Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Ethics Statement
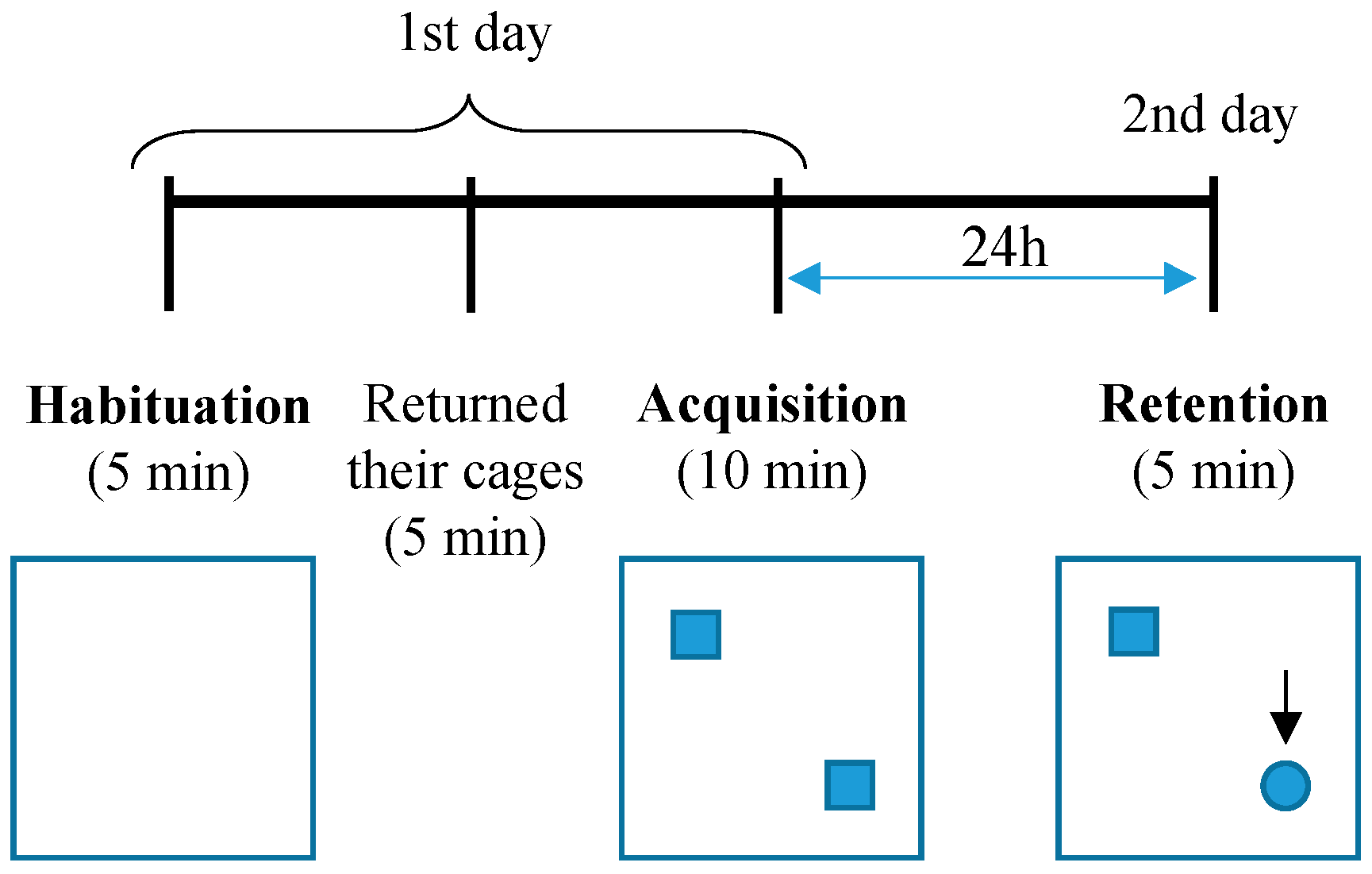
2.2. Novel Object Recognition Test (NOR)
2.3. Tissue Processing and Western Blot Analysis
2.3.1. Brain Protein Extract
2.3.2. Western Blot Analysis
2.4. Immunohistochemical Analysis
2.5. Plaque and Microglial Cell Activation and Neuron Quantification in the Hippocampus
2.6. Statistical Analyses
3. Results
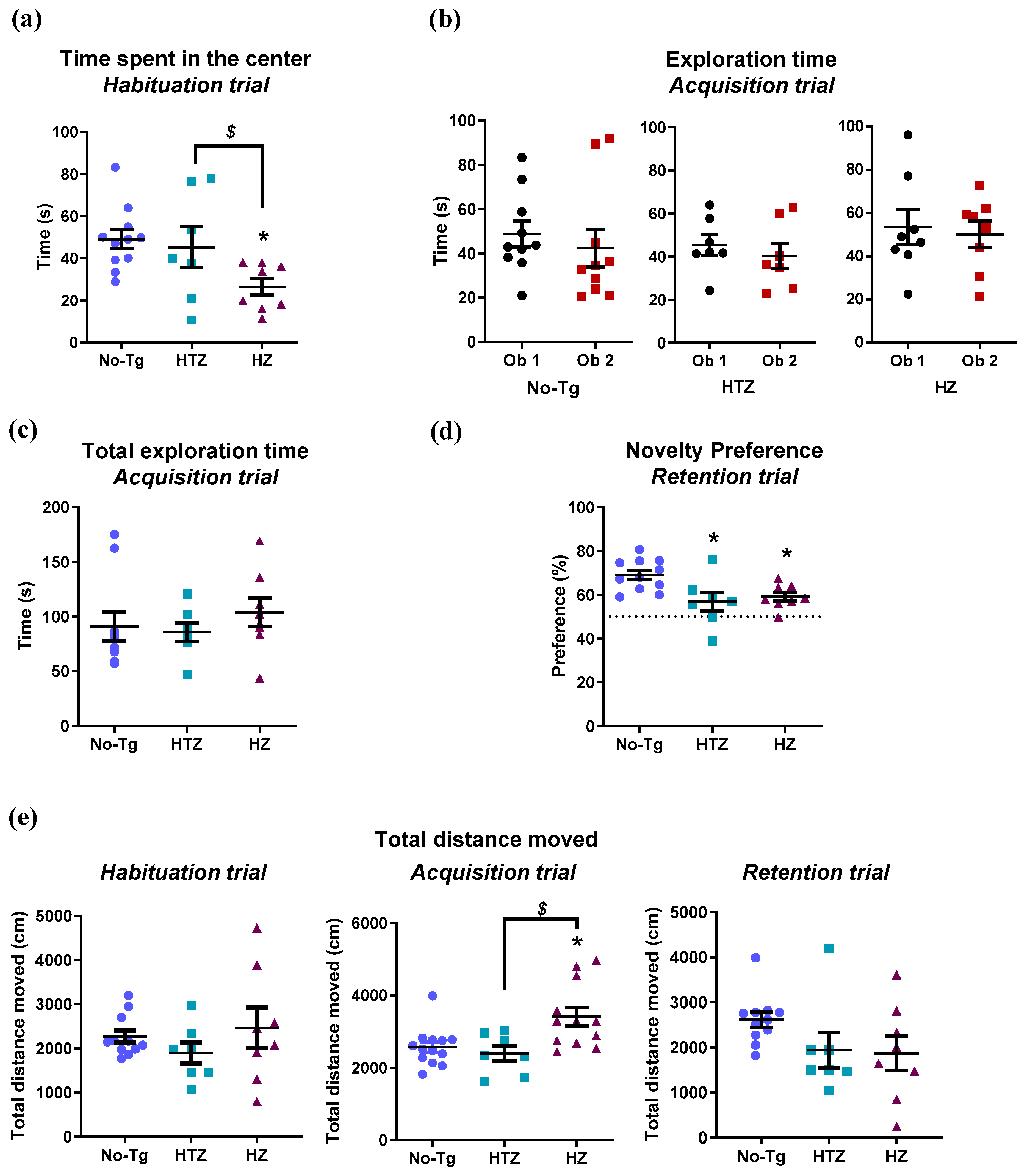
3.1. Both Heterozygous and Homozygous 5xFAD Transgenic Mice Have Impairment in Novel Object Recognition
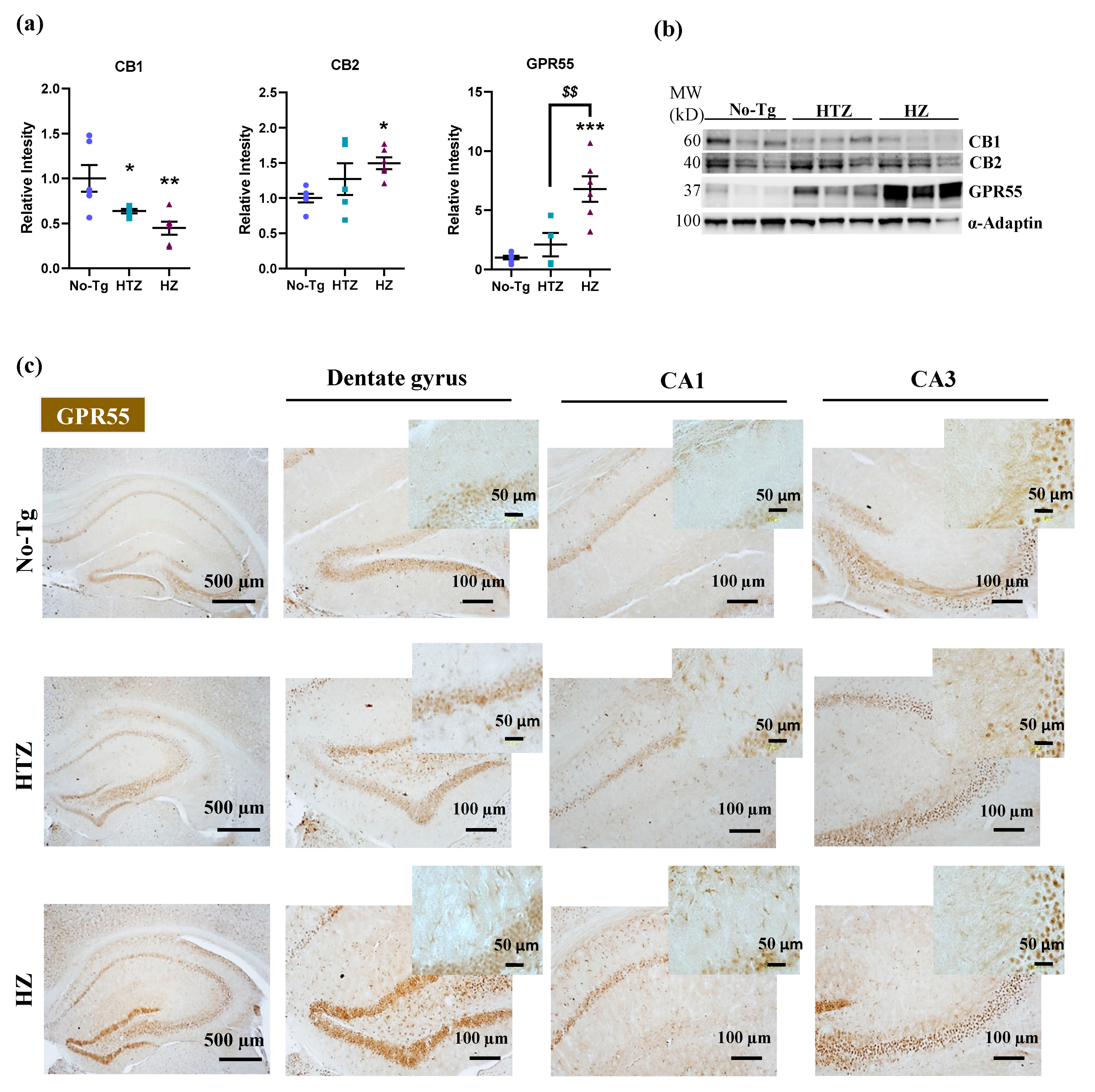
3.2. Hippocampal Alteration of the Expression of CB1, CB2, and GPR55 Receptors in Homozygous 5xFAD Transgenic Mice
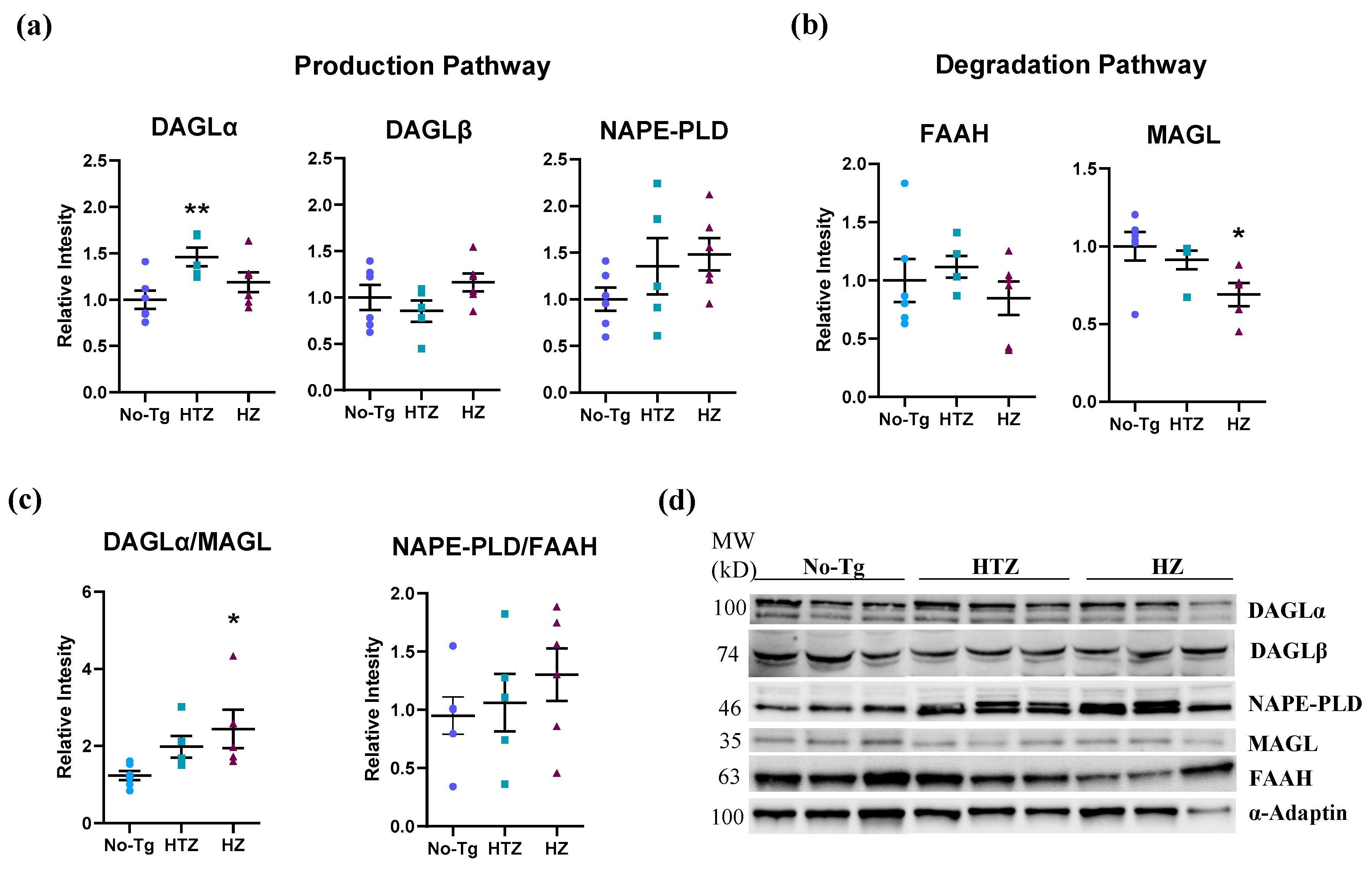
3.3. Alteration in the Endocannabinoid Production and Degradation Pathways in Homozygous 5xFAD Transgenic Mice
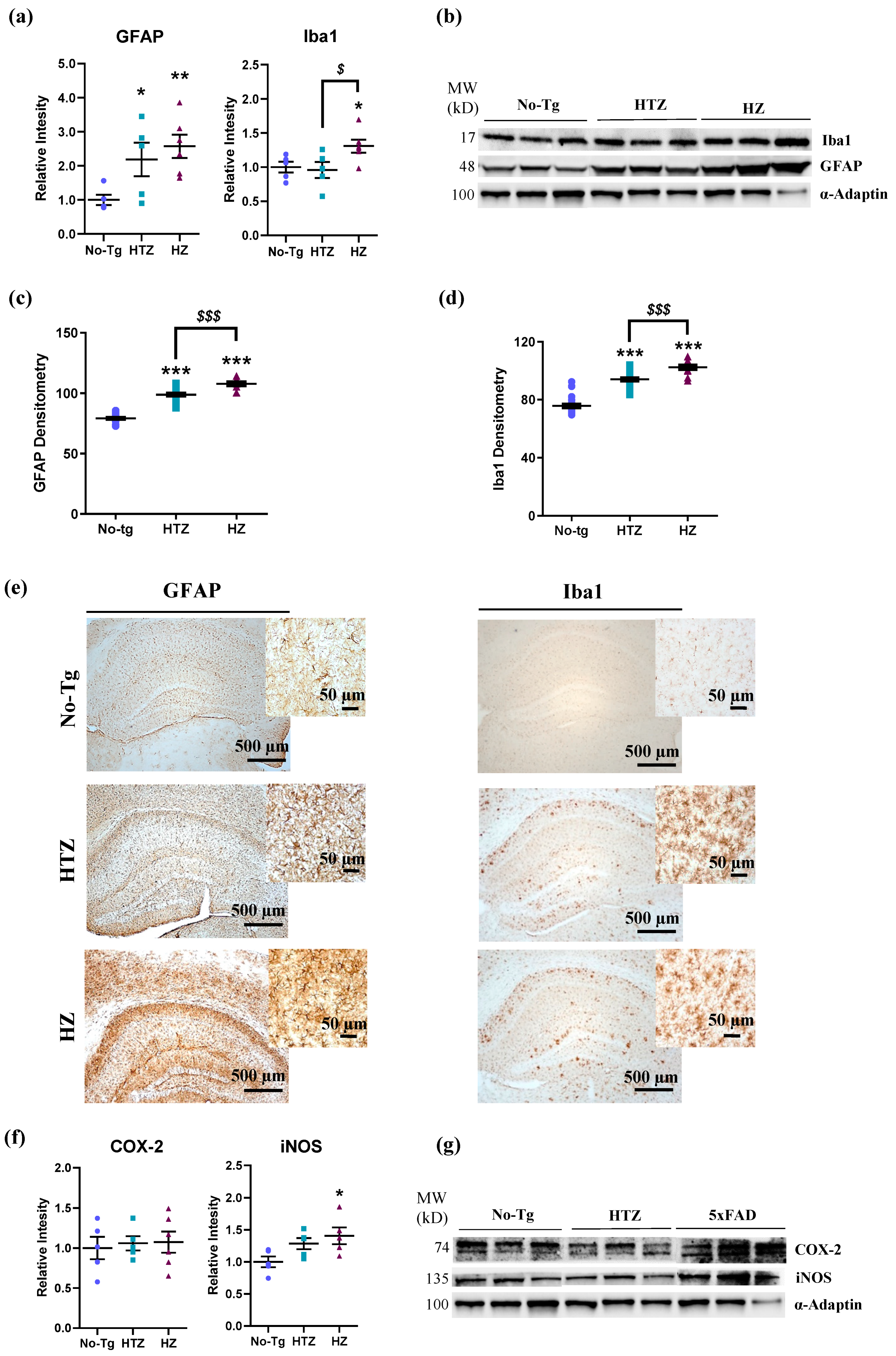
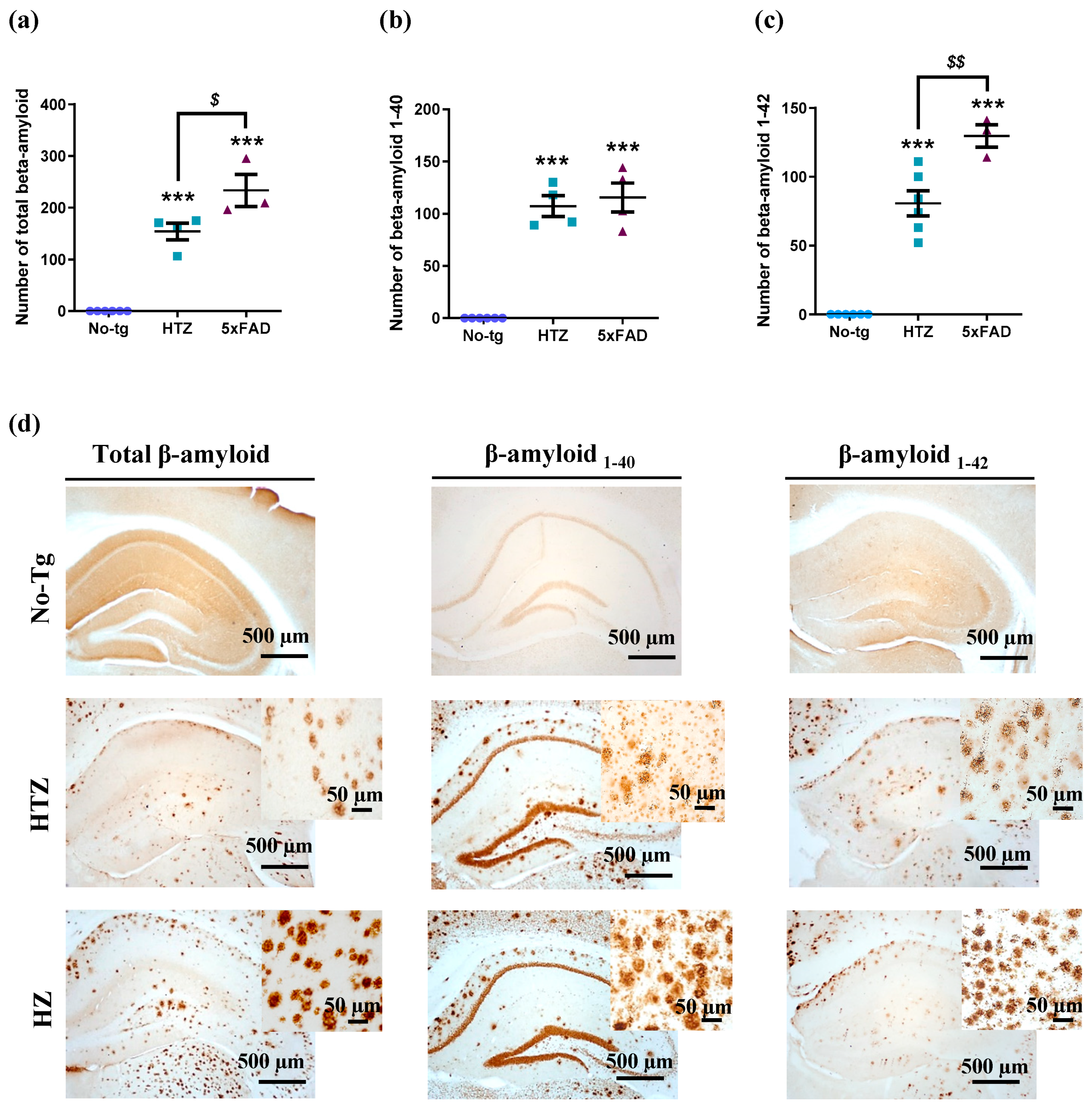
3.4. Neuroinflammatory Response in Both Heterozygous and Homozygous 5xFAD Transgenic Mice—Stronger in the Homozygous Group
3.5. Neuroinflammatory Response in Both Heterozygous and Homozygous 5xFAD Transgenic Mice—Stronger in the Homozygous Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | β-Amyloid |
| Iba1 | Ionized calcium-binding adaptor molecule 1 |
| MAGL | Monoacylglycerol lipase |
| GPR55 | G-protein-coupled receptor 55 |
| CB2 | Cannabinoid receptor type 2 |
| NAPE-PLD | N-Acyl phosphatidylethanolamine-specific phospholipase D |
| GFAP | Glial fibrillary acidic Protein |
| CB1 | Cannabinoid receptor type 1 |
| FAAH | Fatty acid amide hydrolase |
| DAGLβ | Diacylglycerol lipase β |
| COX-2 | Cyclooxygenase 2 |
| DAGLα | Diacylglycerol lipase α |
| iNOS | Inducible nitric oxide synthase |
| LPI | Lysophosphatidylinositol |
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Romano, A.; Lavecchia, A.M.; Cassano, T.; Gaetani, S. The role of endocannabinoid signaling in the molecular mechanisms of neurodegeneration in Alzheimer’s disease. J. Alzheimers. Dis. 2015, 43, 1115–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. J. R. Coll. Physicians Lond. