinsonism by ursolic acid in PD mouse model. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2020, 19, 527–540. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef]
- Schulze, M.; Sommer, A.; Plötz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef]
- Lill, C.M. Genetics of Parkinson’s disease. Mol. Cell. Probes 2016, 30, 386–396. [Google Scholar] [CrossRef]
- Rai, S.N.; Zahra, W.; Singh, S.S.; Birla, H.; Keswani, C.; Dilnashin, H.; Rathore, A.S.; Singh, R.; Singh, R.K.; Singh, S.P. Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotox. Res. 2019, 36, 452–462. [Google Scholar] [CrossRef]
- Schapira, A.H.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; Von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef]
- Apple, D.M.; Solano-Fonseca, R.; Kokovay, E. Neurogenesis in the aging brain. Biochem. Pharmacol. 2017, 141, 77–85. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Agnihotri, S.K.; Sun, L.; Yee, B.K.; Shen, R.; Akundi, R.S.; Zhi, L.; Duncan, M.J.; Cass, W.A.; Büeler, H. PINK1 deficiency is associated with increased deficits of adult hippocampal neurogenesis and lowers the threshold for stress-induced depression in mice. Behav. Brain Res. 2019, 363, 161–172. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Serapide, M.F.; Pluchino, S.; Marchetti, B. Wnt/β-catenin signaling is required to rescue midbrain dopaminergic progenitors and promote neurorepair in ageing mouse model of Parkinson’s disease. Stem Cells 2014, 32, 2147–2163. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Serapide, M.F.; Caniglia, S.; Testa, N.; Leggio, L.; Vivarelli, S.; Iraci, N.; Pluchino, S.; Marchetti, B. Microglia polarization, gene-environment interactions and Wnt/β-catenin signaling: Emerging roles of glia-neuron and glia-stem/neuroprogenitor crosstalk for dopaminergic neurorestoration in aged parkinsonian brain. Front. Aging Neurosci. 2018, 10, 12. [Google Scholar] [CrossRef]
- Singh, S.; Mishra, A.; Bharti, S.; Tiwari, V.; Singh, J.; Shukla, S. Glycogen synthase kinase-3β regulates equilibrium between neurogenesis and gliogenesis in rat model of Parkinson’s disease: A crosstalk with Wnt and notch signaling. Mol. Neurobiol. 2018, 55, 6500–6517. [Google Scholar] [CrossRef]
- Walter, J.; Bolognin, S.; Antony, P.M.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X. Neural stem cells of Parkinson’s disease patients exhibit aberrant mitochondrial morphology and functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef]
- Hirota, Y.; Sawada, M.; Huang, S.-h.; Ogino, T.; Ohata, S.; Kubo, A.; Sawamoto, K. Roles of Wnt Signaling in the Neurogenic Niche of the Adult Mouse Ventricular–Subventricular Zone. Neurochem. Res. 2016, 41, 222–230. [Google Scholar] [CrossRef]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural basis of Wnt recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef]
- Esfandiari, F.; Fathi, A.; Gourabi, H.; Kiani, S.; Nemati, S.; Baharvand, H. Glycogen synthase kinase-3 inhibition promotes proliferation and neuronal differentiation of human-induced pluripotent stem cell-derived neural progenitors. Stem Cells Dev. 2012, 21, 3233–3243. [Google Scholar] [CrossRef]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered differentiation potential of Gaucher’s disease iPSC neuronal progenitors due to Wnt/β-catenin downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef]
- Azim, K.; Rivera, A.; Raineteau, O.; Butt, A.M. GSK3β regulates oligodendrogenesis in the dorsal microdomain of the subventricular zone via Wnt-β-catenin signaling. Glia 2014, 62, 778–779. [Google Scholar] [CrossRef]
- Castelo-Branco, G.; Rawal, N.; Arenas, E. GSK-3β inhibition/β-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 2004, 117, 5731–5737. [Google Scholar] [CrossRef]
