


Transcriptome Responses to Different Environments in Intertidal Zones in the Peanut Worm Sipunculus nudus
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Sample Collection
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. PacBio Iso-Seq Library Preparation and Sequencing
2.4. Illumina RNA-Seq and De Novo Assembly
2.5. Transcriptome Assembly, Annotation, and Functional Enrichment
2.6. Differentially Expressed Genes and Enrichment Analysis
2.7. Quantitative Real-Time PCR Analysis
3. Results
3.1. SMRT Sequencing, Illumina HiSeq Sequencing, and Assembly
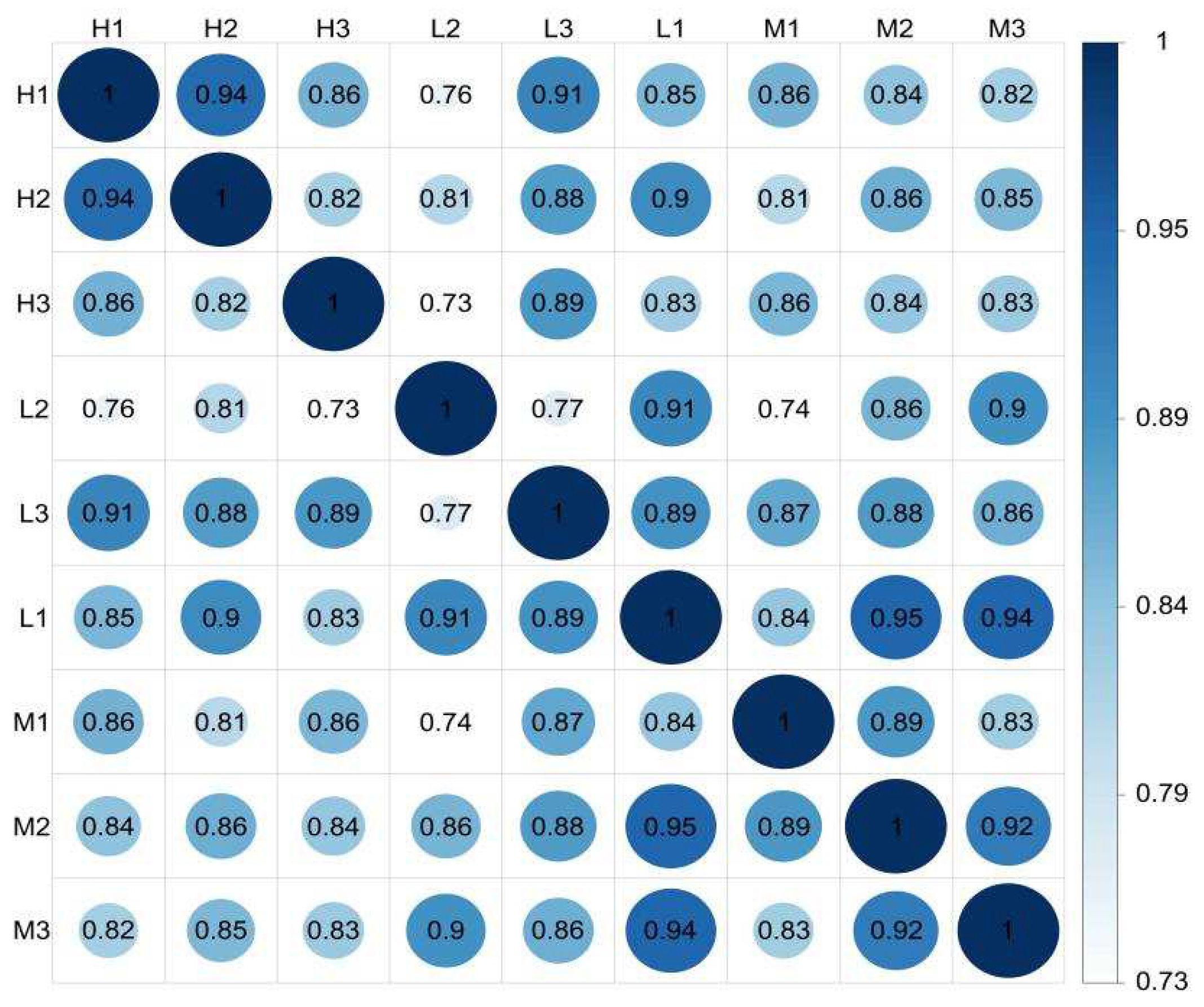
3.2. Expression Levels of Unigenes
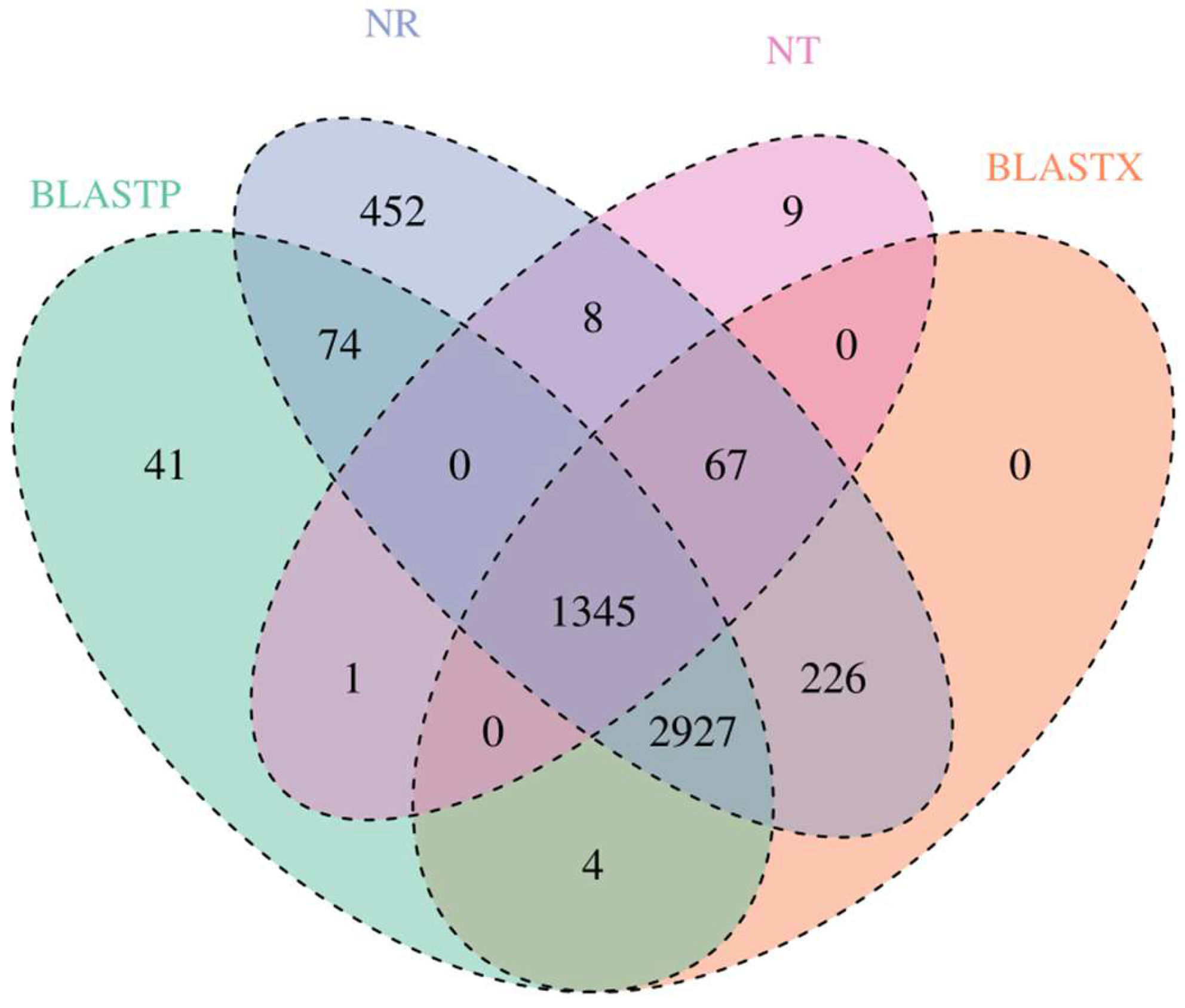
3.3. Gene Annotation and Function Classification
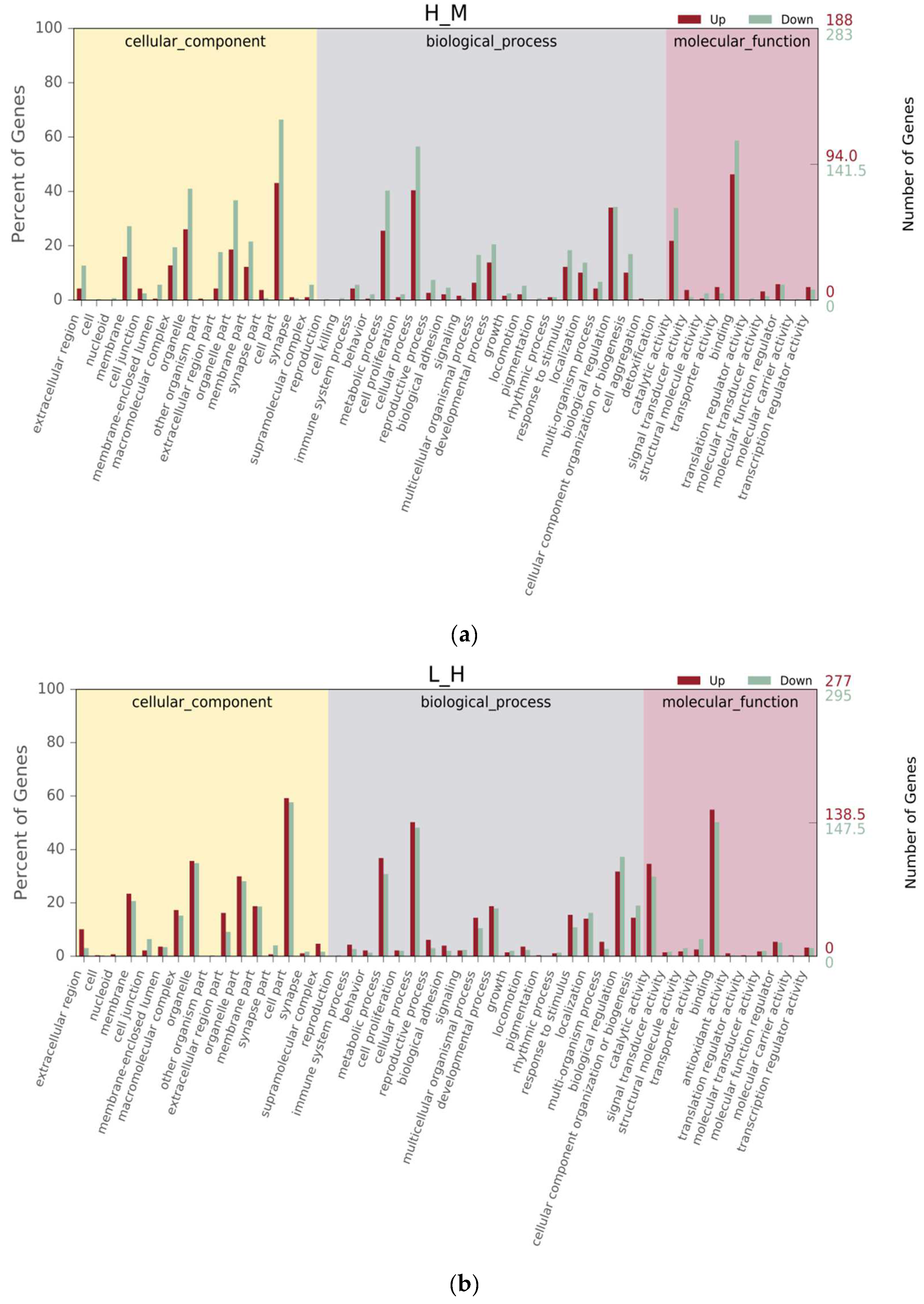
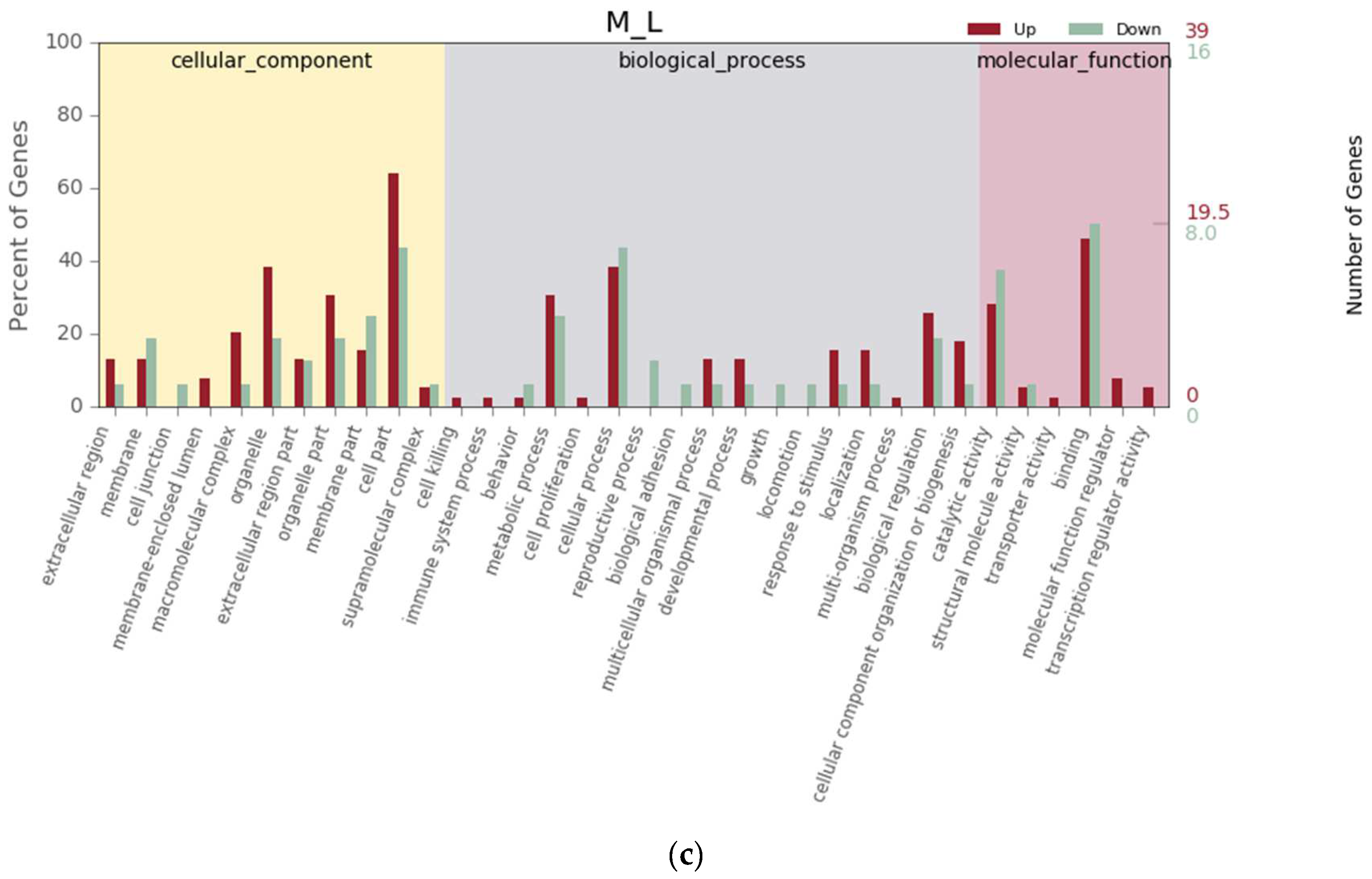
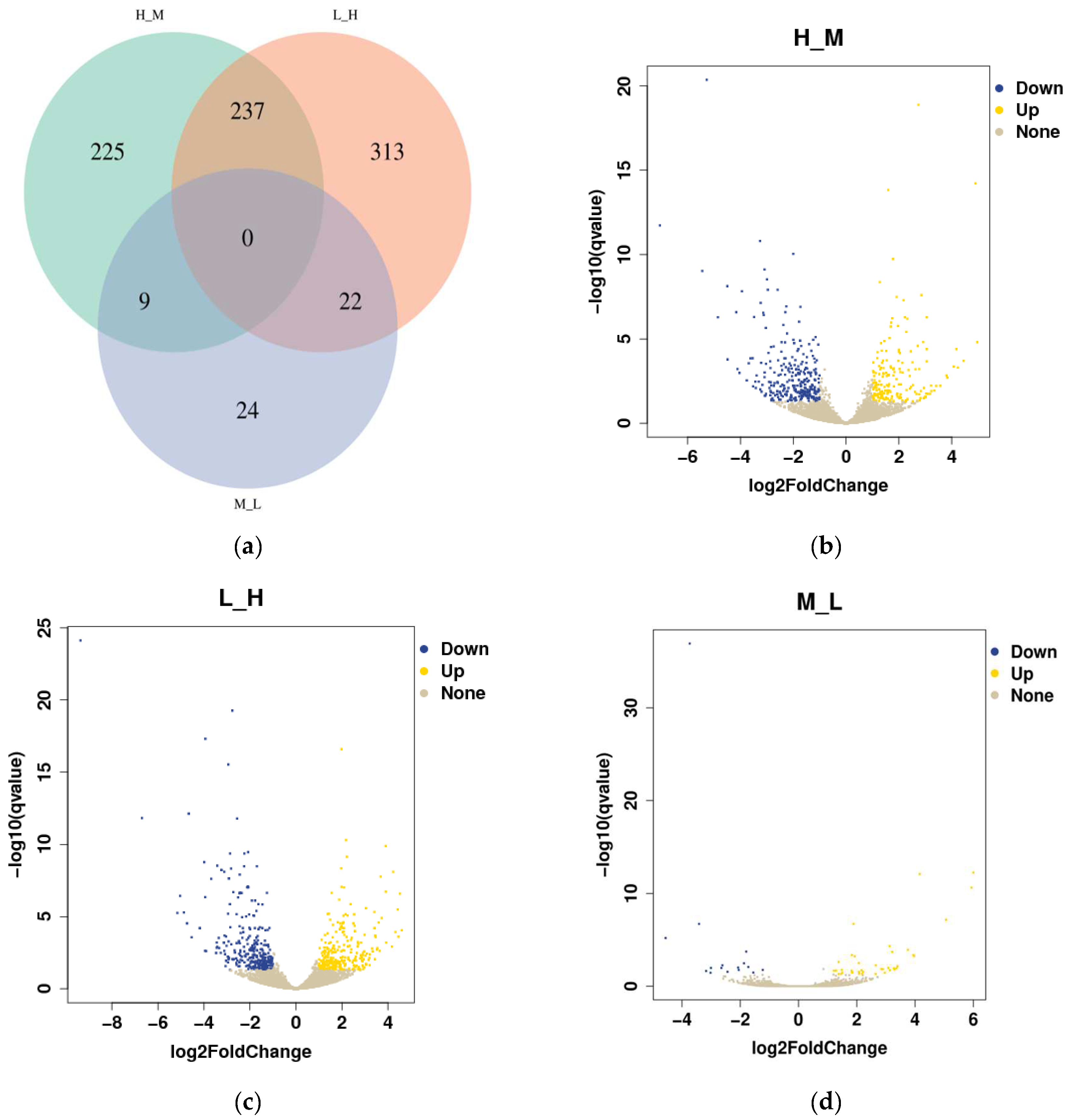
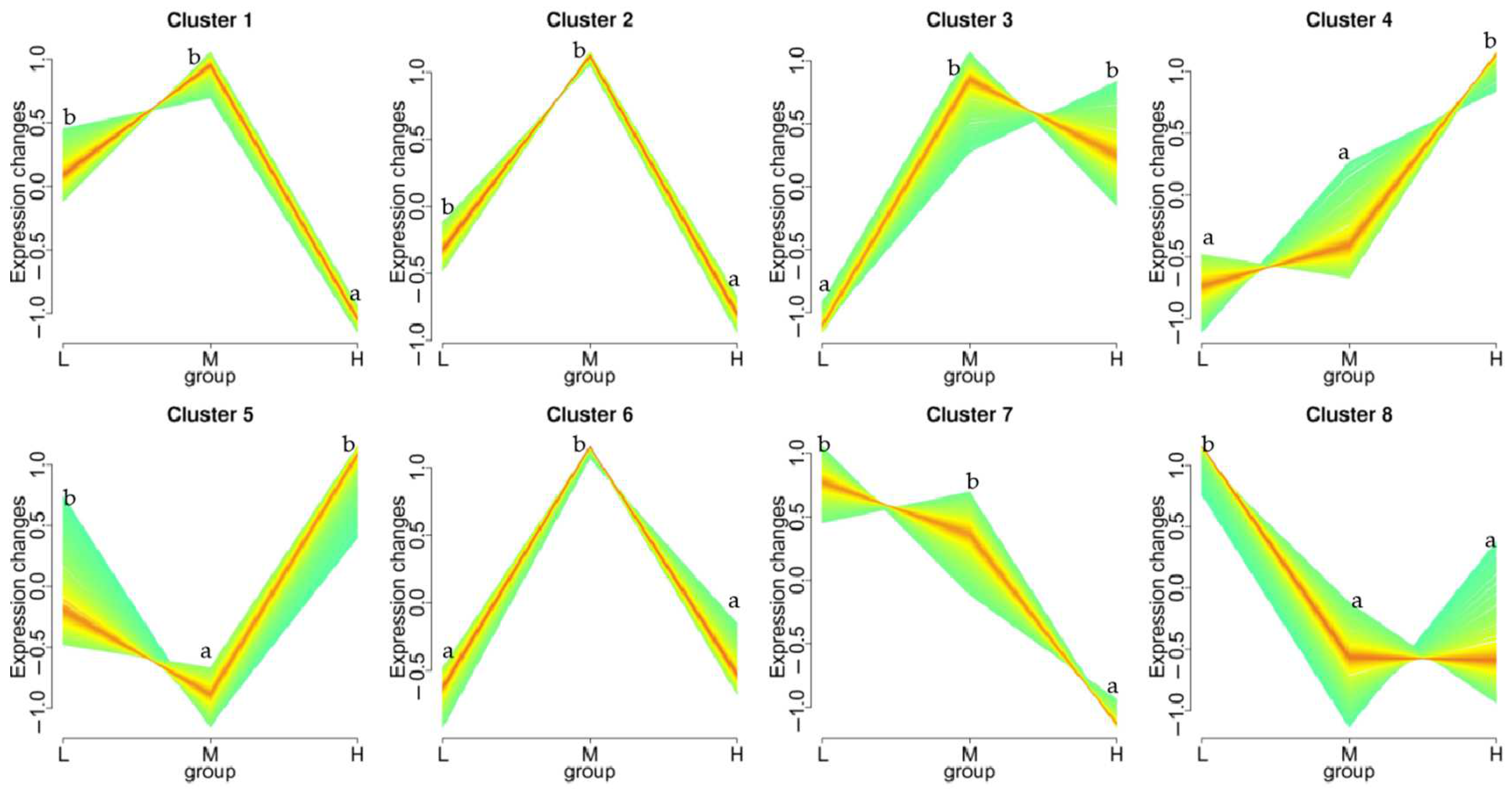
3.4. Differential Gene Expression Analysis
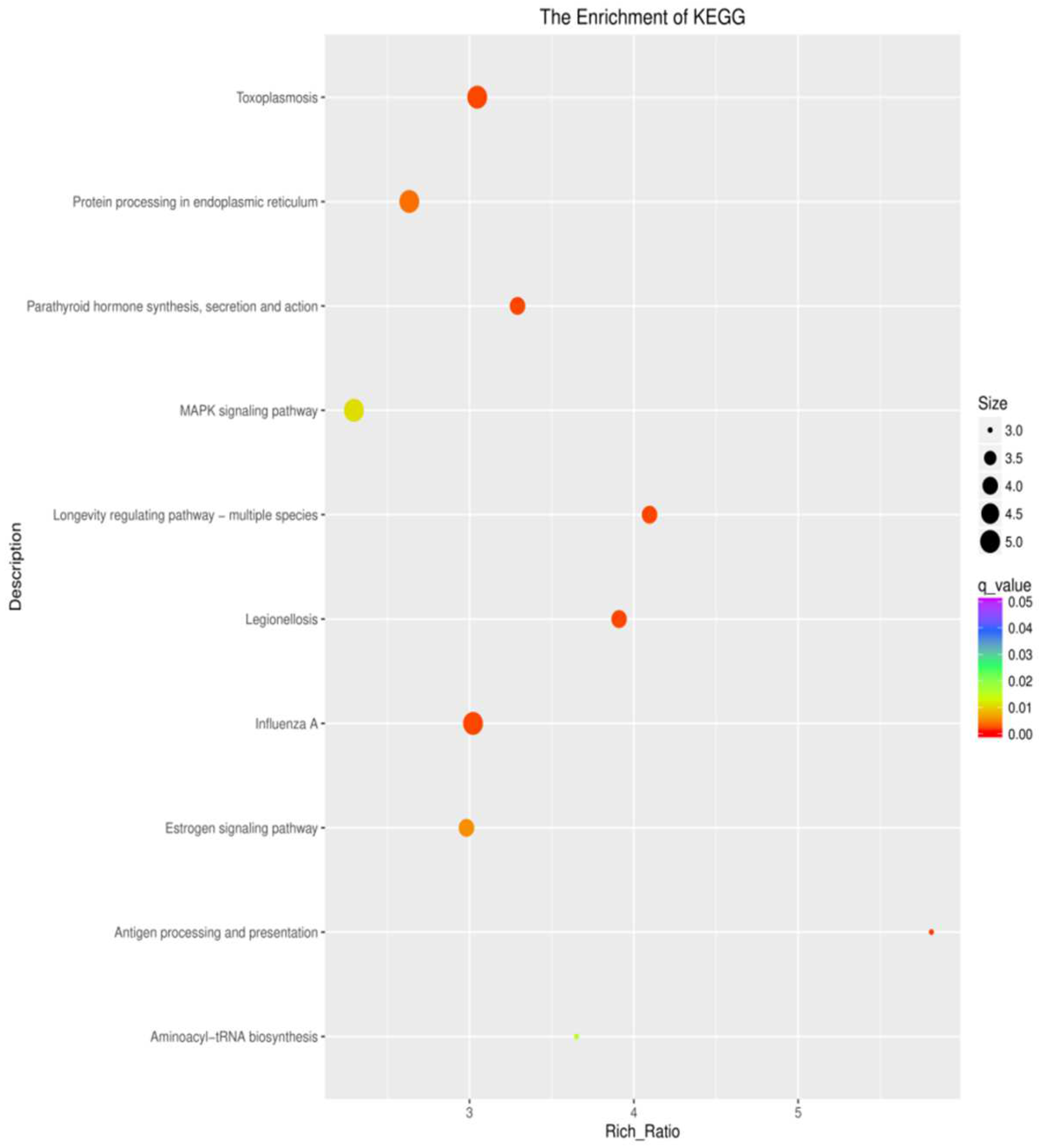
3.5. KEGG Pathway Enrichment Analysis of the Genes
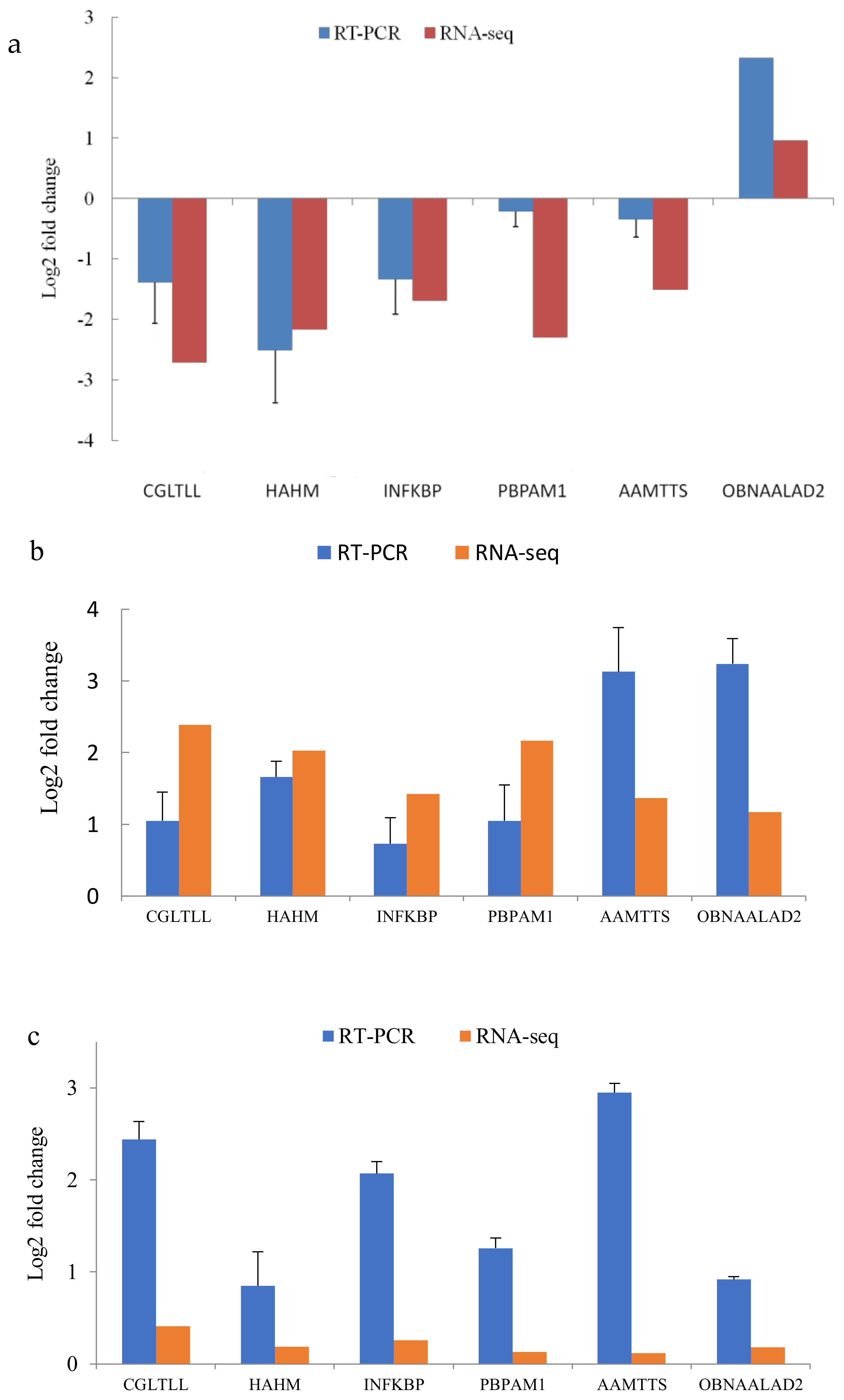
3.6. Validation of the Transcriptome with qRT–PCR
4. Discussion
4.1. Transcriptomic Characteristics of Sipunculus nudus in Tidal Flats
4.2. Analysis of Differentially Expressed Genes in KEGG Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.W.; Liao, M.L.; Meng, X.L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 6, 1274–1279. [Google Scholar] [CrossRef]
- Fields, P.A.; Eraso, A. A year in the salt marsh: Seasonal changes in gill protein expression in the temperate intertidal mussel Geukensia demissa. Mar. Environ. Res. 2020, 161, 105088. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.L.; Dong, Y.W.; Somero, G.N. Thermal adaptation of mRNA secondary structure: Stability versus lability. Proc. Natl. Acad. Sci. USA 2021, 45, e2113324118. [Google Scholar] [CrossRef]