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D. Late-onset Alzheimer Disease. Contin. Minneap. Minn. 2019, 25, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB 2 receptors in the mouse brain: Relevance for Alzheimer’s disease. J. Neuroinflammation 2018, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Aso, E.; Ferrer, I. Cannabinoids for treatment of Alzheimer’s disease: Moving toward the clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol Lipase Is a Therapeutic Target for Alzheimer’s Disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, J.; Davies, P. Targeting the endocannabinoid system in Alzheimer’s disease. J. Alzheimers. Dis. 2008, 15, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.C.; MacKie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.C.; Lee, F.S.; Gee, D.G. The Role of the Endocannabinoid System and Genetic Variation in Adolescent Brain Development. Neuropsychopharmacology 2018, 43, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Marsicano, G.; Lafenêtre, P. Roles of the endocannabinoid system in learning and memory. Curr. Top. Behav. Neurosci. 2009, 1, 201–230. [Google Scholar] [PubMed]
- Lisboa, S.F.; Gomes, F.V.; Terzian, A.L.B.; Aguiar, D.C.; Moreira, F.A.; Resstel, L.B.M.; Guimarães, F.S. The Endocannabinoid System and Anxiety. Vitam. Horm. 2017, 103, 193–279. [Google Scholar]
- Bahr, B.A.; Karanian, D.A.; Makanji, S.S.; Makriyannis, A. Targeting the endocannabinoid system in treating brain disorders. Expert Opin. Investig. Drugs 2006, 15, 351–365. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005. [Google Scholar] [CrossRef]
- Rivera, P.; Arrabal, S.; Cifuentes, M.; Grondona, J.M.; Pérez-Martín, M.; Rubio, L.; Vargas, A.; Serrano, A.; Pavón, F.J.; Suárez, J.; et al. Localization of the cannabinoid CB1 receptor and the 2-AG synthesizing (DAGLα) and degrading (MAGL, FAAH) enzymes in cells expressing the Ca2+-binding proteins calbindin, calretinin, and parvalbumin in the adult rat hippocampus. Front. Neuroanat. 2014, 8, 56. [Google Scholar] [CrossRef]
- De Fonseca, F.R.; Ramos, J.A.; Bonnin, A.; Fernández-Ruiz, J.J. Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport 1993, 4, 135–138. [Google Scholar] [CrossRef]
