
Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
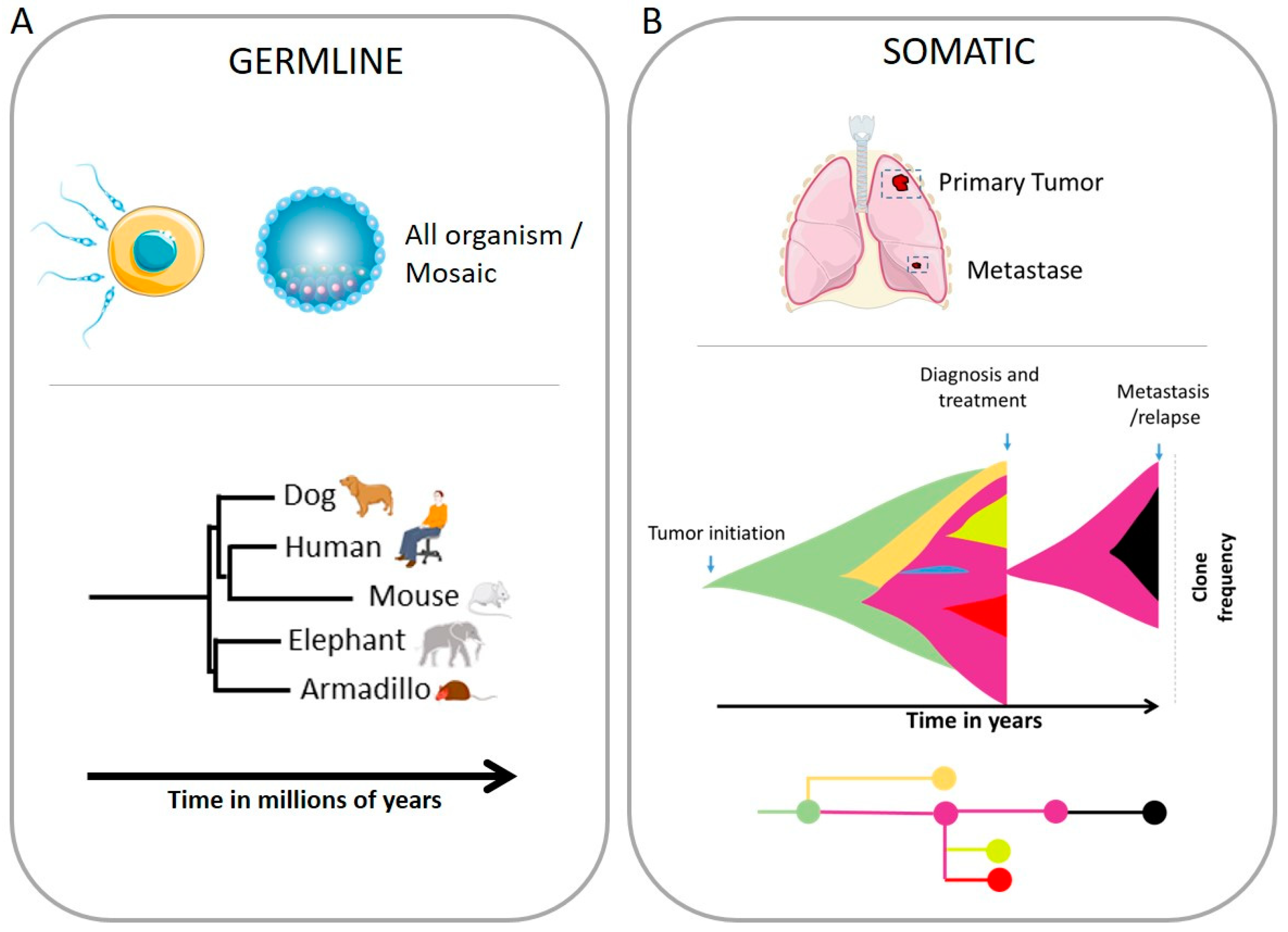
Abstract
:Simple Summary
Abstract
1. Introduction
2. Different Patterns of Structure Variants and Chromosomal Instability
2.1. Aneuploidy
2.2. Common Chromosomal Changes
2.3. Complex Genomic Rearrangements
2.4. ecDNA
3. CIN, Evolution, and Fitness
4. Chromosomal Instability during Evolution
4.1. SNPs
4.2. Genome Size Variability and Chromosomal Instability
4.3. Chromosomal Variance in Human
5. Chromosomal Instability in Cancer
5.1. Tumor Clonal Heterogeneity
5.2. Different Chromosomal Changes Observed in Tumor Evolution
5.3. Punctuated Burst and Genomic Evolution
5.4. CIN and Tumor Progression
6. Possible Origins of Chromosomal Instability and Its Study
6.1. Mitotic-Cell Cycle Errors
6.2. Transient Nuclear Envelope Rupture (NER)
6.3. Double-Stranded Breaks
6.4. Replication Stress
6.5. Implication of Viral Infection
6.6. Expression of Meiotic-Specific Proteins
6.7. P53 and Other Factors Linked to Tumors
6.8. Study of CIN in Genome Evolution of Cancer
- Whole-genome sequencing: Whole-genome sequencing provides a comprehensive view of the entire genome, including chromosomal rearrangements and copy number alterations that result from chromosomal instability.