- Nie, H.T.; Jiang, K.Y.; Li, N.; Li, D.D.; Yan, X.W. Transcriptomic analysis of Ruditapes philippinarum under aerial exposure and reimmersion reveals genes involved in stress response and recovery capacity of the Manila clam. Aquaculture 2020, 524, 735271. [Google Scholar] [CrossRef]
- Li, Q.; Gu, L.L.; Song, J.X.; Li, C.J.; Zhang, Y.H.; Pang, Y.Z.; Zhang, B. Physiological and transcriptome analyses highlight multiple pathways involved in drought stress in Medicago falcata. PLoS ONE 2022, 17, e0266542. [Google Scholar] [CrossRef]
- Nancollas, S.J.; Todgham, A.E. The influence of stochastic temperature fluctuations in shaping the physiological performance of the California mussel, Mytilus californianus. J. Exp. Biol. 2022, 14, 243729. [Google Scholar] [CrossRef]
- Rivera, H.E.; Aichelman, H.E.; Fifer, J.E.; Kriefall, N.G.; Wuitchik, D.M.; Wuitchik, S.J.S.; Davies, S.W.A. A framework for understanding gene expression plasticity and its influence on stress tolerance. Mol. Ecol. 2022, 6, 1381–1397. [Google Scholar] [CrossRef]
- Liu, Y.L.; Li, L.; Huang, B.Y.; Wang, W.; Zhang, G.F. RNAi based transcriptome suggests genes potentially regulated by HSF1 in the Pacific oyster Crassostrea gigas under thermal stress. BMC Genom. 2019, 20, 639. [Google Scholar] [CrossRef]
- Evans, T.G.; Chan, F.; Menge, B.A.; Hofmann, G.E. Transcriptomic responses to ocean acidification in larval sea urchins from a naturally variable pH environment. Mol. Ecol. 2013, 22, 1609–1625. [Google Scholar] [CrossRef]
- Pespeni, M.H.; Barney, B.T.; Palumbi, S.R. Differences in the regulation of growth and biomineralization genes revealed through long-termcommon-garden acclimation and experimentalgenomics in the purple sea urchin. Evolution 2013, 67, 1901–1914. [Google Scholar] [CrossRef]
- Gracey, A.Y.; Chaney, M.L.; Boomhower, J.P.; Tyburczy, W.R.; Connor, K.; Somero, G.N. Rhythms of gene expression in a fluctuating intertidal environment. Curr. Biol. 2008, 18, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.R.; Jiang, D.W.; Jia, F.X.; Chen, M.H. Study on anti-senescence effect of Sipunculus nudus preparation. Chin. J. Mar. Drugs 2004, 1, 30–32, (In Chinese with English abstract). [Google Scholar]
