
A Decade of Discovery—Eukaryotic Replisome Disassembly at Replication Termination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
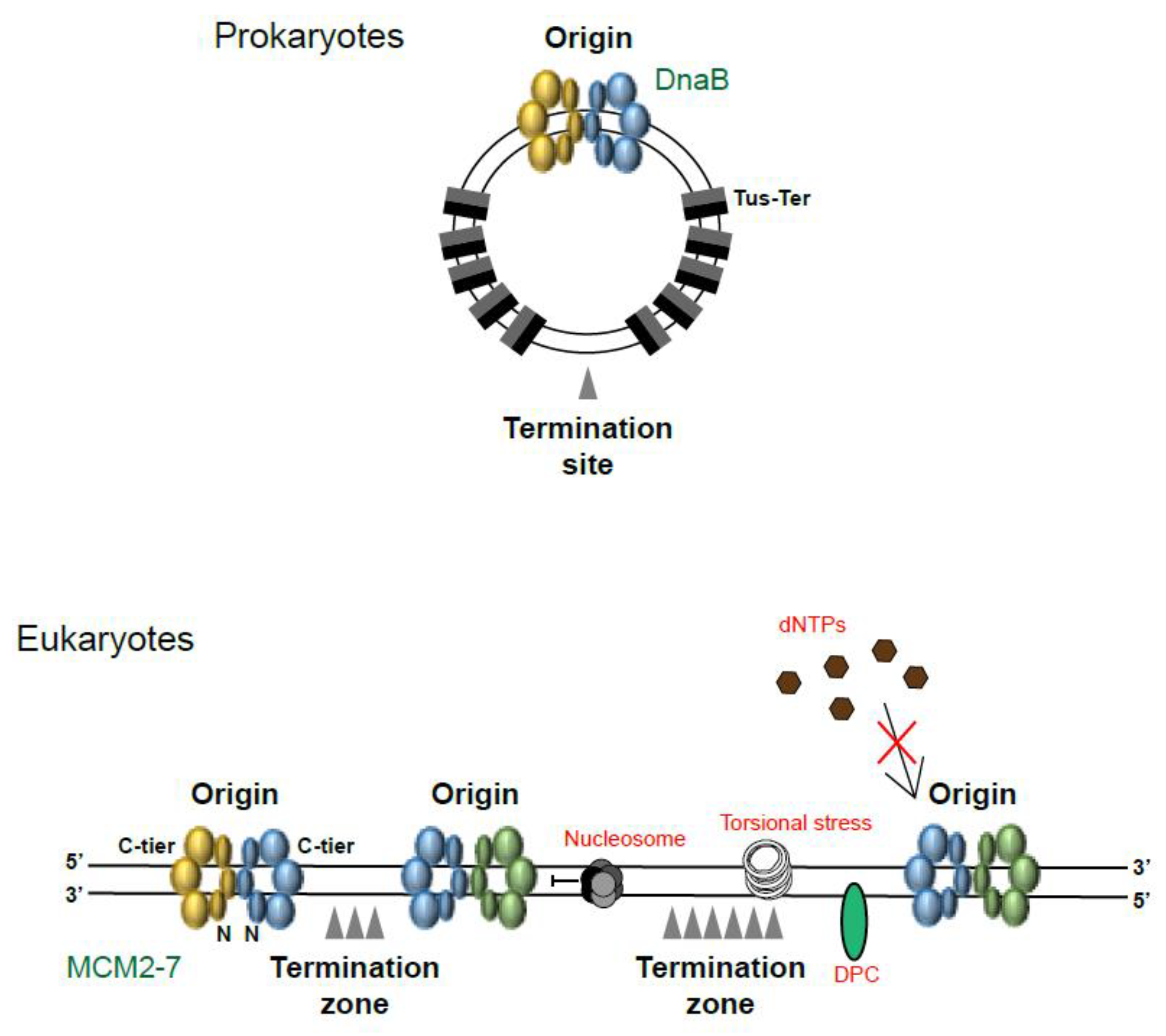
1. Where and When Does Replication Termination Take Place?
2. Replication Fork Convergence
3. Completion of DNA Synthesis
4. Replisome Disassembly
4.1. Cullin Ubiquitin Ligases Driving MCM7 Ubiquitylation
4.2. Recruitment of Ubiquitin Ligase to Terminated Helicases