- Comparative genomic hybridization (CGH): CGH is a technique that compares the copy number of genomic regions between a reference genome and a test genome. This technique can identify regions of gain or loss that result from chromosomal instability.
- Fluorescence in situ hybridization (FISH): FISH is a technique that uses fluorescent probes to bind to specific genomic regions. This technique can be used to visualize chromosomal rearrangements and copy number alterations.
- Single-cell sequencing: Single-cell sequencing can provide insight into the genomic heterogeneity within a tumor and can reveal subpopulations of cells with different patterns of chromosomal instability.
- Cell culture and xenograft models: Cell culture and xenograft models can be used to study the effects of CIN on tumor growth and response to treatment.
- Bioinformatic analysis: Bioinformatic tools can be used to analyze genomic data and identify patterns of chromosomal instability, including chromosomal breakpoints, copy number alterations, and structural variants.
7. Germ Cells and Meiotic Program as Drivers for Genome Evolution
7.1. Chromosomal Axis and Synapsis in Genome Stability and Evolution
7.2. Impact of Three-Dimensional Chromatin Structure on the Control of Gene Expression
7.3. DNA Recombination and Repair as a Mayor Source for Evolution
7.3.1. Recombinational Hotspots as a Factor for Genome Instability and Evolution
7.3.2. Factors and Complexes Define Recombinational Hotspots
Cohesin Complexes and Synaptonemal Complex Axial Elements
Protein PRDM9
Chromosome Organization as a Modulator of Recombinant Landscape
7.3.3. Pathways of Repair of Meiotic DSB to Chiasma Formation
7.3.4. Defects in Meiotic Recombination Leads to Chromosome Rearrangements
Large Chromosomal Rearrangements as Drivers for Speciation
Microchromosomes
Whole Genome Duplications and Gain in Chromosomes
Chromothripsis
7.3.5. Repetitive DNA Elements as Contributors for Chromosome Evolution and Speciation
7.3.6. Telomeres
8. Consequences of Chromosomal Instability Other Than Cancer in Humans
8.1. Infertility
8.2. Rare Diseases
8.2.1. Impairment of DNA Damage Repair Pathways
8.2.2. Germline SV and CCR
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heng, H.H. The genome-centric concept: Resynthesis of evolutionary theory. Bioessays 2009, 31, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Liu, G.; Bremer, S.; Ye, K.J.; Stevens, J.; Ye, C.J. Clonal and non-clonal chromosome aberrations and genome variation and aberration. Genome 2006, 49, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Larkin, D.M.; der Wind, A.E.-V.; Bourque, G.; Tesler, G.; Auvil, L.; Beever, J.E.; Chowdhary, B.P.; Galibert, F.; Gatzke, L. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005, 309, 613–617. [Google Scholar] [CrossRef]
- Navarro, A.; Barton, N.H. Chromosomal speciation and molecular divergence–accelerated evolution in rearranged chromosomes. Science 2003, 300, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gatinois, V.; Puechberty, J.; Geneviève, D.; Lefort, G. Chromothripsis: Potential origin in gametogenesis and preimplantation cell divisions. A review. Fertil. Steril. 2014, 102, 1785–1796. [Google Scholar] [CrossRef]
- Capilla, L.; Garcia Caldés, M.; Ruiz-Herrera, A. Mammalian Meiotic Recombination: A Toolbox for Genome Evolution. Cytogenet. Genome Res. 2016, 150, 1–16. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef]
- Mattioli, K.; Oliveros, W.; Gerhardinger, C.; Andergassen, D.; Maass, P.G.; Rinn, J.L.; Melé, M. Cis and trans effects differentially contribute to the evolution of promoters and enhancers. Genome Biol. 2020, 21, 210. [Google Scholar] [CrossRef]
- Moriyama, Y.; Koshiba-Takeuchi, K. Significance of whole-genome duplications on the emergence of evolutionary novelties. Brief Funct. Genom. 2018, 17, 329–338. [Google Scholar] [CrossRef]