- Dong, L.F.; Zhang, Q.; Tong, T.; Xu, M.Z.; Chen, J.H. Amino acid composition of peanut worm Sipunculus nudus at different growth stages. South China Fish. Sci. 2012, 8, 60–65, (In Chinese with English abstract). [Google Scholar]
- Jiang, D.W.; Shen, X.R.; Jia, F.X.; Chu, Z.Y.; Li, S.L. Nutrient analysis and immune regulation study on extract of Sipunculidae. Chin. J. Biochem. Pharm. 2004, 25, 96–97, (In Chinese with English abstract). [Google Scholar]
- Li, J.W.; Guo, Y.J.; Hu, R.P.; Chen, S.W.; Xie, X.Y.; Zhu, C.B.; Li, T. Comparative analysis of nutritional composition of Sipunculus nudus Linnaeus in different aquaculture modes. South China Fish. Sci. 2018, 14, 80–87, (In Chinese with English abstract). [Google Scholar]
- Li, J.W.; Xie, X.Y.; Zhu, C.B.; Guo, Y.J.; Chen, S.W. Edible peanut worm (Sipunculus nudus) in the Beibu Gulf: Resource, aquaculture, ecological impact and counterplan. J. Ocean Univ. China Ocean. Coast. Sea Res. 2017, 5, 823–830. [Google Scholar] [CrossRef]
- Mark, A.S.; Monika, K. A deep burrowing sipunculan of ecological and geochemical importance. Deep-Sea Res. Part I 2009, 56, 2057–2064. [Google Scholar] [CrossRef]
- Li, J.W.; Hu, R.P.; Guo, Y.J.; Chen, S.W.; Xie, X.Y.; Qin, J.G.; Ma, Z.H.; Zhu, C.B. Bioturbation of peanut worms Sipunculus nudus on the composition of prokaryotic communities in a tidal flat as revealed by 16S rRNA gene sequences. Microbiol. Open 2019, 8, e802. [Google Scholar] [CrossRef]
- Li, J.W.; Zhu, C.B.; Guo, Y.J.; Xie, X.Y.; Huang, G.Q.; Chen, S.W. Experimental study of bioturbation by Sipunculus nudus in a polyculture system. Aquaculture 2015, 437, 175–181. [Google Scholar] [CrossRef]