4.3. Restricting MCM7 Ubiquitylation to Termination
4.4. p97/Cdc48/VCP
4.4.1. p97 Cofactors
4.4.2. Processing Ubiquitylated CMG by p97
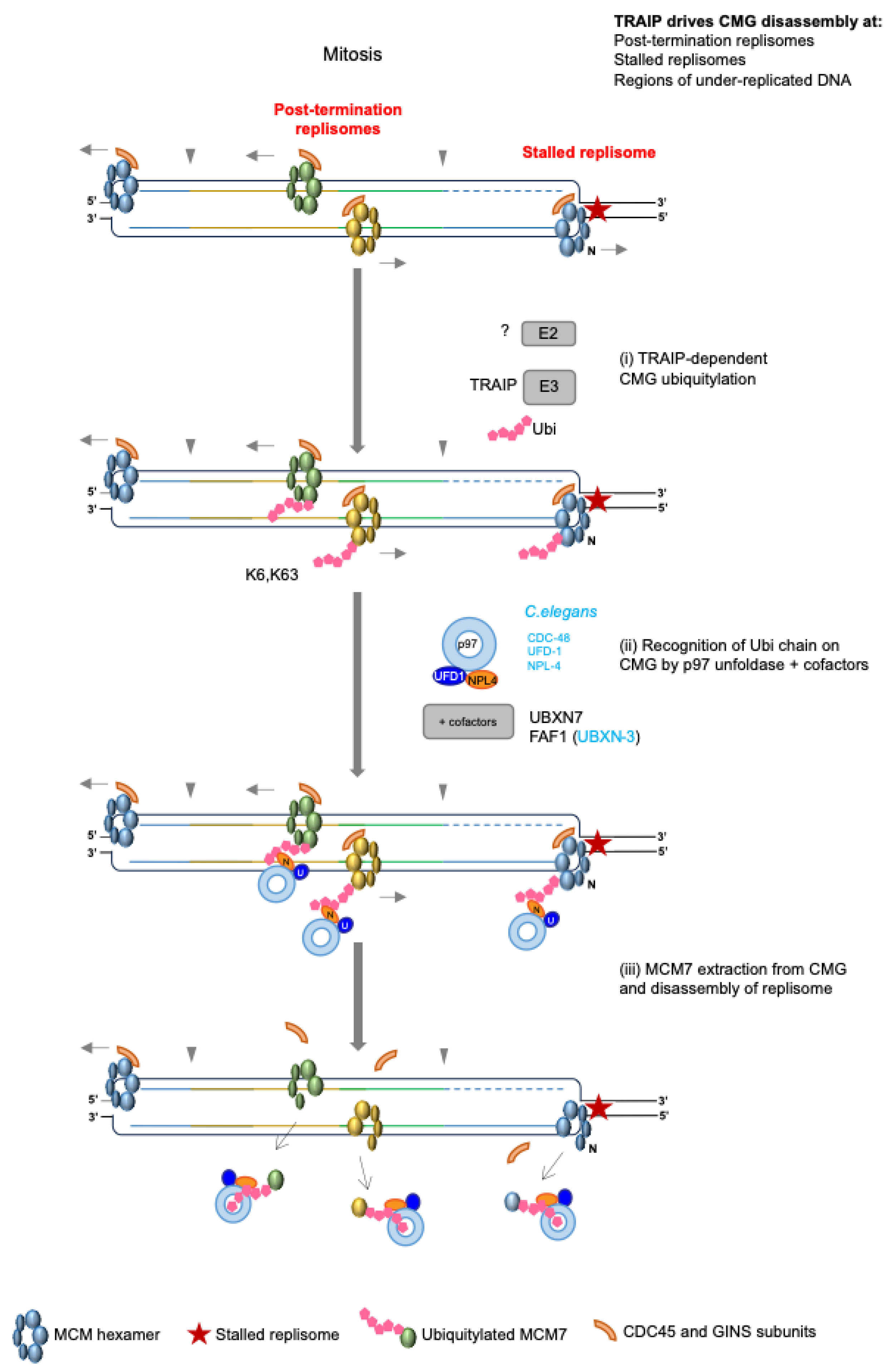
4.5. Replisome Disassembly in Mitosis—The Back-Up Pathway
Role of TRAIP during Incomplete Replication
5. TRAIP Activity in S-Phase
6. Consequences of Defective Replisome Disassembly
7. Future Directions
- 1.