- Reznick, D.N.; Ricklefs, R.E. Darwin’s bridge between microevolution and macroevolution. Nature 2009, 457, 837–842. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarro, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A pan-cancer compendium of chromosomal instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, J.; Turpin, R.; Gautier, M. Chromosomic diagnosis of mongolism. Arch. Fr. Pediatr. 1959, 16, 962–963. [Google Scholar] [PubMed]
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Zhang, C.Z.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet. 2015, 49, 183–211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Bafna, V.; Mischel, P.S. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Cancer 2019, 19, 283–288. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Fioretos, T.; Surace, C.; Lonoce, A.; Mastrorilli, A.; Strombeck, B.; D’Addabbo, P.; Iacovelli, F.; Minervini, C.; Aventin, A.; et al. MYC-containing double minutes in hematologic malignancies: Evidence in favor of the episome model and exclusion of MYC as the target gene. Hum. Mol. Genet. 2006, 15, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevicius, J.; Lukinavicius, G.; Elder, K.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860–2877.e2822. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef]
- Li, R.; Zhu, J. Effects of aneuploidy on cell behaviour and function. Nat. Rev. Mol. Cell Biol. 2022, 23, 250–265. [Google Scholar] [CrossRef]
- Zhu, J.; Tsai, H.J.; Gordon, M.R.; Li, R. Cellular Stress Associated with Aneuploidy. Dev. Cell 2018, 44, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Salgueiro, L.; Buccitelli, C.; Rowald, K.; Somogyi, K.; Kandala, S.; Korbel, J.O.; Sotillo, R. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol. Med. 2020, 12, e10941. [Google Scholar] [CrossRef]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Batlle, E. Targeting the Microenvironment in Advanced Colorectal Cancer. Trends Cancer 2016, 2, 495–504. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, G.; Yang, W.; Jin, W.; Gong, J.; Xu, X.; Niu, X. Animal-SNPAtlas: A comprehensive SNP database for multiple animals. Nucleic Acids Res. 2022, 51, D816–D826. [Google Scholar] [CrossRef] [PubMed]
- Burraco, P.; Orizaola, G. Ionizing radiation and melanism in Chornobyl tree frogs. Evol. Appl. 2022, 15, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Klunk, J.; Vilgalys, T.P.; Demeure, C.E.; Cheng, X.; Shiratori, M.; Madej, J.; Beau, R.; Elli, D.; Patino, M.I.; Redfern, R.; et al. Evolution of immune genes is associated with the Black Death. Nature 2022, 611, 312–319. [Google Scholar] [CrossRef]
- Enard, D. Ancient DNA reveals rapid natural selection during the Black Death. Nature 2022, 611, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Bubonic plague left lingering scars on the human genome. Nature 2022. [Google Scholar] [CrossRef]
- Holland, L.Z.; Ocampo Daza, D. A new look at an old question: When did the second whole genome duplication occur in vertebrate evolution? Genome Biol. 2018, 19, 209. [Google Scholar] [CrossRef]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 166. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, S.M.; Neuhauss, S.C. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genom. 2014, 289, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef]
- Clark, J.W.; Donoghue, P.C.J. Whole-Genome Duplication and Plant Macroevolution. Trends Plant Sci. 2018, 23, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, H.; Schubert, M.; Seguin-Orlando, A.; Ginolhac, A.; Petersen, L.; Fumagalli, M.; Albrechtsen, A.; Petersen, B.; Korneliussen, T.S.; Vilstrup, J.T.; et al. Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc. Natl. Acad. Sci. USA 2014, 111, 18655–18660. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Stock, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef]