- Arciniegas Ruiz, S.M.; Eldar-Finkelman, H. Glycogen synthase kinase-3 inhibitors: Preclinical and clinical focus on CNS-A decade onward. Front. Mol. Neurosci. 2022, 14, 349. [Google Scholar] [CrossRef]
- Song, J.L.; Nigam, P.; Tektas, S.S.; Selva, E. microRNA regulation of Wnt signaling pathways in development and disease. Cell Signal. 2015, 27, 1380–1391. [Google Scholar] [CrossRef]
- Anderegg, A.; Lin, H.-P.; Chen, J.-A.; Caronia-Brown, G.; Cherepanova, N.; Yun, B.; Joksimovic, M.; Rock, J.; Harfe, B.D.; Johnson, R. An Lmx1b-miR135a2 regulatory circuit modulates Wnt1/Wnt signaling and determines the size of the midbrain dopaminergic progenitor pool. PLoS Genet. 2013, 9, e1003973. [Google Scholar] [CrossRef]
- Anderegg, A.; Awatramani, R. Making a mes: A transcription factor-microRNA pair governs the size of the midbrain and the dopaminergic progenitor pool. Neurogenesis 2015, 2, e998101. [Google Scholar] [CrossRef]
- Chmielarz, P.; Konovalova, J.; Najam, S.S.; Alter, H.; Piepponen, T.P.; Erfle, H.; Sonntag, K.C.; Schütz, G.; Vinnikov, I.A.; Domanskyi, A. Dicer and microRNAs protect adult dopamine neurons. Cell Death Dis. 2017, 8, e2813. [Google Scholar] [CrossRef]
- Zhang, W.M.; Zhang, Z.R.; Yang, X.T.; Zhang, Y.G.; Gao, Y.S. Overexpression of miR-21 promotes neural stem cell proliferation and neural differentiation via the Wnt/β-catenin signaling pathway in vitro. Mol. Med. Rep. 2018, 17, 330–335. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke; National Institutes of Health. Parkinson’s Disease: Challenges, Progress, and Promise; National Institute of Neurological Disorders and Stroke, National Institutes of Health: Bethesda, MD, USA, 2004. [Google Scholar]
- Ntetsika, T.; Papathoma, P.-E.; Markaki, I. Novel targeted therapies for Parkinson’s disease. Mol. Med. 2021, 27, 17. [Google Scholar] [CrossRef]
- Mishra, A.; Chandravanshi, L.P.; Trigun, S.K.; Krishnamurthy, S. Ambroxol modulates 6-Hydroxydopamine-induced temporal reduction in Glucocerebrosidase (GCase) enzymatic activity and Parkinson’s disease symptoms. Biochem. Pharmacol. 2018, 155, 479–493. [Google Scholar] [CrossRef]
- Giovannini, D.; Andreola, F.; Spitalieri, P.; Krasnowska, E.K.; Colini Baldeschi, A.; Rossi, S.; Sangiuolo, F.; Cozzolino, M.; Serafino, A. Natriuretic peptides are neuroprotective on in vitro models of PD and promote dopaminergic differentiation of hiPSCs-derived neurons via the Wnt/β-catenin signaling. Cell Death Discov. 2021, 7, 330. [Google Scholar] [CrossRef]
- Serafino, A.; Giovannini, D.; Rossi, S.; Cozzolino, M. Targeting the Wnt/β-catenin pathway in neurodegenerative diseases: Recent approaches and current challenges. Expert Opin. Drug Discov. 2020, 15, 803–822. [Google Scholar] [CrossRef]
- Willis, C.M.; Nicaise, A.M.; Peruzzotti-Jametti, L.; Pluchino, S. The neural stem cell secretome and its role in brain repair. Brain Res. 2020, 1729, 146615. [Google Scholar] [CrossRef]
- Tepekoy, F.; Akkoyunlu, G.; Demir, R. The role of Wnt signaling members in the uterus and embryo during pre-implantation and implantation. J. Assist. Reprod. Genet. 2015, 32, 337–346. [Google Scholar] [CrossRef]
- Kele, J.; Andersson, E.R.; Villaescusa, J.C.; Cajanek, L.; Parish, C.L.; Bonilla, S.; Toledo, E.M.; Bryja, V.; Rubin, J.S.; Shimono, A.; et al. SFRP1 and SFRP2 dose-dependently regulate midbrain dopamine neuron development in vivo and in embryonic stem cells. Stem Cells 2012, 30, 865–875. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, B.K.; Rathore, A.S.; Zahra, W.; Keswani, C.; Birla, H.; Singh, S.S.; Dilnashin, H.; Singh, S.P. Quality control in Huntington’s disease: A therapeutic target. Neurotox. Res. 2019, 36, 612–626. [Google Scholar] [CrossRef]