- Is there a role for deubiquitylating enzymes (DUBs) in preventing premature replisome disassembly?
- 2.
- What are the full consequences of inhibiting replisome disassembly in S-phase, making LRR1 an essential gene? Why is TRAIP unable to substitute for its loss?
- 3.
- What about dormant MCM2-7 helicases—how are they removed from DNA? While we know that they encircle DNA in a very stable manner and can be pushed ahead of replication forks, the mechanism of their unloading from chromatin is still a mystery.
- 4.
- Can the process of replisome disassembly be explored in the context of human health and disease? Replication initiation and elongation are frequently targeted in cancer therapy. Could we now also exploit our knowledge of replication termination to benefit human health?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGuffee, S.R.; Smith, D.J.; Whitehouse, I. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol. Cell 2013, 50, 123–135. [Google Scholar] [CrossRef]
- Berghuis, B.A.; Raducanu, V.S.; Elshenawy, M.M.; Jergic, S.; Depken, M.; Dixon, N.E.; Hamdan, S.M.; Dekker, N.H. What is all this fuss about Tus? Comparison of recent findings from biophysical and biochemical experiments. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 49–63. [Google Scholar] [CrossRef]
- Rudolph, C.J.; Corocher, T.-A.; Grainge, I.; Duggin, I.G. Termination of DNA Replication in Prokaryotes. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2001; pp. 1–15. [Google Scholar]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A.; Lloyd, R.G. Avoiding chromosome pathology when replication forks collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef]
- Merrikh, H.; Zhang, Y.; Grossman, A.D.; Wang, J.D. Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 2012, 10, 449–458. [Google Scholar] [CrossRef]
- Weaver, D.T.; Fields-Berry, S.C.; DePamphilis, M.L. The termination region for SV40 DNA replication directs the mode of separation for the two sibling molecules. Cell 1985, 41, 565–575. [Google Scholar] [CrossRef]
- Hyrien, O.; Goldar, A. Mathematical modelling of eukaryotic DNA replication. Chromosome Res. 2010, 18, 147–161. [Google Scholar] [CrossRef]
- Kane, D.A.; Kimmel, C.B. The zebrafish midblastula transition. Development 1993, 119, 447–456. [Google Scholar] [CrossRef]
- Siefert, J.C.; Clowdus, E.A.; Sansam, C.L. Cell cycle control in the early embryonic development of aquatic animal species. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2015, 178, 8–15. [Google Scholar] [CrossRef]
- Walter, J.; Newport, J.W. Regulation of replicon size in Xenopus egg extracts. Science 1997, 275, 993–995. [Google Scholar] [CrossRef]
- Collart, C.; Allen, G.E.; Bradshaw, C.R.; Smith, J.C.; Zegerman, P. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 2013, 341, 893–896. [Google Scholar] [CrossRef]
- Hand, R. Regulation of DNA replication on subchromosomal units of mammalian cells. J. Cell Biol. 1975, 64, 89–97. [Google Scholar] [CrossRef]
- Zhu, J.; Newlon, C.S.; Huberman, J.A. Localization of a DNA replication origin and termination zone on chromosome III of Saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 4733–4741. [Google Scholar] [CrossRef]
- Hyrien, O.; Mechali, M. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J. 1993, 12, 4511–4520. [Google Scholar] [CrossRef]
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208. [Google Scholar] [CrossRef]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737. [Google Scholar] [CrossRef]
- Zhao, P.A.; Sasaki, T.; Gilbert, D.M. High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 2020, 21, 76. [Google Scholar] [CrossRef]
- Miotto, B.; Ji, Z.; Struhl, K. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. USA 2016, 113, E4810–E4819. [Google Scholar] [CrossRef]