- Helmuth, B.; Mieszkowska, N.; Moore, P.; Hawkins, S.J. Living on the edge of two changing worlds: Forecasting the responses of rocky intertidal ecosystems to climate change. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 373–404. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.G.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 4, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 1, 323. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 1, 5. [Google Scholar] [CrossRef]
- Wage, J.; Lerebours, A.; Hardege, J.D.; Rotchell, J.M. Exposure to low pH induces molecular level changes in the marine worm, Platynereis dumerilii. Ecotoxicol. Environ. Saf. 2016, 124, 105–110. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 4, 402–408. [Google Scholar] [CrossRef]
- Li, X.X.; Tan, Y.; Sun, Y.X.; Wang, J.; Dong, Y.W. Microhabitat temperature variation combines with physiological variation to enhance thermal resilience of the intertidal mussel Mytilisepta virgate. Funct. Ecol. 2022, 11, 2497–2507. [Google Scholar] [CrossRef]
- Tan, J.; Wang, X.; Wang, L.; Zhou, X.Q.; Liu, C.L.; Ge, J.L.; Bian, L.; Chen, S.Q. Transcriptomic responses to air exposure stress in coelomocytes of the sea cucumber, Apostichopus japonicus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 42, 100963. [Google Scholar] [CrossRef]
- Wang, P.P.; Wang, J.; Su, Y.Q.; Mao, Y.; Zhang, J.S.; Wu, C.W.; Ke, Q.Z.; Han, K.H.; Zheng, W.Q.; Xu, N.D. Transcriptome analysis of the Larimichthys crocea liver in response to Cryptocaryon irritans. Fish Shellfish Immun. 2016, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Ho, L.P.; Yang, C.H.; Kao, T.Y.; Chou, H.Y.; Pai, T.W. Comparison of grouper infection with two different iridoviruses using transcriptome sequencing and multiple reference species selection. Fish Shellfish Immun. 2017, 71, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Shi, X.L.; Guo, H.Y.; Bai, Y.Z.; Shen, C.C.; Zhang, Y.P.; Wang, Z.F. Comparative transcriptome analysis of the gills of Procambarus clarkii provides novel insights into the immune-related mechanism of copper stress tolerance. Fish Shellfish Immun. 2020, 96, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tian, T.; Xiao, K.; Zeng, Q.K.; Tan, C.; Du, H.J. Pathogenic infection and immune-related gene expression of Chinese sturgeon (Acipenser sinensis) challenged by Citrobacter freundii. Dev. Comp. Immun. 2021, 114, 103872. [Google Scholar] [CrossRef] [PubMed]