- Carbonell-Bejerano, P.; Royo, C.; Torres-Perez, R.; Grimplet, J.; Fernandez, L.; Franco-Zorrilla, J.M.; Lijavetzky, D.; Baroja, E.; Martinez, J.; Garcia-Escudero, E.; et al. Catastrophic Unbalanced Genome Rearrangements Cause Somatic Loss of Berry Color in Grapevine. Plant Physiol. 2017, 175, 786–801. [Google Scholar] [CrossRef]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Porubsky, D.; Hops, W.; Ashraf, H.; Hsieh, P.; Rodriguez-Martin, B.; Yilmaz, F.; Ebler, J.; Hallast, P.; Maria Maggiolini, F.A.; Harvey, W.T.; et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 2022, 185, 1986–2005.e1926. [Google Scholar] [CrossRef]
- Roosen, M.; Ode, Z.; Bunt, J.; Kool, M. The oncogenic fusion landscape in pediatric CNS neoplasms. Acta Neuropathol. 2022, 143, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e314. [Google Scholar] [CrossRef]
- Nattestad, M.; Goodwin, S.; Ng, K.; Baslan, T.; Sedlazeck, F.J.; Rescheneder, P.; Garvin, T.; Fang, H.; Gurtowski, J.; Hutton, E.; et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res. 2018, 28, 1126–1135. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e7. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef] [PubMed]
- De Falco, A.; Caruso, F.; Su, X.D.; Iavarone, A.; Ceccarelli, M. A variational algorithm to detect the clonal copy number substructure of tumors from scRNA-seq data. Nat. Commun. 2023, 14, 1074. [Google Scholar] [CrossRef]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e12. [Google Scholar] [CrossRef]
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef]
- Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; Swanton, C. The evolution of the unstable cancer genome. Curr. Opin. Genet. Dev. 2014, 24, 61–67. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef]
- Jing, A.; Vizeacoumar, F.S.; Parameswaran, S.; Haave, B.; Cunningham, C.E.; Wu, Y.; Arnold, R.; Bonham, K.; Freywald, A.; Han, J.; et al. Expression-based analyses indicate a central role for hypoxia in driving tumor plasticity through microenvironment remodeling and chromosomal instability. NPJ Syst. Biol. Appl. 2018, 4, 38. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439.e2424. [Google Scholar] [CrossRef]
- Spurr, L.F.; Weichselbaum, R.R.; Pitroda, S.P. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers. Nat. Genet. 2022, 54, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Comaills, V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int. J. Mol. Sci. 2021, 22, 7281. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, F.; Wu, J.; Cheng, Y.; Cao, Y.; Wu, X.; Ma, M.; Tang, F.; Liu, Z.; Liu, H.; et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy 2021, 17, 3976–3991. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef]
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Gauthier, B.R.; Lorenzo, P.I.; Comaills, V. Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success? Cancers 2021, 14, 83. [Google Scholar] [CrossRef]
- Weigelin, B.; den Boer, A.T.; Wagena, E.; Broen, K.; Dolstra, H.; de Boer, R.J.; Figdor, C.G.; Textor, J.; Friedl, P. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat. Commun. 2021, 12, 5217. [Google Scholar] [CrossRef] [PubMed]
- Nader, G.P.F.; Aguera-Gonzalez, S.; Routet, F.; Gratia, M.; Maurin, M.; Cancila, V.; Cadart, C.; Palamidessi, A.; Ramos, R.N.; San Roman, M.; et al. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 2021, 184, 5230–5246.e5222. [Google Scholar] [CrossRef]
- Tang, S.; Stokasimov, E.; Cui, Y.; Pellman, D. Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature 2022, 606, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.; Mazzagatti, A.; De Angelis, S.; Johnson, S.C.; Bakker, B.; Spierings, D.C.J.; Wardenaar, R.; Maniati, E.; Wang, J.; Boemo, M.A.; et al. Replication stress generates distinctive landscapes of DNA copy number alterations and chromosome scale losses. Genome Biol. 2022, 23, 223. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef]