- Conti, C.; Sacca, B.; Herrick, J.; Lalou, C.; Pommier, Y.; Bensimon, A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 2007, 18, 3059–3067. [Google Scholar] [CrossRef]
- Brewer, B.J.; Fangman, W.L. A replication fork barrier at the 3’ end of yeast ribosomal RNA genes. Cell 1988, 55, 637–643. [Google Scholar] [CrossRef]
- Labib, K.; Hodgson, B. Replication fork barriers: Pausing for a break or stalling for time? EMBO Rep. 2007, 8, 346–353. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Sedlackova, H.; Rask, M.B.; Gupta, R.; Choudhary, C.; Somyajit, K.; Lukas, J. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature 2020, 587, 297–302. [Google Scholar] [CrossRef]
- Hennion, M.; Arbona, J.M.; Lacroix, L.; Cruaud, C.; Theulot, B.; Tallec, B.L.; Proux, F.; Wu, X.; Novikova, E.; Engelen, S.; et al. FORK-seq: Replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. Genome Biol. 2020, 21, 125. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, X.; d’Aubenton-Carafa, Y.; Thermes, C.; Chen, C.L. OKseqHMM: A genome-wide replication fork directionality analysis toolkit. Nucleic Acids Res. 2023, 51, e22. [Google Scholar] [CrossRef]
- Dewar, J.M.; Budzowska, M.; Walter, J.C. The mechanism of DNA replication termination in vertebrates. Nature 2015, 525, 345–350. [Google Scholar] [CrossRef]
- Heintzman, D.R.; Campos, L.V.; Byl, J.A.W.; Osheroff, N.; Dewar, J.M. Topoisomerase II Is Crucial for Fork Convergence during Vertebrate Replication Termination. Cell Rep. 2019, 29, 422–436.e5. [Google Scholar] [CrossRef]
- Sundin, O.; Varshavsky, A. Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell 1980, 21, 103–114. [Google Scholar] [CrossRef]
- Sundin, O.; Varshavsky, A. Arrest of segregation leads to accumulation of highly intertwined catenated dimers: Dissection of the final stages of SV40 DNA replication. Cell 1981, 25, 659–669. [Google Scholar] [CrossRef]
- Ishimi, Y.; Sugasawa, K.; Hanaoka, F.; Eki, T.; Hurwitz, J. Topoisomerase II plays an essential role as a swivelase in the late stage of SV40 chromosome replication in vitro. J. Biol. Chem. 1992, 267, 462–466. [Google Scholar] [CrossRef]
- Postow, L.; Crisona, N.J.; Peter, B.J.; Hardy, C.D.; Cozzarelli, N.R. Topological challenges to DNA replication: Conformations at the fork. Proc. Natl. Acad. Sci. USA 2001, 98, 8219–8226. [Google Scholar] [CrossRef]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The Causes and Consequences of Topological Stress during DNA Replication. Genes 2016, 7, 134. [Google Scholar] [CrossRef]
- Schalbetter, S.A.; Mansoubi, S.; Chambers, A.L.; Downs, J.A.; Baxter, J. Fork rotation and DNA precatenation are restricted during DNA replication to prevent chromosomal instability. Proc. Natl. Acad. Sci. USA 2015, 112, E4565–E4570. [Google Scholar] [CrossRef]
- Baxter, J.; Diffley, J.F. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol. Cell 2008, 30, 790–802. [Google Scholar] [CrossRef]
- Baxter, J.; Sen, N.; Martinez, V.L.; De Carandini, M.E.; Schvartzman, J.B.; Diffley, J.F.; Aragon, L. Positive supercoiling of mitotic DNA drives decatenation by topoisomerase II in eukaryotes. Science 2011, 331, 1328–1332. [Google Scholar] [CrossRef]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and mechanism of DNA topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef]
- Hiasa, H.; Marians, K.J. Two distinct modes of strand unlinking during theta-type DNA replication. J. Biol. Chem. 1996, 271, 21529–21535. [Google Scholar] [CrossRef]
- Deegan, T.D.; Baxter, J.; Ortiz Bazan, M.A.; Yeeles, J.T.P.; Labib, K.P.M. Pif1-Family Helicases Support Fork Convergence during DNA Replication Termination in Eukaryotes. Mol. Cell 2019, 74, 231–244 e239. [Google Scholar] [CrossRef]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 2010, 39, 595–605. [Google Scholar] [CrossRef]