- Benito, C.; Tolón, R.M.; Pazos, M.R.; Núñez, E.; Castillo, A.I.; Romero, J. Cannabinoid CB2 receptors in human brain inflammation. Br. J. Pharmacol. 2008, 153, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, B.G.; Blázquez, C.; Gómez Del Pulgar, T.; Guzmán, M.; De Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef]
- Thompson, M.D.; Sakurai, T.; Rainero, I.; Maj, M.C.; Kukkonen, J.P. Orexin receptor multimerization versus functional interactions: Neuropharmacological implications for opioid and cannabinoid signalling and pharmacogenetics. Pharmaceuticals 2017, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Kruk-Slomka, M.; Dzik, A.; Budzynska, B.; Biala, G. Endocannabinoid System: The Direct and Indirect Involvement in the Memory and Learning Processes—A Short Review. Mol. Neurobiol. 2017, 54, 8332–8347. [Google Scholar] [CrossRef] [Green Version]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Brusco, A.; Tagliaferro, P.A.; Saez, T.; Onaivi, E.S. Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Ann. N. Y. Acad. Sci. 2008, 1139, 450–457. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann. N. Y. Acad. Sci. 2008, 1139, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 cannabinoid receptor agonist ameliorates alzheimer-like phenotype in AβPP/PS1 mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 791–804. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 2012, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Shi, J.; Wang, B.; Li, J.; Jia, H. CB2 cannabinoid receptor agonist ameliorates novel object recognition but not spatial memory in transgenic APP/PS1 mice. Neurosci. Lett. 2019, 707, 134286. [Google Scholar] [CrossRef]
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; Van Der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflammation 2018, 15, 322. [Google Scholar] [CrossRef]
- Shi, Q.X.; Yang, L.K.; Shi, W.L.; Wang, L.; Zhou, S.M.; Guan, S.Y.; Zhao, M.G.; Yang, Q. The novel cannabinoid receptor GPR55 mediates anxiolytic-like effects in the medial orbital cortex of mice with acute stress. Mol. Brain 2017, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Méndez-Díaz, M.; Prospéro-García, O. Possible role of hippocampal GPR55 in spatial learning and memory in rats. Acta Neurobiol. Exp. (Wars) 2018, 78, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K. Mechanisms of CB1 receptor signaling: Endocannabinoid modulation of synaptic strength. Int. J. Obes. 2006, 30, S19–S23. [Google Scholar] [CrossRef] [Green Version]