- Bates, G.; Tabrizi, S.; Jones, L. Huntington’s Disease; Oxford Monographs on Medical G; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Mason, S.L.; Barker, R.A. Novel targets for Huntington’s disease: Future prospects. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 25–36. [Google Scholar]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef]
- Bäckman, L.; Robins-Wahlin, T.; Lundin, A.; Ginovart, N.; Farde, L. Cognitive deficits in Huntington’s disease are predicted by dopaminergic PET markers and brain volumes. Brain J. Neurol. 1997, 120, 2207–2217. [Google Scholar] [CrossRef]
- Starkstein, S.; Brandt, J.; Bylsma, F.; Peyser, C.; Folstein, M.; Folstein, S. Neuropsychological correlates of brain atrophy in Huntington’s disease: A magnetic resonance imaging study. Neuroradiology 1992, 34, 487–489. [Google Scholar] [CrossRef]
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington disease. Cogn. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Priller, J.; Ecker, D.; Landwehrmeyer, B.; Craufurd, D. A Europe-wide assessment of current medication choices in Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 1788. [Google Scholar] [CrossRef]
- Frank, S. Tetrabenazine: The first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatr. Dis. Treat. 2010, 6, 657–665. [Google Scholar] [CrossRef]
- Dean, M.; Sung, V.W. Review of deutetrabenazine: A novel treatment for chorea associated with Huntington’s disease. Drug Des. Dev. Ther. 2018, 12, 313–319. [Google Scholar] [CrossRef]
- Mason, S.L.; Barker, R.A. Advancing pharmacotherapy for treating Huntington’s disease: A review of the existing literature. Expert Opin. Pharmacother. 2016, 17, 41–52. [Google Scholar] [CrossRef]
- Bonomo, R.; Elia, A.E.; Bonomo, G.; Romito, L.M.; Mariotti, C.; Devigili, G.; Cilia, R.; Giossi, R.; Eleopra, R. Deep brain stimulation in Huntington’s disease: A literature review. Neurol. Sci. 2021, 42, 4447–4457. [Google Scholar] [CrossRef]
- Ghatak, S.; Raha, S. Micro RNA-214 contributes to proteasome independent downregulation of beta catenin in Huntington’s disease knock-in striatal cell model STHdhQ111/Q111. Biochem. Biophys. Res. Commun. 2015, 459, 509–514. [Google Scholar] [CrossRef]
- Liu, T.; Im, W.; Mook-Jung, I.; Kim, M. MicroRNA-124 slows down the progression of Huntington’s disease by promoting neurogenesis in the striatum. Neural Regen. Res. 2015, 10, 786–791. [Google Scholar]
- Godin, J.D.; Poizat, G.; Hickey, M.A.; Maschat, F.; Humbert, S. Mutant huntingtin-impaired degradation of β-catenin causes neurotoxicity in Huntington’s disease. EMBO J. 2010, 29, 2433–2445. [Google Scholar] [CrossRef]
- Smith-Geater, C.; Hernandez, S.J.; Lim, R.G.; Adam, M.; Wu, J.; Stocksdale, J.T.; Wassie, B.T.; Gold, M.P.; Wang, K.Q.; Miramontes, R. Aberrant development corrected in adult-onset huntington’s disease iPSC-derived neuronal cultures via WNT signaling modulation. Stem Cell Rep. 2020, 14, 406–419. [Google Scholar] [CrossRef]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef]
- Jiang, X.; Guan, Y.; Zhao, Z.; Meng, F.; Wang, X.; Gao, X.; Liu, J.; Chen, Y.; Zhou, F.; Zhou, S. Potential roles of the WNT signaling pathway in amyotrophic lateral sclerosis. Cells 2021, 10, 839. [Google Scholar] [CrossRef]
- Mehta, P.; Kaye, W.; Raymond, J.; Punjani, R.; Larson, T.; Cohen, J.; Muravov, O.; Horton, K. Prevalence of amyotrophic lateral sclerosis—United States, 2015. Morb. Mortal. Wkly. Rep. 2018, 67, 1285. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.-X. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Sun, X.-D.; Li, L.; Liu, F.; Huang, Z.-H.; Bean, J.C.; Jiao, H.-F.; Barik, A.; Kim, S.-M.; Wu, H.; Shen, C. Lrp4 in astrocytes modulates glutamatergic transmission. Nat. Neurosci. 2016, 19, 1010–1018. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and therapeutic potential of astrocytes in amyotrophic lateral sclerosis. Curr. Pharm. Des. 2017, 23, 5010–5021. [Google Scholar] [CrossRef]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef]