- Groelly, F.J.; Dagg, R.A.; Petropoulos, M.; Rossetti, G.G.; Prasad, B.; Panagopoulos, A.; Paulsen, T.; Karamichali, A.; Jones, S.E.; Ochs, F.; et al. Mitotic DNA synthesis is caused by transcription-replication conflicts in BRCA2-deficient cells. Mol. Cell 2022, 82, 3382–3397.e7. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Tamura, N.; Shaikh, N.; Muliaditan, D.; Soliman, T.N.; McGuinness, J.R.; Maniati, E.; Moralli, D.; Durin, M.-A.; Green, C.M.; Balkwill, F.R.; et al. Specific Mechanisms of Chromosomal Instability Indicate Therapeutic Sensitivities in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4946–4959. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E deregulation promotes loss of specific genomic regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef]
- Pentzold, C.; Shah, S.A.; Hansen, N.R.; Le Tallec, B.; Seguin-Orlando, A.; Debatisse, M.; Lisby, M.; Oestergaard, V.H. FANCD2 binding identifies conserved fragile sites at large transcribed genes in avian cells. Nucleic Acids Res. 2018, 46, 1280–1294. [Google Scholar] [CrossRef]
- Macheret, M.; Bhowmick, R.; Sobkowiak, K.; Padayachy, L.; Mailler, J.; Hickson, I.D.; Halazonetis, T.D. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell. Res. 2020, 30, 997–1008. [Google Scholar] [CrossRef]
- Bakker, B.; Taudt, A.; Belderbos, M.E.; Porubsky, D.; Spierings, D.C.; de Jong, T.V.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; Bradley, A.; et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 2016, 17, 115. [Google Scholar] [CrossRef]
- Soto, M.; Raaijmakers, J.A.; Bakker, B.; Spierings, D.C.J.; Lansdorp, P.M.; Foijer, F.; Medema, R.H. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep. 2017, 19, 2423–2431. [Google Scholar] [CrossRef]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Kulmann-Leal, B.; Ziliotto, M.; Chies, J.A.B. HIV Infection, Chromosome Instability, and Micronucleus Formation. Viruses 2023, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.G.; Demeulemeester, J.; Otero, P.; Jolly, C.; Garcia-Souto, D.; Pequeno-Valtierra, A.; Zamora, J.; Tojo, M.; Temes, J.; Baez-Ortega, A.; et al. Aberrant integration of Hepatitis B virus DNA promotes major restructuring of human hepatocellular carcinoma genome architecture. Nat. Commun. 2021, 12, 6910. [Google Scholar] [CrossRef] [PubMed]
- Tubio, J.M.; Estivill, X. Cancer: When catastrophe strikes a cell. Nature 2011, 470, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, J.W.; Koster, J.; Lodder, P.; Repping, S.; Hamer, G. Massive expression of germ cell-specific genes is a hallmark of cancer and a potential target for novel treatment development. Oncogene 2018, 37, 5694–5700. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, J.; Larcombe, L.; McFarlane, R.J. Meta-analysis of expression of l(3)mbt tumor-associated germline genes supports the model that a soma-to-germline transition is a hallmark of human cancers. Int. J. Cancer 2014, 134, 2359–2365. [Google Scholar] [CrossRef]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. J. Ski. Cancer 2013, 2013, 190109. [Google Scholar] [CrossRef]
- Folco, H.; Chalamcharla, V.R.; Sugiyama, T.; Thillainadesan, G.; Zofall, M.; Balachandran, V.; Dhakshnamoorthy, J.; Mizuguchi, T.; Grewal, S.I. Untimely expression of gametogenic genes in vegetative cells causes uniparental disomy. Nature 2017, 543, 126–130. [Google Scholar] [CrossRef]
- Houle, A.A.; Gibling, H.; Lamaze, F.C.; Edgington, H.A.; Soave, D.; Fave, M.J.; Agbessi, M.; Bruat, V.; Stein, L.D.; Awadalla, P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 2018, 28, 1611–1620. [Google Scholar] [CrossRef]
- Baslan, T.; Morris, J.P.t.; Zhao, Z.; Reyes, J.; Ho, Y.J.; Tsanov, K.M.; Bermeo, J.; Tian, S.; Zhang, S.; Askan, G.; et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 2022, 608, 795–802. [Google Scholar] [CrossRef]
