Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive

 ,
, 
Abstract
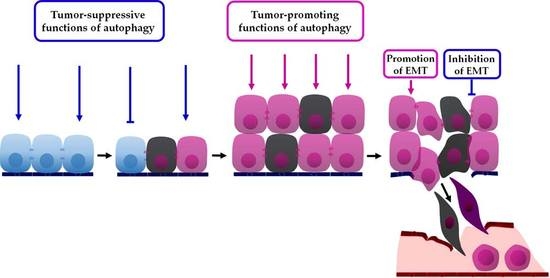
:
1. Introduction
2. The Autophagic Pathway
3. Autophagy in Cancer
4. Epithelial to Mesenchymal Transition in Cancer
5. Cytokines in the Tumor Microenvironment and Their Effects on EMT
6. Autophagy and Its Effects on Epithelial to Mesenchymal Transition in Cancer Cells
7. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy modulation in cancer: Current knowledge on action and therapy. Oxid. Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Cotzomi-Ortega, I.; Aguilar-Alonso, P.; Reyes-Leyva, J.; Maycotte, P. Autophagy and its role in protein secretion: Implications for cancer therapy. Mediat. Inflamm. 2018, 2018, 4231591. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Singla, M.; Bhattacharyya, S. Autophagy as a potential therapeutic target during epithelial to mesenchymal transition in renal cell carcinoma: An in vitro study. Biomed. Pharmacother. 2017, 94, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Kroemer, G. Therapeutic modulation of autophagy: Which disease comes first? Cell Death Differ. 2019, 26, 680–689. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef]
- Huang, J.; Klionsky, D.J. Autophagy and human disease. Cell Cycle 2007, 6, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- Harnett, M.M.; Pineda, M.A.; Latre de Late, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From christian de duve to yoshinori ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/sqstm1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Mainz, L.; Rosenfeldt, M.T. Autophagy and cancer—Insights from mouse models. FEBS J. 2018, 285, 792–808. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy, metabolism, and cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar]
- Levy, J.M.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. FOXO3 links autophagy to apoptosis. Autophagy 2018, 14, 1467–1468. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic ras-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy supports breast cancer stem cell maintenance by regulating il6 secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated RAS requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, J.; Staskiewicz, L.; Goodall, M.L.; Dimberg, L.; Frankel, A.E.; Ford, H.L.; Thorburn, A. Non-cell-autonomous effects of autophagy inhibition in tumor cells promote growth of drug-resistant cells. Mol. Pharmacol. 2017, 91, 58–64. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Levy, J.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Levy, J.M.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. Stat3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhu, W.; Huang, W.; Lu, S.S.; Li, X.H.; Xiao, Z.Q.; Yi, H. Rack1 promotes tumorigenicity of colon cancer by inducing cell autophagy. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Li, C.; Zheng, W.; Guo, Q.; Zhang, Y.; Kang, M.; Zhang, B.; Yang, B.; Li, B.; Yang, H.; et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ros in gastric cancer cells. Oncotarget 2015, 6, 39839–39854. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of hcc in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef]
- Dower, C.M.; Bhat, N.; Wang, E.W.; Wang, H.G. Selective reversible inhibition of autophagy in hypoxic breast cancer cells promotes pulmonary metastasis. Cancer Res. 2017, 77, 646–657. [Google Scholar] [CrossRef]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef]
- Hu, S.; Wang, L.; Zhang, X.; Wu, Y.; Yang, J.; Li, J. Autophagy induces transforming growth factor-beta-dependent epithelial-mesenchymal transition in hepatocarcinoma cells through camp response element binding signalling. J. Cell Mol. Med. 2018, 22, 5518–5532. [Google Scholar] [CrossRef]
- Dash, S.; Sarashetti, P.M.; Rajashekar, B.; Chowdhury, R.; Mukherjee, S. Tgf-beta2-induced EMT is dampened by inhibition of autophagy and TNF-alpha treatment. Oncotarget 2018, 9, 6433–6449. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liang, Q.Y.; Chen, H.K.; Wu, P.F.; Feng, Z.Y.; Ma, X.M.; Wu, H.R.; Zhou, G.Q. Dram1 regulates the migration and invasion of hepatoblastoma cells via autophagy-emt pathway. Oncol. Lett 2018, 16, 2427–2433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, H.; Liu, D.; Hill, C.; Ertay, A.; Li, J.; Zou, Y.; Miller, P.; White, E.; Downward, J.; et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in Ras-mutated cancer cells. Autophagy 2019, 15, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Yin, L.; Deng, G.; Guo, C.; Han, Y.; Li, Y.; Cai, C.; Fu, Y.; Liu, S.; Zeng, S. Knockdown of beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J. Cell Biochem. 2018, 119, 7022–7031. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar] [PubMed]