- Chipika, R.H.; Finegan, E.; Li Hi Shing, S.; Hardiman, O.; Bede, P. Tracking a fast-moving disease: Longitudinal markers, monitoring, and clinical trial endpoints in ALS. Front. Neurol. 2019, 10, 229. [Google Scholar] [CrossRef]
- Bharti, K.; Graham, S.J.; Benatar, M.; Briemberg, H.; Chenji, S.; Dupré, N.; Dionne, A.; Frayne, R.; Genge, A.; Korngut, L. Functional alterations in large-scale resting-state networks of amyotrophic lateral sclerosis: A multi-site study across Canada and the United States. PLoS ONE 2022, 17, e0269154. [Google Scholar] [CrossRef]
- Tan, H.H.; Westeneng, H.J.; Nitert, A.D.; van Veenhuijzen, K.; Meier, J.M.; van der Burgh, H.K.; van Zandvoort, M.J.; van Es, M.A.; Veldink, J.H.; van den Berg, L.H. MRI clustering reveals three ALS subtypes with unique neurodegeneration patterns. Ann. Neurol. 2022, 92, 1030–1045. [Google Scholar] [CrossRef]
- Alves, C.J.; De Santana, L.P.; Dos Santos, A.J.D.; De Oliveira, G.P.; Duobles, T.; Scorisa, J.M.; Martins, R.S.; Maximino, J.R.; Chadi, G. Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 2011, 1394, 90–104. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Oskarsson, B.; Moore, D.; Mozaffar, T.; Ravits, J.; Wiedau-Pazos, M.; Parziale, N.; Joyce, N.C.; Mandeville, R.; Goyal, N.; Cudkowicz, M.E. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: A randomized, double-blind crossover trial. Muscle Nerve 2018, 58, 42–48. [Google Scholar] [CrossRef]
- Bedlack, R.S.; Pastula, D.M.; Hawes, J.; Heydt, D. Open-label pilot trial of levetiracetam for cramps and spasticity in patients with motor neuron disease. Amyotroph. Lateral Scler. 2009, 10, 210–215. [Google Scholar] [CrossRef]
- Miller, R.; Jackson, C.; Kasarskis, E.; England, J.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.; Mitsumoto, H.; Rosenfeld, J. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1227–1233. [Google Scholar]
- Stone, C.A.; O’Leary, N. Systematic review of the effectiveness of botulinum toxin or radiotherapy for sialorrhea in patients with amyotrophic lateral sclerosis. J. Pain Symptom Manag. 2009, 37, 246–258. [Google Scholar] [CrossRef]
- Brooks, B.; Thisted, R.; Appel, S.H.; Bradley, W.; Olney, R.; Berg, J.; Pope, L.; Smith, R. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: A randomized trial. Neurology 2004, 63, 1364–1370. [Google Scholar] [CrossRef]
- Miller, R.G.; Jackson, C.E.; Kasarskis, E.J.; England, J.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.; Mitsumoto, H.; Rosenfeld, J. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, and respiratory therapies (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1218–1226. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015, 359, 215–223. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, C.; Mancuso, R.; Del Valle, J.; Navarro, X.; Rodríguez, F.J. Wnt signaling alteration in the spinal cord of amyotrophic lateral sclerosis transgenic mice: Special focus on frizzled-5 cellular expression pattern. PLoS ONE 2016, 11, e0155867. [Google Scholar] [CrossRef]
- Yu, L.; Guan, Y.; Wu, X.; Chen, Y.; Liu, Z.; Du, H.; Wang, X. Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem. Res. 2013, 38, 1904–1913. [Google Scholar] [CrossRef]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691. [Google Scholar]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early changes of neuromuscular transmission in the SOD1 (G93A) mice model of ALS start long before motor symptoms onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1 G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16. [Google Scholar] [CrossRef]