- Gowran, A.; Noonan, J.; Campbell, V.A. The multiplicity of action of cannabinoids: Implications for treating neurodegeneration. CNS Neurosci. Ther. 2011. [Google Scholar] [CrossRef]
- Vázquez, C.; Tolón, R.M.; Grande, M.T.; Caraza, M.; Moreno, M.; Koester, E.C.; Villaescusa, B.; Ruiz-Valdepeñas, L.; Fernández-Sánchez, F.J.; Cravatt, B.F.; et al. Endocannabinoid regulation of amyloid-induced neuroinflammation. Neurobiol. Aging 2015. [Google Scholar] [CrossRef]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.Q.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer’s Disease. Cell Rep. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ativie, F.; Albayram, O.; Bach, K.; Pradier, B.; Zimmer, A.; Bilkei-Gorzo, A. Enhanced microglial activity in FAAH(-/-) animals. Life Sci. 2015. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006. [Google Scholar] [CrossRef]
- Ohno, M. Genetic and pharmacological basis for therapeutic inhibition of beta- and gamma-secretases in mouse models of Alzheimer’s memory deficits. Rev. Neurosci. 2006, 17, 429–454. [Google Scholar] [CrossRef]
- Ohno, M.; Chang, L.; Tseng, W.; Oakley, H.; Citron, M.; Klein, W.L.; Vassar, R.; Disterhoft, J.F. Temporal memory deficits in Alzheimer’s mouse models: Rescue by genetic deletion of BACE1. Eur. J. Neurosci. 2006. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Aβ42 accumulation and Caspase-3 activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.K.; Han, D.; Park, J.; Choi, H.; Park, J.-C.; Cha, M.-Y.; Woo, J.; Byun, M.S.; Lee, D.Y.; Kim, Y.; et al. Deep proteome profiling of the hippocampus in the 5XFAD mouse model reveals biological process alterations and a novel biomarker of Alzheimer’s disease. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Devi, L.; Ohno, M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 2010, 113, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Creighton, S.D.; Mendell, A.L.; Palmer, D.; Kalisch, B.E.; MacLusky, N.J.; Prado, V.F.; Prado, M.A.M.; Winters, B.D. Dissociable cognitive impairments in two strains of transgenic Alzheimer’s disease mice revealed by a battery of object-based tests. Sci. Rep. 2019, 9, 57. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Sarroca, S.; Ivanova, A.; Puigoriol-Illamola, D.; Aguado, F.; Camins, A.; Sanfeliu, C.; Pallàs, M. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY) 2016, 8, 664–684. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Silva-Peña, D.; García-Marchena, N.; Alén, F.; Araos, P.; Rivera, P.; Vargas, A.; García-Fernández, M.I.; Martín-Velasco, A.I.; Villanúa, M.Á.; Castilla-Ortega, E.; et al. Alcohol-induced cognitive deficits are associated with decreased circulating levels of the neurotrophin BDNF in humans and rats. Addict. Biol. 2019. [Google Scholar] [CrossRef]
- Miedel, C.J.; Patton, J.M.; Miedel, A.N.; Miedel, E.S.; Levenson, J.M. Assessment of spontaneous alternation, novel object recognition and limb clasping in transgenic mouse models of amyloid-β and tau neuropathology. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla-Ortega, E.; Hoyo-Becerra, C.; Pedraza, C.; Chun, J.; Rodríguez De Fonseca, F.; Estivill-Torrús, G.; Santín, L.J. Aggravation of chronic stress effects on hippocampal neurogenesis and spatial memory in LPA1 receptor knockout mice. PLoS ONE 2011, 6, e25522. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Talarico, G.; Trebbastoni, A.; Bruno, G.; de Lena, C. Modulation of the Cannabinoid System: A New Perspective for the Treatment of the Alzheimer’s Disease. Curr. Neuropharmacol. 2018. [Google Scholar] [CrossRef]
- Richard, B.C.; Kurdakova, A.; Baches, S.; Bayer, T.A.; Weggen, S.; Wirths, O. Gene dosage dependent aggravation of the neurological phenotype in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015. [Google Scholar] [CrossRef]
- Ruehle, S.; Rey, A.A.; Remmers, F.; Lutz, B. The endocannabinoid system in anxiety, fear memory and habituation. J. Psychopharmacol. 2012. [Google Scholar] [CrossRef]
- Kruk-Slomka, M.; Biala, G. CB1 receptors in the formation of the different phases of memory-related processes in the inhibitory avoidance test in mice. Behav. Brain Res. 2016. [Google Scholar] [CrossRef]