- Santaguida, S.; Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cel Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fang, X.; Baker, D.J.; Guo, L.; Gao, X.; Wei, Z.; Han, S.; van Deursen, J.M.; Zhang, P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14188–14193. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e1375. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Wormann, S.M.; Zhang, A.; Thege, F.I.; Cowan, R.W.; Rupani, D.N.; Wang, R.; Manning, S.L.; Gates, C.; Wu, W.; Levin-Klein, R.; et al. APOBEC3A drives deaminase domain-independent chromosomal instability to promote pancreatic cancer metastasis. Nat. Cancer 2021, 2, 1338–1356. [Google Scholar] [CrossRef]
- Vera-Rodriguez, M.; Chavez, S.L.; Rubio, C.; Reijo Pera, R.A.; Simon, C. Prediction model for aneuploidy in early human embryo development revealed by single-cell analysis. Nat. Commun. 2015, 6, 7601. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kleckner, N.; Storlazzi, A.; Zickler, D. Coordinate variation in meiotic pachytene SC length and total crossover/chiasma frequency under conditions of constant DNA length. TRENDS Genet. 2003, 19, 623–628. [Google Scholar] [CrossRef]
- Ruiz-Herrera, A.; Vozdova, M.; Fernández, J.; Sebestova, H.; Capilla, L.; Frohlich, J.; Vara, C.; Hernández-Marsal, A.; Sipek, J.; Robinson, T.J.; et al. Recombination correlates with synaptonemal complex length and chromatin loop size in bovids-insights into mammalian meiotic chromosomal organization. Chromosoma 2017, 126, 615–631. [Google Scholar] [CrossRef]
- Ohkura, H. Meiosis: An overview of key differences from mitosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a015859. [Google Scholar] [CrossRef]
- Bannister, L.A.; Reinholdt, L.G.; Munroe, R.J.; Schimenti, J.C. Positional cloning and characterization of mouse mei8, a disrupted allele of the meiotic cohesin Rec8. Genesis 2004, 40, 184–194. [Google Scholar] [CrossRef]
- Revenkova, E.; Eijpe, M.; Heyting, C.; Hodges, C.A.; Hunt, P.A.; Liebe, B.; Scherthan, H.; Jessberger, R. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat. Cell Biol. 2004, 6, 555–562. [Google Scholar] [CrossRef]
- Winkel, K.; Alsheimer, M.; Öllinger, R.; Benavente, R. Protein SYCP2 provides a link between transverse filaments and lateral elements of mammalian synaptonemal complexes. Chromosoma 2009, 118, 259–267. [Google Scholar] [CrossRef]
- Yang, F.; De La Fuente, R.; Leu, N.A.; Baumann, C.; McLaughlin, K.J.; Wang, P.J. Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J. Cell Biol. 2006, 173, 497–507. [Google Scholar] [CrossRef]
- Henderson, K.A.; Keeney, S. Synaptonemal complex formation: Where does it start? BioEssays 2005, 27, 995–998. [Google Scholar] [CrossRef]
- Page, S.L.; Hawley, R.S. The genetics and molecular biology of the synaptonemal complex. Annu. Rev. Cell Dev. Biol. 2004, 20, 525–558. [Google Scholar] [CrossRef]
- Romanienko, P.J.; Camerini-Otero, R.D. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 2000, 6, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet. 2001, 2, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, A.C.; Berman, J. Shift and adapt: The costs and benefits of karyotype variations. Curr. Opin. Microbiol. 2015, 26, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Martinez, D.A.; Sakthikumar, S.; Anderson, M.Z.; Berlin, A.; Gujja, S.; Zeng, Q.; Zisson, E.; Wang, J.M.; Greenberg, J.M.; et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015, 25, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.; Mariotti, M.; Tyshkovskiy, A.; Zhou, X.; Hulke, M.L.; Ma, S.; Gerashchenko, M.V.; Koren, A.; Gladyshev, V.N. Molecular signatures of aneuploidy-driven adaptive evolution. Nat. Commun. 2020, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Knight, E.; Bomblies, K. The meiotic cohesin subunit REC8 contributes to multigenic adaptive evolution of autopolyploid meiosis in Arabidopsis arenosa. PLoS Genet. 2022, 18, e1010304. [Google Scholar] [CrossRef]