- Chen, Y.; Guan, Y.; Liu, H.; Wu, X.; Yu, L.; Wang, S.; Zhao, C.; Du, H.; Wang, X. Activation of the Wnt/β-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem. Biophys. Res. Commun. 2012, 420, 397–403. [Google Scholar] [CrossRef]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Yang, W.; Leystra-Lantz, C.; Strong, M.J. Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 2008, 1196, 131–139. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef]
- Martínez-González, L.; Gonzalo-Consuegra, C.; Gómez-Almería, M.; Porras, G.; de Lago, E.; Martín-Requero, Á.; Martínez, A. Tideglusib, a Non-ATP Competitive Inhibitor of GSK-3β as a Drug Candidate for the Treatment of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 8975. [Google Scholar] [CrossRef]
- Pinto, C.; Medinas, D.B.; Fuentes-Villalobos, F.; Maripillán, J.; Castro, A.F.; Martínez, A.D.; Osses, N.; Hetz, C.; Henríquez, J.P. β-catenin aggregation in models of ALS motor neurons: GSK3β inhibition effect and neuronal differentiation. Neurobiol. Dis. 2019, 130, 104497. [Google Scholar] [CrossRef]
- González-Fernández, C.; Gonzalez, P.; Andres-Benito, P.; Ferrer, I.; Rodríguez, F.J. Wnt signaling alterations in the human spinal cord of amyotrophic lateral sclerosis cases: Spotlight on Fz2 and Wnt5a. Mol. Neurobiol. 2019, 56, 6777–6791. [Google Scholar] [CrossRef]
- Pereira, C.; Schaer, D.J.; Bachli, E.B.; Kurrer, M.O.; Schoedon, G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 504–510. [Google Scholar] [CrossRef]
- Halleskog, C.; Dijksterhuis, J.P.; Kilander, M.B.C.; Becerril-Ortega, J.; Villaescusa, J.C.; Lindgren, E.; Arenas, E.; Schulte, G. Heterotrimeric G protein-dependent WNT-5A signaling to ERK1/2 mediates distinct aspects of microglia proinflammatory transformation. J. Neuroinflamm. 2012, 9, 111. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef]
- Nonneman, A.; Robberecht, W.; Den Bosch, L.V. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2014, 4, 223–239. [Google Scholar] [CrossRef]
- Ortega, F.; Gascón, S.; Masserdotti, G.; Deshpande, A.; Simon, C.; Fischer, J.; Dimou, L.; Chichung Lie, D.; Schroeder, T.; Berninger, B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 2013, 15, 602–613. [Google Scholar] [CrossRef]
- Azim, K.; Butt, A.M. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef]
- Rodríguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular junction dysfunction in amyotrophic lateral sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef]
- Hughes, B.W.; Kusner, L.L.; Kaminski, H.J. Molecular architecture of the neuromuscular junction. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2006, 33, 445–461. [Google Scholar] [CrossRef]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef]
- Yumoto, N.; Kim, N.; Burden, S.J. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 2012, 489, 438–442. [Google Scholar] [CrossRef]
- Ruegg, M.A.; Bixby, J.L. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998, 21, 22–27. [Google Scholar] [CrossRef]
- Henriquez, J.P.; Webb, A.; Bence, M.; Bildsoe, H.; Sahores, M.; Hughes, S.M.; Salinas, P.C. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. USA 2008, 105, 18812–18817. [Google Scholar] [CrossRef]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nürnberg, G.; Cenani, A. LRP4 mutations alter Wnt/β-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef]
- Li, X.-M.; Dong, X.-P.; Luo, S.-W.; Zhang, B.; Lee, D.-H.; Ting, A.K.; Neiswender, H.; Kim, C.-H.; Carpenter-Hyland, E.; Gao, T.-M. Retrograde regulation of motoneuron differentiation by muscle β-catenin. Nat. Neurosci. 2008, 11, 262–268. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Kwan, T.; Kazamel, M.; Thoenes, K.; Si, Y.; Jiang, N.; King, P.H. Wnt antagonist FRZB is a muscle biomarker of denervation atrophy in amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 16679. [Google Scholar] [CrossRef]