- Lyu, L.K.; Wen, H.S.; Li, Y.; Li, J.F.; Zhao, J.; Zhang, S.M. Deep transcriptomic analysis of black rockfish (Sebastes schlegelii) Provides new insights on responses to acute temperature stress. Sci. Rep. 2017, 8, 9113. [Google Scholar] [CrossRef]
- Cao, F.J.; Zhong, R.Z.; Yang, C.Y.; Hao, R.J.; Wang, Q.H.; Liao, Y.S.; Deng, Y.W. Transcriptmic analysis of differentially expressed genes in the larval settlement and metamorphosis of peanut worm Sipunculus nudus. Aquacult. Rep. 2020, 18, 100475. [Google Scholar] [CrossRef]
- Yan, X.W.; Nie, H.T.; Huo, Z.M.; Ding, J.F.; Li, Z.Z.; Yan, L.L.; Jiang, L.W.; Mu, Z.Q.; Wang, H.M.; Meng, X.Y.; et al. Clam genome sequence clarifies the molecular basis of Its benthic adaptation and extraordinary shell color diversity. iScience 2019, 19, 1225–1237. [Google Scholar] [CrossRef]
- Perkins, T.L.; Clements, K.; Baas, J.H.; Jago, C.F.; Jones, D.L.; Malham, S.K.; McDonald, J.E. Sediment composition influences spatial variation in the abundance of human pathogen indicator bacteria within an estuarine environment. PLoS ONE 2014, 11, e112951. [Google Scholar] [CrossRef]
- Bernáldez-Sarabia, J.; Lizárraga-Partida, M.L.; Hernández-López, E.L.; Gasperin-Bulbarela, J.; Licea-Navarro, A.F.; Guerrero, A.; Sánchez-Castrejón, E.; Franco-Moreno, A. Distribution of pathogenic vibrios and Vibrio spp. in the water column and sediment samples from the southern Gulf of Mexico. Mar. Pollut. Bull. 2021, 173B, 113116. [Google Scholar] [CrossRef]