- De Oliveira Alvares, L.; Genro, B.P.; Diehl, F.; Quillfeldt, J.A. Differential role of the hippocampal endocannabinoid system in the memory consolidation and retrieval mechanisms. Neurobiol. Learn. Mem. 2008. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; De Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991. [Google Scholar] [CrossRef]
- Hampson, R.E.; Deadwyler, S.A. Role of cannabinoid receptors in memory storage. Neurobiol. Dis. 1998. [Google Scholar] [CrossRef]
- Riedel, G.; Davies, S.N. Cannabinoid function in learning, memory and plasticity. Handb. Exp. Pharmacol. 2005. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. CB2 Cannabinoid Receptor As Potential Target against Alzheimer’s Disease. Front. Neurosci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Dong, Z.; Liu, S. β-caryophyllene ameliorates the Alzheimer-like phenotype in APP/PS1 mice through CB2 receptor activation and the PPARγ pathway. Pharmacology 2014. [Google Scholar] [CrossRef] [PubMed]
- Kramar, C.; Loureiro, M.; Renard, J.; Laviolette, S.R. Palmitoylethanolamide Modulates GPR55 Receptor Signaling in the Ventral Hippocampus to Regulate Mesolimbic Dopamine Activity, Social Interaction, and Memory Processing. Cannabis Cannabinoid Res. 2017. [Google Scholar] [CrossRef]
- Wu, C.S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013. [Google Scholar] [CrossRef] [Green Version]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017. [Google Scholar] [CrossRef]
- Hill, J.D.; Zuluaga-Ramirez, V.; Gajghate, S.; Winfield, M.; Persidsky, Y. Activation of GPR55 increases neural stem cell proliferation and promotes early adult hippocampal neurogenesis. Br. J. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Deliu, E.; Zhang, X.Q.; Hoffman, N.E.; Carter, R.L.; Grisanti, L.A.; Brailoiu, G.C.; Madesh, M.; Cheung, J.Y.; Force, T.; et al. Differential activation of cultured neonatal cardiomyocytes by plasmalemmal versus intracellular G protein-coupled receptor 55. J. Biol. Chem. 2013. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Zhu, M.X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China Life Sci. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michno, W.; Wehrli, P.M.; Zetterberg, H.; Blennow, K.; Hanrieder, J. GM1 locates to mature amyloid structures implicating a prominent role for glycolipid-protein interactions in Alzheimer pathology. Biochim. Biophys. Acta Proteins Proteom. 2019. [Google Scholar] [CrossRef] [Green Version]