- Su, Z.; Li, G.; Liu, C.; Ren, S.; Deng, T.; Zhang, S.; Tian, Y.; Liu, Y.; Qiu, Y. Autophagy inhibition impairs the epithelial-mesenchymal transition and enhances cisplatin sensitivity in nasopharyngeal carcinoma. Oncol. Lett. 2017, 13, 4147–4154. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. Dedd interacts with pi3kc3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef]
- Ahn, J.S.; Ann, E.J.; Kim, M.Y.; Yoon, J.H.; Lee, H.J.; Jo, E.H.; Lee, K.; Lee, J.S.; Park, H.S. Autophagy negatively regulates tumor cell proliferation through phosphorylation dependent degradation of the notch1 intracellular domain. Oncotarget 2016, 7, 79047–79063. [Google Scholar] [CrossRef]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lai, T.H.; Pham, T.M.; Kim, D.R. Control of the epithelial-to-mesenchymal transition and cancer metastasis by autophagy-dependent snai1 degradation. Cells 2019, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting snail degradation. Cell Death Dis. 2015, 6, e1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bu, S.; Xin, D.; Li, B.; Wang, L.; Lai, D. Autophagy is indispensable for the self-renewal and quiescence of ovarian cancer spheroid cells with stem cell-like properties. Oxid. Med. Cell Longev. 2018, 2018, 7010472. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy differentially regulates distinct breast cancer stem-like cells in murine models via EGFR/Stat3 and Tgfß/smad signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol. Cell 2017, 68, 281.e5–292.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.H. Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: A comprehensive overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Sistigu, A.; Di Modugno, F.; Manic, G.; Nistico, P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. 2017, 36, 67–77. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell 2018, 45, 696.e698–711.e698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bhaijee, F.; Ishaq, N.; Pepper, D.J.; Backus, K.; Brown, A.S.; Zhou, X.; Miele, L. Correlation of Notch1, pAKT and nuclear NF-κB expression in triple negative breast cancer. Am. J. Cancer Res. 2013, 3, 230–239. [Google Scholar] [PubMed]
- Savagner, P. Epithelial-mesenchymal transitions: From cell plasticity to concept elasticity. Curr Top. Dev. Biol. 2015, 112, 273–300. [Google Scholar] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681.e684–695.e684. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; Yao, Z.; Wang, L. Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression. J. Biomed. Res. 2018, 32, 81–90. [Google Scholar]
- Liu, X.; Li, J.; Cadilha, B.L.; Markota, A.; Voigt, C.; Huang, Z.; Lin, P.P.; Wang, D.D.; Dai, J.; Kranz, G.; et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci. Adv. 2019, 5, eaav4275. [Google Scholar] [CrossRef] [Green Version]
- Corallino, S.; Malabarba, M.G.; Zobel, M.; Di Fiore, P.P.; Scita, G. Epithelial-to-mesenchymal plasticity harnesses endocytic circuitries. Front. Oncol. 2015, 5, 45. [Google Scholar] [CrossRef]
- Sung, C.O.; Park, C.K.; Kim, S.H. Classification of epithelial-mesenchymal transition phenotypes in esophageal squamous cell carcinoma is strongly associated with patient prognosis. Mod. Pathol. 2011, 24, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Epithelial-mesenchymal transition phenotype is associated with clinicopathological factors that indicate aggressive biological behavior and poor clinical outcomes in invasive breast cancer. J. Breast Cancer 2015, 18, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.N.; Xiang, B.D.; Wu, F.X.; Ye, J.Z.; Zhong, J.H.; Wang, Y.Y.; Chen, Y.Y.; Chen, Z.S.; Ma, L.; Chen, J.; et al. Circulating tumor cells undergoing emt provide a metric for diagnosis and prognosis of patients with hepatocellular carcinoma. Cancer Res. 2018, 78, 4731–4744. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco. Targets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Hirayama, M.; Ikeda, T.; Yuno, A.; Matsumura, K.; Fukuma, D.; Araki, K.; Mizuta, H.; Nakayama, H.; et al. Soluble IL6R expressed by myeloid cells reduces tumor-specific Th1 differentiation and drives tumor progression. Cancer Res. 2017, 77, 2279–2291. [Google Scholar] [CrossRef]