- Hassard, F.; Andrew, A.; Jones, D.; Parsons, L.; Jones, V.; Cox, B.A.; Daldorph, P.; Brett, H.; McDonald, J.E.; Malham, S.K. Physicochemical factors influence the abundance and culturability of human enteric pathogen and fecal indicator organisms in estuarine water and sediment. Front. Microbiol. 2017, 8, 1996. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signaling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Jeong, C.B.; Lee, Y.H.; Cui, Y.H.; Kim, D.H.; Lee, M.C.; Kim, H.S.; Han, J.; Hwang, D.S.; Lee, S.J. Cross-reactivities of mammalian MAPKs antibodies in rotifer and copepod: Application in mechanistic studies in aquatic ecotoxicology. Mar. Pollut. Bull. 2017, 2, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Schumacher, B. Insights from the worm: The C. elegans model for innate immunity. Semin. Immunol. 2014, 4, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Achache, H.; Falk, R.; Lerner, N.; Beatus, T.; Tzur, Y.B. Oocyte aging is controlled by mitogen-activated protein kinase signaling. Aging Cell 2021, 6, e13386. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Soll, D. Aminoacyl-Trna Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Guo, R.; Li, Y.H.; Kang, G.L.; Wu, Y.; Cheng, J.; Jia, J.; Wang, W.X.; Li, Z.H.; Wang, A.Q.; et al. Contribution of upregulated aminoacyl-tRNA biosynthesis to metabolic dysregulation in gastric cancer. J. Gastroenterol. Hepatol. 2021, 11, 3113–3126. [Google Scholar] [CrossRef]
- Wang, R.R.; Qian, J.M.; Ji, D.; Liu, X.Y.; Dong, R.R. Transcriptome Analysis Reveals Effect of Dietary Probiotics on Immune Response Mechanism in Southern Catfish (Silurus meridionalis) in Response to Plesiomonas shigelloides. Animals 2023, 13, 449. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Deng, S.Y.; Zhang, J.Y.; Zhu, G.Q.; Zhou, J.; Ye, W.J.; Wang, Q.W.; Wang, Y.; Zou, B.W.; Zhang, P.; et al. Single nucleotide polymorphisms within NFKBIA are associated with nasopharyngeal carcinoma susceptibility in Chinese Han population. Cytokine 2021, 138, 155356. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Kinzy, T.G. eEF1A: Thinking outside the ribosome. J. Biol. Chem. 2010, 28, 21209–21213. [Google Scholar] [CrossRef]