- Campos, L.V.; Van Ravenstein, S.X.; Vontalge, E.J.; Greer, B.H.; Heintzman, D.R.; Kavlashvili, T.; McDonald, W.H.; Rose, K.L.; Eichman, B.F.; Dewar, J.M. RTEL1 and MCM10 overcome topological stress during vertebrate replication termination. Cell Rep. 2023, 42, 112109. [Google Scholar] [CrossRef]
- Choudhary, R.; Niska-Blakie, J.; Adhil, M.; Liberi, G.; Achar, Y.J.; Giannattasio, M.; Foiani, M. Sen1 and Rrm3 ensure permissive topological conditions for replication termination. Cell Rep. 2023, 42, 112747. [Google Scholar] [CrossRef]
- Low, E.; Chistol, G.; Zaher, M.S.; Kochenova, O.V.; Walter, J.C. The DNA replication fork suppresses CMG unloading from chromatin before termination. Genes Dev. 2020, 34, 1534–1545. [Google Scholar] [CrossRef]
- Georgescu, R.; Yuan, Z.; Bai, L.; de Luna Almeida Santos, R.; Sun, J.; Zhang, D.; Yurieva, O.; Li, H.; O’Donnell, M.E. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. USA 2017, 114, E697–E706. [Google Scholar] [CrossRef]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef]
- Kang, Y.H.; Galal, W.C.; Farina, A.; Tappin, I.; Hurwitz, J. Properties of the human Cdc45/Mcm2-7/GINS helicase complex and its action with DNA polymerase epsilon in rolling circle DNA synthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 6042–6047. [Google Scholar] [CrossRef]
- Santamaria, D.; de la Cueva, G.; Martinez-Robles, M.L.; Krimer, D.B.; Hernandez, P.; Schvartzman, J.B. DnaB helicase is unable to dissociate RNA-DNA hybrids. Its implication in the polar pausing of replication forks at ColE1 origins. J. Biol. Chem. 1998, 273, 33386–33396. [Google Scholar] [CrossRef]
- Bruning, J.G.; Marians, K.J. Replisome bypass of transcription complexes and R-loops. Nucleic Acids Res. 2020, 48, 10353–10367. [Google Scholar] [CrossRef]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef]
- Segawa, M.; Sugano, S.; Yamaguchi, N. Association of simian virus 40 T antigen with replicating nucleoprotein complexes of simian virus 40. J. Virol. 1980, 35, 320–330. [Google Scholar] [CrossRef]
- Chen, M.C.; Birkenmeier, E.; Salzman, N.P. Simian virus 40 DNA replication: Characterization of gaps in the termination region. J. Virol. 1976, 17, 614–621. [Google Scholar] [CrossRef]
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Tack, L.C.; DePamphilis, M.L. Analysis of simian virus 40 chromosome-T-antigen complexes: T-antigen is preferentially associated with early replicating DNA intermediates. J. Virol. 1983, 48, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346, 1253596. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Mukherjee, P.; Tatham, M.H.; Hay, R.; Labib, K. Ufd1-Npl4 Recruit Cdc48 for Disassembly of Ubiquitylated CMG Helicase at the End of Chromosome Replication. Cell Rep. 2017, 18, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Mimura, S.; Komata, M.; Kishi, T.; Shirahige, K.; Kamura, T. SCF(Dia2) regulates DNA replication forks during S-phase in budding yeast. EMBO J. 2009, 28, 3693–3705. [Google Scholar] [CrossRef]
- Jang, S.M.; Redon, C.E.; Thakur, B.L.; Bahta, M.K.; Aladjem, M.I. Regulation of cell cycle drivers by Cullin-RING ubiquitin ligases. Exp. Mol. Med. 2020, 52, 1637–1651. [Google Scholar] [CrossRef]
- Morohashi, H.; Maculins, T.; Labib, K. The amino-terminal TPR domain of Dia2 tethers SCF(Dia2) to the replisome progression complex. Curr. Biol. 2009, 19, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Maculins, T.; Nkosi, P.J.; Nishikawa, H.; Labib, K. Tethering of SCF(Dia2) to the Replisome Promotes Efficient Ubiquitylation and Disassembly of the CMG Helicase. Curr. Biol. 2015, 25, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Moreno, S.P.; Knebel, A.; Johnson, C.; Hastie, C.J.; Gartner, A.; Gambus, A.; Labib, K. CUL-2(LRR-1) and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol. 2017, 19, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Low, E.; Mann, M.; Raschle, M.; Walter, J.C. CRL2(Lrr1) promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Fujisawa, R.; Ainsworth, J.; Nishimura, K.; Lie, A.L.M.; Lacaud, G.; Labib, K.P. CUL2(LRR1), TRAIP and p97 control CMG helicase disassembly in the mammalian cell cycle. EMBO Rep. 2021, 22, e52164. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Koberlin, M.S.; Ratnayeke, N.; Liu, C.; Deshpande, M.; Gerhardt, J.; Meyer, T. LRR1-mediated replisome disassembly promotes DNA replication by recycling replisome components. J. Cell Biol. 2021, 220, e202009147. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Ruiz, J.H.; Scaramuzza, S.; Nath, S.; Henklewska, M.; Natsume, T.; Romero, F.; Kanemaki, M.T.; Gambus, A. Characterising replisome disassembly in human cells. bioRxiv 2022, 2022.2007.2012.499744. [Google Scholar] [CrossRef]
- Okumura, F.; Matsuzaki, M.; Nakatsukasa, K.; Kamura, T. The Role of Elongin BC-Containing Ubiquitin Ligases. Front. Oncol. 2012, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Yang, H. The structure and regulation of Cullin 2 based E3 ubiquitin ligases and their biological functions. Cell Div. 2016, 11, 7. [Google Scholar] [CrossRef]
- Mukherjee, P.P.; Labib, K.P.M. In Vitro Reconstitution Defines the Minimal Requirements for Cdc48-Dependent Disassembly of the CMG Helicase in Budding Yeast. Cell Rep. 2019, 28, 2777–2783.e4. [Google Scholar] [CrossRef]
- Xia, Y.; Fujisawa, R.; Deegan, T.D.; Sonneville, R.; Labib, K.P.M. TIMELESS-TIPIN and UBXN-3 promote replisome disassembly during DNA replication termination in Caenorhabditis elegans. EMBO J. 2021, 40, e108053. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Ainsworth, J.; Polo Rivera, C.; Macartney, T.; Labib, K.P.M. Reconstitution of human CMG helicase ubiquitylation by CUL2LRR1 and multiple E2 enzymes. Biochem. J. 2021, 478, 2825–2842. [Google Scholar] [CrossRef]
- Jenkyn-Bedford, M.; Jones, M.L.; Baris, Y.; Labib, K.P.M.; Cannone, G.; Yeeles, J.T.P.; Deegan, T.D. A conserved mechanism for regulating replisome disassembly in eukaryotes. Nature 2021, 600, 743–747. [Google Scholar] [CrossRef]
- Zhou, H.; Zaher, M.S.; Walter, J.C.; Brown, A. Structure of CRL2Lrr1, the E3 ubiquitin ligase that promotes DNA replication termination in vertebrates. Nucleic Acids Res. 2021, 49, 13194–13206. [Google Scholar] [CrossRef] [PubMed]
- Deegan, T.D.; Mukherjee, P.P.; Fujisawa, R.; Polo Rivera, C.; Labib, K. CMG helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. Elife 2020, 9, e60371. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Vrtis, K.B.; Dewar, J.M.; Chistol, G.; Wu, R.A.; Graham, T.G.W.; Walter, J.C. Single-strand DNA breaks cause replisome disassembly. Mol. Cell 2021, 81, 1309–1318.e6. [Google Scholar] [CrossRef]
- Eickhoff, P.; Kose, H.B.; Martino, F.; Petojevic, T.; Abid Ali, F.; Locke, J.; Tamberg, N.; Nans, A.; Berger, J.M.; Botchan, M.R.; et al. Molecular Basis for ATP-Hydrolysis-Driven DNA Translocation by the CMG Helicase of the Eukaryotic Replisome. Cell Rep. 2019, 28, 2673–2688.e8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Georgescu, R.; Bai, L.; Zhang, D.; Li, H.; O’Donnell, M.E. DNA unwinding mechanism of a eukaryotic replicative CMG helicase. Nat. Commun. 2020, 11, 688. [Google Scholar] [CrossRef] [PubMed]
- Baretic, D.; Jenkyn-Bedford, M.; Aria, V.; Cannone, G.; Skehel, M.; Yeeles, J.T.P. Cryo-EM Structure of the Fork Protection Complex Bound to CMG at a Replication Fork. Mol. Cell 2020, 78, 926–940.e13. [Google Scholar] [CrossRef]
- Bodnar, N.; Rapoport, T. Toward an understanding of the Cdc48/p97 ATPase. F1000Res 2017, 6, 1318. [Google Scholar] [CrossRef]