- Elkabets, M.; Ribeiro, V.S.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A. Il-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Tesi, R.J. Mdsc; the most important cell you have never heard of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmani, A.; Delisle, J. Tgf-β in t cell biology: Implications for cancer immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. Il-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of map3k7 as a target for cancer therapy. Drug Resist. Updat. 2017, 33–35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Franco-Barraza, J.; Valdivia-Silva, J.E.; Zamudio-Meza, H.; Castillo, A.; Garcia-Zepeda, E.A.; Benitez-Bribiesca, L.; Meza, I. Actin cytoskeleton participation in the onset of Il-1beta induction of an invasive mesenchymal-like phenotype in epithelial MCF-7 cells. Arch. Med. Res. 2010, 41, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking Il-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and cancer progression: The emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Taher, M.Y.; Davies, D.M.; Maher, J. The role of the interleukin (Il)-6/Il-6 receptor axis in cancer. Biochem Soc. Trans. 2018, 46, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Heo, T.H.; Wahler, J.; Suh, N. Potential therapeutic implications of Il-6/Il-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Apte, R.N. Il-1 in colon inflammation, colon carcinogenesis and invasiveness of colon cancer. Cancer Microenviron. 2015, 8, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.M.; Mariano, V.S.; Pastrez, P.R.A.; Pinto, M.C.; Castro, A.G.; Syrjanen, K.J.; Longatto-Filho, A. High systemic Il-6 is associated with worse prognosis in patients with non-small cell lung cancer. PLoS ONE 2017, 12, e0181125. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lian, H.; Jia, Q.; Wan, Y. Proteome screening of pleural effusions identifies Il1a as a diagnostic biomarker for non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2015, 457, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Malhotra, P.S.; Thomas, G.R.; Ondrey, F.G.; Duffey, D.C.; Smith, C.W.; Enamorado, I.; Yeh, N.T.; Kroog, G.S.; Rudy, S.; et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin. Cancer Res. 1999, 5, 1369–1379. [Google Scholar]
- Choudhary, M.; France, T.; Teknos, T.; Kumar, P. Interleukin-6 role in head and neck squamous cell carcinoma progression. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 90e97. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.S.; Chen, W.C.; Lu, C.H.; Chen, M.F. The prognosis of head and neck squamous cell carcinoma related to immunosuppressive tumor microenvironment regulated by Il-6 signaling. Oral Oncol. 2019, 91, 47–55. [Google Scholar] [CrossRef]
- Nomura, A.; Gupta, V.K.; Dauer, P.; Sharma, N.S.; Dudeja, V.; Merchant, N.; Saluja, A.K.; Banerjee, S. NFkappab-mediated invasiveness in CD133(+) pancreatic tics is regulated by autocrine and paracrine activation of Il1 signaling. Mol. Cancer Res. 2018, 16, 162–172. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the Il-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Loffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermuller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wu, D.; Gao, H.; Balic, J.J.; Tsykin, A.; Han, T.S.; Liu, Y.D.; Kennedy, C.L.; Li, J.K.; Mao, J.Q.; et al. Clinical utility of a STAT3-regulated miRNA-200 family signature with prognostic potential in early gastric cancer. Clin. Cancer Res. 2018, 24, 1459–1472. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via Il6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.; Tan, L.; Bolland, D.E.; Coffman, L.G.; Peterson, L.F.; Talpaz, M.; Neamati, N.; Buckanovich, R.J. Leukemia inhibitory factor functions in parallel with interleukin-6 to promote ovarian cancer growth. Oncogene 2019, 38, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, M.; Sacchetti, A.; Cansoy, M.; Joosten, R.; Teeuwssen, M.; Heijmans-Antonissen, C.; Ewing-Graham, P.C.; Burger, C.W.; Blok, L.J.; Fodde, R. Il6/JAK1/STAT3 signaling blockade in endometrial cancer affects the ALDHhi/CD126+ stem-like component and reduces tumor burden. Cancer Res. 2015, 75, 3608–3622. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Potent EMT and CSC phenotypes are induced by Oncostatin-M in pancreatic cancer. Mol. Cancer Res. 2017, 15, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Palena, C.; Hamilton, D.H.; Fernando, R.I. Influence of Il-8 on the epithelial-mesenchymal transition and the tumor microenvironment. Future Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, J.; Chen, Y.; Xiong, N.; Li, L.; Zhang, J.; Yang, H.; Wu, C.; Zeng, H.; Liu, Y. Mcp-1-induced ERK/GSK-3beta/Snail signaling facilitates the epithelial-mesenchymal transition and promotes the migration of MCF-7 human breast carcinoma cells. Cell Mol. Immunol. 2017, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castanon, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.; Hansen, R.J.; Maycotte, P.; Thorburn, A.; Gustafson, D.L. Role of autophagy inhibition in metastatic disease utilizing mouse models. Mol. Cancer Res. 2014, 12. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T.; et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraya, A.A.; Piao, S.; Xu, X.; Zhang, G.; Herlyn, M.; Gimotty, P.; Levine, B.; Amaravadi, R.K.; Speicher, D.W. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sample, A.; Shea, C.R.; Soltani, K.; Macleod, K.F.; He, Y.Y. Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy 2017, 13, 2086–2103. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int J. Mol. Sci 2017, 18, 1279. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Beneficial Effects of Autophagy Inhibition | Cancer-Related Feature | Counter-Productive Effects of Autophagy Inhibition |
|---|---|---|
| RAS-transformed cancer cells [36,37] | Proliferation/ cancer Progression | |
| K-rasG12D or BrafV600E;atg5/7flox/flox mouse lung cancer models [24,25,38] | MEFs with Atg gene KO; epidermal squamous cell carcinoma mouse xenografts [39] | |
| MMTV-PyMT mice with FIP200−/− in mammary epithelial cells [40] | Dying autophagy-deficient cell lines induced proliferation of resistant cells in response to targeted therapy [41] | |
| KRAS mutant pancreatic cancer [42] | Mice with autophagy inhibition together with p53−/− had increased pancreatic ductal adenocarcinoma frequency [23] | |
| BRAFV600E central nervous system tumors [43] | ||
| Triple negative breast cancer cell lines [44] | ||
| RACK1-induced autophagy in CRC cells [45] | ||
| RAS transformed cancer cells [37] | Migration/ invasion/ EMT or metastasis establishment | Gastric cancer cell lines and mouse xenografts [46] |
| HCC cell lines and xenografts [47] | Loss of ULK1 to suppress autophagy in the MDA-MB-231 breast cancer cell line during hypoxia [48] | |
| Starvation- [49,50], TGF-β2- [51], or DRAM1- induced [52] autophagy in hepatic carcinoma cell lines | RAS-mutated cancer cells [53] | |
| TGFβ1- or rapamycin-induced autophagy in non-small cell lung cancer cells [54] | Glioblastoma cell lines [55] | |
| Rapamycin-induced autophagy in CRC cell lines [56] | Ovarian cancer cell lines [57] | |
| Cisplatin-induced autophagy in nasopharyngeal carcinoma cells [58] | DEDD-induced autophagy in breast cancer cell lines [59] | |
| MEFs with Atg gene KO; epidermal squamous cell carcinoma mouse xenografts [39] | ||
| MDA-MB-231 breast cancer cell line [60] | ||
| H1299 lung or HeLa cervical cancer cell lines [61] | ||
| Liver-specific autophagy-deficiency or TGFβ−treated immortalized hepatocytes [62] | ||
| MMTV-PyMT mice with FIP200−/− conditional KO in mammary epithelial cells [40] | Immunoediting | Gastric cancer cell lines [35] |
| K-rasG12D;Atg5flox/flox mouse lung cancer model [24] | Colorectal or osteosarcoma cancer cell lines treated with immunogenic chemotherapy [34] | |
| Ovarian cancer spheroids [63] | Tumor-initiating cells/CSCs | |
| Breast cancer stem cells [32,64,65] | ||
| Hepatic cancer stem cells [66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas-Sanchez, G.; Cotzomi-Ortega, I.; Pazos-Salazar, N.G.; Reyes-Leyva, J.; Maycotte, P. Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive. Biology 2019, 8, 71. https://doi.org/10.3390/biology8040071
Rojas-Sanchez G, Cotzomi-Ortega I, Pazos-Salazar NG, Reyes-Leyva J, Maycotte P. Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive. Biology. 2019; 8(4):71. https://doi.org/10.3390/biology8040071
Chicago/Turabian StyleRojas-Sanchez, Guadalupe, Israel Cotzomi-Ortega, Nidia G. Pazos-Salazar, Julio Reyes-Leyva, and Paola Maycotte. 2019. "Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive" Biology 8, no. 4: 71. https://doi.org/10.3390/biology8040071