- Vera, M.; Pani, B.; Griffiths, L.A.; Muchardt, C.; Abbott, C.M.; Singer, R.H.; Nudler, E. The translation elongation factor eEF1A1 couples transcription to translation during heat shock response. eLife 2014, 3, e03164. [Google Scholar] [CrossRef]
- Scaggiante, B.; Dapas, B.; Cristiano, L.; Grassi, G. The Role of the Eef1A Family in Human Cancers. Oncogene Proteins: New Research; Chapter 6; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2008; pp. 177–184. [Google Scholar]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 1, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.N.; Nishio, N.; Ito, S.; Tanaka, Y. Growth arrest and DNA damage-inducible protein (GADD34) enhanced liver inflammation and tumorigenesis in a diethylnitrosamine (DEN)-treated murine model. Cancer Immun. Immunother. 2015, 6, 777–789. [Google Scholar] [CrossRef]
- Li, J.W.; Chen, S.W.; Wu, P.; Zhu, C.B.; Hu, R.P.; Li, T.; Guo, Y.J. Insights into the relationship between intestinal microbiota of the aquaculture worm Sipunculus nudus and surrounding sediments. Fishes 2023, 8, 32. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) Shaping the outcome of MAP kinase signaling. FEBS J. 2013, 2, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.J.; Rose, A.L.; Lu, Z.Y. Reactive oxygen species in the world ocean and their impacts on marine ecosystems. Redox Biol. 2022, 52, 102285. [Google Scholar] [CrossRef]
- Lornejad-Schäfer, M.R.; Schäfer, C.; Schöffl, H.; Frank, J. Cytoprotective role of mitogen-activated protein kinase phosphatase-1 in light-damaged human retinal pigment epithelial cells. Photochem. Photobiol. 2009, 3, 834–842. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TN (%) | TOC (%) | TS (%) | Salinity (‰) | Body Weight (g) | |
|---|---|---|---|---|---|
| H | 0.002 ± 0.001 | 0.0586 ± 0.005 | 0.600 ± 0.211 | 24 ± 0.3 | 9.8 ± 1.4 |
| M | 0.004 ± 0.001 | 0.099 ± 0.0028 | 0.335 ± 0.073 | 23 ± 0.2 | 8.6 ± 1.3 |
| L | 0.011 ± 0.001 | 0.2559 ± 0.049 | 0.337 ± 0.105 | 23 ± 0.5 | 7.8 ± 1.3 |
| Parameters | PacBio Iso-Seq | Illumina RNA-Seq |