- Davies, J.M.; Brunger, A.T.; Weis, W.I. Improved structures of full-length p97, an AAA ATPase: Implications for mechanisms of nucleotide-dependent conformational change. Structure 2008, 16, 715–726. [Google Scholar] [CrossRef]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Fujisawa, R.; Polo Rivera, C.; Labib, K.P.M. Multiple UBX proteins reduce the ubiquitin threshold of the mammalian p97-UFD1-NPL4 unfoldase. Elife 2022, 11, e76763. [Google Scholar] [CrossRef]
- Kochenova, O.V.; Mukkavalli, S.; Raman, M.; Walter, J.C. Cooperative assembly of p97 complexes involved in replication termination. Nat. Commun. 2022, 13, 6591. [Google Scholar] [CrossRef]
- Tarcan, Z.; Poovathumkadavil, D.; Skagia, A.; Gambus, A. The p97 segregase cofactor Ubxn7 facilitates replisome disassembly during S-phase. J. Biol. Chem. 2022, 298, 102234. [Google Scholar] [CrossRef]
- Alexandru, G.; Graumann, J.; Smith, G.T.; Kolawa, N.J.; Fang, R.; Deshaies, R.J. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 2008, 134, 804–816. [Google Scholar] [CrossRef]
- Bandau, S.; Knebel, A.; Gage, Z.O.; Wood, N.T.; Alexandru, G. UBXN7 docks on neddylated cullin complexes using its UIM motif and causes HIF1alpha accumulation. BMC Biol. 2012, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Shaw, A.; Zhang, X.; Kondo, H.; Lally, J.; Freemont, P.S.; Matthews, S. Solution structure and interaction surface of the C-terminal domain from p47: A major p97-cofactor involved in SNARE disassembly. J. Mol. Biol. 2001, 311, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, N.O.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735.e9. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Li, H.; Peterle, D.; Paulo, J.A.; Ficarro, S.B.; Wales, T.E.; Marto, J.A.; Gygi, S.P.; Engen, J.R.; Rapoport, T.A. Translocation of polyubiquitinated protein substrates by the hexameric Cdc48 ATPase. Mol. Cell 2022, 82, 570–584.e8. [Google Scholar] [CrossRef] [PubMed]
- Ernst, R.; Mueller, B.; Ploegh, H.L.; Schlieker, C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell 2009, 36, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Priego Moreno, S.; Jones, R.M.; Poovathumkadavil, D.; Scaramuzza, S.; Gambus, A. Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci. Alliance 2019, 2, e201900390. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wu, R.A.; Sonneville, R.; Kochenova, O.V.; Labib, K.; Pellman, D.; Walter, J.C. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol. Cell 2019, 73, 915–929.e6. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife 2019, 8, e48686. [Google Scholar] [CrossRef]
- Lange, S.M.; McFarland, M.R.; Lamoliatte, F.; Kwaśna, D.; Shen, L.; Wallace, I.; Cole, I.; Armstrong, L.A.; Knebel, A.; Johnson, C.; et al. Comprehensive approach to study branched ubiquitin chains reveals roles for K48-K63 branches in VCP/p97-related processes. bioRxiv 2023, 2023.2001.2010.523363. [Google Scholar] [CrossRef]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef]
- Wu, W.; Barwacz, S.A.; Bhowmick, R.; Lundgaard, K.; Goncalves Dinis, M.M.; Clausen, M.; Kanemaki, M.T.; Liu, Y. Mitotic DNA synthesis in response to replication stress requires the sequential action of DNA polymerases zeta and delta in human cells. Nat. Commun. 2023, 14, 706. [Google Scholar] [CrossRef]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 2019, 567, 267–272. [Google Scholar] [CrossRef]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.C.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e14. [Google Scholar] [CrossRef]
- Long, D.T.; Joukov, V.; Budzowska, M.; Walter, J.C. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 2014, 56, 174–185. [Google Scholar] [CrossRef]