- Pérez-García, M.J.; Burden, S.J. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep. 2012, 2, 497–502. [Google Scholar] [CrossRef]
- Sengupta-Ghosh, A.; Dominguez, S.L.; Xie, L.; Barck, K.H.; Jiang, Z.; Earr, T.; Imperio, J.; Phu, L.; Budayeva, H.G.; Kirkpatrick, D.S. Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis. 2019, 124, 340–352. [Google Scholar] [CrossRef]
- Cantor, S.; Zhang, W.; Delestree, N.; Remedio, L.; Mentis, G.Z.; Burden, S.J. Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. Elife 2018, 7, e34375. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Zouvelou, V.; Evangelakou, P.; Tsonis, A.; Thomaidis, T.; Lauria, G.; Andreetta, F. LRP 4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2014, 1, 80–87. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis–a review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Balasa, R.; Barcutean, L.; Balasa, A.; Motataianu, A.; Roman-Filip, C.; Manu, D. The action of TH17 cells on blood brain barrier in multiple sclerosis and experimental autoimmune encephalomyelitis. Hum. Immunol. 2020, 81, 237–243. [Google Scholar] [CrossRef]
- Ma, J.; Wang, R.; Fang, X.; Ding, Y.; Sun, Z. Critical role of TCF-1 in repression of the IL-17 gene. PLoS ONE 2011, 6, e24768. [Google Scholar] [CrossRef]
- Yu, Q.; Sharma, A.; Ghosh, A.; Sen, J.M. T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J. Immunol. 2011, 186, 3946–3952. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Manoharan, I.; Hong, Y.; Swafford, D.; Majumdar, T.; Taketo, M.M.; Manicassamy, B.; Koni, P.A.; Thangaraju, M.; Sun, Z. Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J. Immunol. 2015, 194, 3295–3304. [Google Scholar] [CrossRef]
- De Sarno, P.; Axtell, R.C.; Raman, C.; Roth, K.A.; Alessi, D.R.; Jope, R.S. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 338–345. [Google Scholar] [CrossRef]
- Yuan, S.; Shi, Y.; Tang, S.-J. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. J. Neuroimmune Pharmacol. 2012, 7, 904–913. [Google Scholar] [CrossRef]
- Fancy, S.P.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef]
- Xie, C.; Li, Z.; Zhang, G.-X.; Guan, Y. Wnt signaling in remyelination in multiple sclerosis: Friend or foe? Mol. Neurobiol. 2014, 49, 1117–1125. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.-Y. Endothelial β-catenin signaling is required for maintaining adult blood–brain barrier integrity and central nervous system homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef]
- Lengfeld, J.E.; Lutz, S.E.; Smith, J.R.; Diaconu, C.; Scott, C.; Kofman, S.B.; Choi, C.; Walsh, C.M.; Raine, C.S.; Agalliu, I. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1168–E1177. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 2120–2133. [Google Scholar] [CrossRef]
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Bowerman, M.; Becker, C.G.; Yáñez-Muñoz, R.J.; Ning, K.; Wood, M.J.; Gillingwater, T.H.; Talbot, K.; Consortium, U.S.R. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 2017, 10, 943–954. [Google Scholar] [CrossRef]
- Fallini, C.; Donlin-Asp, P.G.; Rouanet, J.P.; Bassell, G.J.; Rossoll, W. Deficiency of the Survival of Motor Neuron Protein Impairs mRNA Localization and Local Translation in the Growth Cone of Motor Neurons. J. Neurosci. 2016, 36, 3811–3820. [Google Scholar] [CrossRef]
- Schroth, M.K. Special considerations in the respiratory management of spinal muscular atrophy. Pediatrics 2009, 123, S245–S249. [Google Scholar] [CrossRef]
- Durkin, E.T.; Schroth, M.K.; Helin, M.; Shaaban, A.F. Early laparoscopic fundoplication and gastrostomy in infants with spinal muscular atrophy type I. J. Pediatr. Surg. 2008, 43, 2031–2037. [Google Scholar] [CrossRef]