|---|---|---|
| Sequencing data | ||
| Number of subreads or raw reads | 9,717,992 | 58,212,575 |
| Number of CCS or clean reads | 410,183 | 57,010,224 |
| Full-length or assembled transcriptome | ||
| Number of transcripts | 21,154 | 105,259 |
| Number of nucleotide bases (Mb) | 54.67 | |
| GC content (%) | 43.83 | 39.44 |
| Mean length | 2584.71 | 924.00 |
| Minimum length (bp) | 209 | 201 |
| Maximum length (bp) | 11,923 | 35,077 |
| N50 | 2673 | 1755 |
| Length range of transcripts (bp) | ||
| <400 | 108 | 47,105 |
| 400–1000 | 674 | 30,851 |
| 1000–2000 | 5835 | 14,791 |
| 2000–3000 | 8722 | 6451 |
| >3000 | 5815 | 6061 |
| Sample | Raw Reads | Clean Reads | Low-Quality Reads Rate (%) | Clean Q30 Bases Rate |
|---|---|---|---|---|
| H1 | 58,231,232 | 57,068,540 | 0.52 | 93.97 |
| H2 | 56,276,300 | 55,083,032 | 0.5 | 94.19 |
| H3 | 60,401,216 | 59,155,796 | 0.63 | 93.88 |
| M1 | 63,840,402 | 62,436,042 | 0.87 | 93.09 |
| M2 | 59,975,242 | 58,780,862 | 0.6 | 93.94 |
| M3 | 60,770,110 | 59,506,128 | 0.52 | 94.01 |
| L1 | 46,464,356 | 45,751,916 | 0.57 | 94.88 |
| L2 | 54,854,172 | 53,396,614 | 0.52 | 94.43 |
| L3 | 63,100,148 | 61,913,086 | 0.5 | 94.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wen, J.; Hu, R.; Pei, S.; Li, T.; Shan, B.; Huang, H.; Zhu, C. Transcriptome Responses to Different Environments in Intertidal Zones in the Peanut Worm Sipunculus nudus. Biology 2023, 12, 1182. https://doi.org/10.3390/biology12091182
Li J, Wen J, Hu R, Pei S, Li T, Shan B, Huang H, Zhu C. Transcriptome Responses to Different Environments in Intertidal Zones in the Peanut Worm Sipunculus nudus. Biology. 2023; 12(9):1182. https://doi.org/10.3390/biology12091182
Chicago/Turabian StyleLi, Junwei, Jiufu Wen, Ruiping Hu, Surui Pei, Ting Li, Binbin Shan, Honghui Huang, and Changbo Zhu. 2023. "Transcriptome Responses to Different Environments in Intertidal Zones in the Peanut Worm Sipunculus nudus" Biology 12, no. 9: 1182. https://doi.org/10.3390/biology12091182