- Salas-Lloret, D.; García-Rodríguez, N.; Giebel, L.; Ru, A.D.; Veelen, P.A.V.; Huertas, P.; Vertegaal, A.C.O.; González-Prieto, R. BRCA1/BARD1 ubiquitinates PCNA in unperturbed conditions to promote replication fork stability and continuous DNA synthesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Harley, M.E.; Murina, O.; Leitch, A.; Higgs, M.R.; Bicknell, L.S.; Yigit, G.; Blackford, A.N.; Zlatanou, A.; Mackenzie, K.J.; Reddy, K.; et al. TRAIP promotes DNA damage response during genome replication and is mutated in primordial dwarfism. Nat. Genet. 2016, 48, 36–43. [Google Scholar] [CrossRef]
- Hoffmann, S.; Smedegaard, S.; Nakamura, K.; Mortuza, G.B.; Raschle, M.; de Opakua, A.I.; Oka, Y.; Feng, Y.; Blanco, F.J.; Mann, M.; et al. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J. Cell Biol. 2016, 212, 63–75. [Google Scholar] [CrossRef]
- Feng, W.; Guo, Y.; Huang, J.; Deng, Y.; Zang, J.; Huen, M.S. TRAIP regulates replication fork recovery and progression via PCNA. Cell Discov. 2016, 2, 16016. [Google Scholar] [CrossRef]
- Scaramuzza, S.; Jones, R.M.; Sadurni, M.M.; Reynolds-Winczura, A.; Poovathumkadavil, D.; Farrell, A.; Natsume, T.; Rojas, P.; Cuesta, C.F.; Kanemaki, M.T.; et al. TRAIP resolves DNA replication-transcription conflicts during the S-phase of unperturbed cells. Nat. Commun. 2023, 14, 5071. [Google Scholar] [CrossRef]
- Larsen, N.B.; Gao, A.O.; Sparks, J.L.; Gallina, I.; Wu, R.A.; Mann, M.; Raschle, M.; Walter, J.C.; Duxin, J.P. Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 2019, 73, 574–588.e7. [Google Scholar] [CrossRef]
- Park, E.S.; Choi, S.; Kim, J.M.; Jeong, Y.; Choe, J.; Park, C.S.; Choi, Y.; Rho, J. Early embryonic lethality caused by targeted disruption of the TRAF-interacting protein (TRIP) gene. Biochem. Biophys. Res. Commun. 2007, 363, 971–977. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Mouysset, J.; Deichsel, A.; Moser, S.; Hoege, C.; Hyman, A.A.; Gartner, A.; Hoppe, T. Cell cycle progression requires the CDC-48UFD-1/NPL-4 complex for efficient DNA replication. Proc. Natl. Acad. Sci. USA 2008, 105, 12879–12884. [Google Scholar] [CrossRef]
- Blake, D.; Luke, B.; Kanellis, P.; Jorgensen, P.; Goh, T.; Penfold, S.; Breitkreutz, B.J.; Durocher, D.; Peter, M.; Tyers, M. The F-box protein Dia2 overcomes replication impedance to promote genome stability in Saccharomyces cerevisiae. Genetics 2006, 174, 1709–1727. [Google Scholar] [CrossRef]
- Ohya, Y.; Sese, J.; Yukawa, M.; Sano, F.; Nakatani, Y.; Saito, T.L.; Saka, A.; Fukuda, T.; Ishihara, S.; Oka, S.; et al. High-dimensional and large-scale phenotyping of yeast mutants. Proc. Natl. Acad. Sci. USA 2005, 102, 19015–19020. [Google Scholar] [CrossRef]
- Chapard, C.; Meraldi, P.; Gleich, T.; Bachmann, D.; Hohl, D.; Huber, M. TRAIP is a regulator of the spindle assembly checkpoint. J. Cell Sci. 2014, 127, 5149–5156. [Google Scholar] [CrossRef]
- Park, I.S.; Jo, K.S.; Won, H.S.; Kim, H. Dimerization of TRAF-interacting protein (TRAIP) regulates the mitotic progression. Biochem. Biophys. Res. Commun. 2015, 463, 864–869. [Google Scholar] [CrossRef]
- Merlet, J.; Burger, J.; Tavernier, N.; Richaudeau, B.; Gomes, J.E.; Pintard, L. The CRL2LRR-1 ubiquitin ligase regulates cell cycle progression during C. elegans development. Development 2010, 137, 3857–3866. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, R.M.; Reynolds-Winczura, A.; Gambus, A. A Decade of Discovery—Eukaryotic Replisome Disassembly at Replication Termination. Biology 2024, 13, 233. https://doi.org/10.3390/biology13040233
Jones RM, Reynolds-Winczura A, Gambus A. A Decade of Discovery—Eukaryotic Replisome Disassembly at Replication Termination. Biology. 2024; 13(4):233. https://doi.org/10.3390/biology13040233
Chicago/Turabian StyleJones, Rebecca M., Alicja Reynolds-Winczura, and Agnieszka Gambus. 2024. "A Decade of Discovery—Eukaryotic Replisome Disassembly at Replication Termination" Biology 13, no. 4: 233. https://doi.org/10.3390/biology13040233