- Fondevila, M.F.; Fernandez, U.; Gonzalez-Rellan, M.J.; Da Silva Lima, N.; Buque, X.; Gonzalez-Rodriguez, A.; Alonso, C.; Iruarrizaga-Lejarreta, M.; Delgado, T.C.; Varela-Rey, M.; et al. The L-α-lysophosphatidylinositol/GPR55 system induces the development of non-alcoholic steatosis and steatohepatitis. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid signaling in the brain. Science 2002. [Google Scholar] [CrossRef] [Green Version]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Ortega-Gutiérrez, S.; Van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003. [Google Scholar] [CrossRef] [Green Version]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Chanda, P.K.; Gao, Y.; Mark, L.; Btesh, J.; Strassle, B.W.; Lu, P.; Piesla, M.J.; Zhang, M.Y.; Bingham, B.; Uveges, A.; et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol. Pharmacol. 2010. [Google Scholar] [CrossRef]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011. [Google Scholar] [CrossRef]
- Altamura, C.; Ventriglia, M.; Martini, M.G.; Montesano, D.; Errante, Y.; Piscitelli, F.; Scrascia, F.; Quattrocchi, C.; Palazzo, P.; Seccia, S.; et al. Elevation of plasma 2-arachidonoylglycerol levels in alzheimer’s disease patients as a potential protective mechanism against neurodegenerative decline. J. Alzheimer’s Dis. 2015, 46, 497–506. [Google Scholar] [CrossRef]
- Gamba, P.; Testa, G.; Gargiulo, S.; Staurenghi, E.; Poli, G.; Leonarduzzi, G. Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front. Aging Neurosci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.P.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef]
- Ohsawa, K.; Imai, Y.; Sasaki, Y.; Kohsaka, S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 2004. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Zhao, M.L.; Hirano, A.; Dickson, D.W. Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: Association with Hirano bodies, neurofibrillary tangles, and senile plaques. J. Neuropathol. Exp. Neurol. 1999. [Google Scholar] [CrossRef] [Green Version]