- Haaker, G.; Fujak, A. Proximal spinal muscular atrophy: Current orthopedic perspective. Appl. Clin. Genet. 2013, 6, 113–120. [Google Scholar]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M. Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). ACS Publications. J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef]
- Suzukia, Y.; Sano, N.; Shinonaga, C.; Fukuda, M.; Hyodo, M.; Morimoto, T. Successful botulinum toxin treatment of dysphagia in a spinal muscular atrophy type 2 patient. Brain Dev. 2007, 29, 662–665. [Google Scholar] [CrossRef]
- Rudnik-Schöneborn, S.; Botzenhart, E.; Eggermann, T.; Senderek, J.; Schoser, B.G.; Schröder, R.; Wehnert, M.; Wirth, B.; Zerres, K. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics 2007, 8, 137–142. [Google Scholar] [CrossRef]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef]
- Šoltić, D.; Shorrock, H.K.; Allardyce, H.; Wilson, E.L.; Holt, I.; Synowsky, S.A.; Shirran, S.L.; Parson, S.H.; Gillingwater, T.H.; Fuller, H.R. Lamin A/C dysregulation contributes to cardiac pathology in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet. 2019, 28, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Powis, R.A.; Karyka, E.; Boyd, P.; Côme, J.; Jones, R.A.; Zheng, Y.; Szunyogova, E.; Groen, E.J.; Hunter, G.; Thomson, D. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016, 1, e87908. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018, 46, 8326–8346. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.A.; Lee, D.D.; Bartlett, S. Aminoacyl tRNA synthetase complex interacting multifunctional protein 1 simultaneously binds Glutamyl-Prolyl-tRNA synthetase and scaffold protein aminoacyl tRNA synthetase complex interacting multifunctional protein 3 of the multi-tRNA synthetase complex. Int. J. Biochem. Cell Biol. 2018, 99, 197–202. [Google Scholar] [CrossRef]
- James, R.; Chaytow, H.; Ledahawsky, L.M.; Gillingwater, T.H. Revisiting the role of mitochondria in spinal muscular atrophy. Cell. Mol. Life Sci. 2021, 78, 4785–4804. [Google Scholar] [CrossRef]
- Wiese, K.E.; Nusse, R.; van Amerongen, R. Wnt signalling: Conquering complexity. Development 2018, 145, 165902. [Google Scholar] [CrossRef]
- Bernkopf, D.B.; Jalal, K.; Brückner, M.; Knaup, K.X.; Gentzel, M.; Schambony, A.; Behrens, J. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018, 217, 1383–1394. [Google Scholar] [CrossRef]
- Rauschenberger, V.; Bernkopf, D.B.; Krenn, S.; Jalal, K.; Heller, J.; Behrens, J.; Gentzel, M.; Schambony, A. The phosphatase Pgam5 antagonizes Wnt/β-Catenin signaling in embryonic anterior-posterior axis patterning. Development 2017, 144, 2234–2247. [Google Scholar] [CrossRef]
- Wu, H.; Xue, D.; Chen, G.; Han, Z.; Huang, L.; Zhu, C.; Wang, X.; Jin, H.; Wang, J.; Zhu, Y. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 2014, 10, 1712–1725. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramakrishna, K.; Nalla, L.V.; Naresh, D.; Venkateswarlu, K.; Viswanadh, M.K.; Nalluri, B.N.; Chakravarthy, G.; Duguluri, S.; Singh, P.; Rai, S.N.; et al. WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: Current Status and Future Perspective. Diseases 2023, 11, 89. https://doi.org/10.3390/diseases11030089
Ramakrishna K, Nalla LV, Naresh D, Venkateswarlu K, Viswanadh MK, Nalluri BN, Chakravarthy G, Duguluri S, Singh P, Rai SN, et al. WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: Current Status and Future Perspective. Diseases. 2023; 11(3):89. https://doi.org/10.3390/diseases11030089
Chicago/Turabian StyleRamakrishna, Kakarla, Lakshmi Vineela Nalla, Dumala Naresh, Kojja Venkateswarlu, Matte Kasi Viswanadh, Buchi N. Nalluri, Guntupalli Chakravarthy, Sajusha Duguluri, Payal Singh, Sachchida Nand Rai, and et al. 2023. "WNT-β Catenin Signaling as a Potential Therapeutic Target for Neurodegenerative Diseases: Current Status and Future Perspective" Diseases 11, no. 3: 89. https://doi.org/10.3390/diseases11030089