- Manuel, I.; De San Román, E.G.; Giralt, M.T.; Ferrer, I.; Rodríguez-Puertas, R. Type-1 cannabinoid receptor activity during Alzheimer’s disease progression. J. Alzheimer’s Dis. 2014. [Google Scholar] [CrossRef] [Green Version]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pearson’s Correlation | CB1 | CB2 | GPR55 |
|---|---|---|---|
| Time spent in the center: habituation trial | 0.589 | −0.523 | −0.750 |
| 0.72550 | <0.0001 *** | <0.0001 *** | |
| Locomotion (distance moved): acquisition trial | −0.479 | 0.445 | 0.680 |
| 0.00210 ** | 0.45070 | 0.0249 * | |
| Percentage novelty preference: retention trial | 0.634 | −0.483 | −0.525 |
| 0.03950 * | 0.00340 ** | 0.0126 * |
| Pearson’s Correlation | CB1 | CB2 | GPR55 |
|---|---|---|---|
| Iba1 | −0.513 | 0.470 | −0.704 |
| 0.003 ** | 0.9685 | 0.0089 ** | |
| GFAP | −0.722 | 0.583 | −0.656 |
| <0.0001 *** | 0.0062 ** | 0.1453 | |
| COX2 | −0.717 | 0.576 | −0.696 |
| 0.0006 *** | 0.0446 * | 0.0029 ** | |
| iNOS | −0.724 | 0.583 | −0.729 |
| <0.0001 *** | 0.716 | 0.0065 ** |
| Pearson’s Correlation | CB1 | CB2 | GPR55 |
|---|---|---|---|
| Aβ40 | −0.700 | 0.553 | 0.626 |
| <0.0001 *** | <0.0001 *** | 0.4077 | |
| Aβ42 | −0.723 | 0.587 | 0.712 |
| <0.0001 *** | <0.0001 *** | 0.4806 | |
| Aβtotal | −0.717 | 0.575 | 0.681 |
| <0.0001 *** | <0.0001 *** | 0.4652 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Vera, D.; Rosell-Valle, C.; López-Gambero, A.J.; Navarro, J.A.; Zambrana-Infantes, E.N.; Rivera, P.; Santín, L.J.; Suarez, J.; Rodríguez de Fonseca, F. Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity. Biology 2020, 9, 377. https://doi.org/10.3390/biology9110377
Medina-Vera D, Rosell-Valle C, López-Gambero AJ, Navarro JA, Zambrana-Infantes EN, Rivera P, Santín LJ, Suarez J, Rodríguez de Fonseca F. Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity. Biology. 2020; 9(11):377. https://doi.org/10.3390/biology9110377
Chicago/Turabian StyleMedina-Vera, Dina, Cristina Rosell-Valle, Antonio J. López-Gambero, Juan A. Navarro, Emma N. Zambrana-Infantes, Patricia Rivera, Luis J. Santín, Juan Suarez, and Fernando Rodríguez de Fonseca. 2020. "Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity" Biology 9, no. 11: 377. https://doi.org/10.3390/biology9110377