- Matveevsky, S.; Bakloushinskaya, I.; Tambovtseva, V.; Atsaeva, M.; Grishaeva, T.; Bogdanov, A.; Kolomiets, O. Nonhomologous Chromosome Interactions in Prophase I: Dynamics of Bizarre Meiotic Contacts in the Alay Mole Vole Ellobius alaicus (Mammalia, Rodentia). Genes 2022, 13, 2196. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Le Dily, F.; Garcia, F.; Salvà-Castro, J.; Gómez, H.L.; Julià, E.; Moutinho, C.; Aiese Cigliano, R.; et al. Three-Dimensional Genomic Structure and Cohesin Occupancy Correlate with Transcriptional Activity during Spermatogenesis. Cell Rep. 2019, 28, 352–367.e9. [Google Scholar] [CrossRef] [PubMed]
- Alavattam, K.G.; Maezawa, S.; Sakashita, A.; Khoury, H.; Barski, A.; Kaplan, N.; Namekawa, S.H. Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat. Struct. Mol. Biol. 2019, 26, 175–184. [Google Scholar] [CrossRef] [PubMed]
- de Massy, B. Initiation of meiotic recombination: How and where? Conservation and specificities among eukaryotes. Annu. Rev. Genet. 2013, 47, 563–599. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.; Keeney, S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016634. [Google Scholar] [CrossRef]
- Bompadre, O.; Andrey, G. Chromatin topology in development and disease. Curr. Opin. Genet. Dev. 2019, 55, 32–38. [Google Scholar] [CrossRef]
- Farré, M.; Robinson, T.J.; Ruiz-Herrera, A. An Integrative Breakage Model of genome architecture, reshuffling and evolution: The Integrative Breakage Model of genome evolution, a novel multidisciplinary hypothesis for the study of genome plasticity. Bioessays 2015, 37, 479–488. [Google Scholar] [CrossRef]
- Franke, M.; Gómez-Skarmeta, J.L. An evolutionary perspective of regulatory landscape dynamics in development and disease. Curr. Opin. Cell Biol. 2018, 55, 24–29. [Google Scholar] [CrossRef]
- Veller, C.; Kleckner, N.; Nowak, M.A. A rigorous measure of genome-wide genetic shuffling that takes into account crossover positions and Mendel’s second law. Proc. Natl. Acad. Sci. USA 2019, 116, 1659–1668. [Google Scholar] [CrossRef]
- Kumar, R.; Bourbon, H.-M.; de Massy, B. Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes. Dev. 2010, 24, 1266–1280. [Google Scholar] [CrossRef]
- Lange, J.; Yamada, S.; Tischfield, S.E.; Pan, J.; Kim, S.; Zhu, X.; Socci, N.D.; Jasin, M.; Keeney, S. The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 2016, 167, 695–708.e16. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V.; Kruglyak, L. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000419. [Google Scholar] [CrossRef] [PubMed]
- Barlow, A.L.; Benson, F.E.; West, S.C.; Hulten, M.A. Distribution of the Rad51 recombinase in human and mouse spermatocytes. EMBO J. 1997, 16, 5207–5215. [Google Scholar] [CrossRef]
- Cole, F.; Kauppi, L.; Lange, J.; Roig, I.; Wang, R.; Keeney, S.; Jasin, M. Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat. Cell Biol. 2012, 14, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.; Bowden, R.; Tumian, A.; Bontrop, R.E.; Freeman, C.; MacFie, T.S.; McVean, G.; Donnelly, P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 2010, 327, 876–879. [Google Scholar] [CrossRef]
- Smagulova, F.; Gregoretti, I.V.; Brick, K.; Khil, P.; Camerini-Otero, R.D.; Petukhova, G.V. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 2011, 472, 375–378. [Google Scholar] [CrossRef]
- Cappelletti, E.; Piras, F.M.; Badiale, C.; Bambi, M.; Santagostino, M.; Vara, C.; Masterson, T.A.; Sullivan, K.F.; Nergadze, S.G.; Ruiz-Herrera, A.; et al. CENP-A binding domains and recombination patterns in horse spermatocytes. Sci. Rep. 2019, 9, 15800. [Google Scholar] [CrossRef]
- Brick, K.; Smagulova, F.; Khil, P.; Camerini-Otero, R.D.; Petukhova, G.V. Genetic recombination is directed away from functional genomic elements in mice. Nature 2012, 485, 642–645. [Google Scholar] [CrossRef]
- Parvanov, E.D.; Petkov, P.M.; Paigen, K. Prdm9 controls activation of mammalian recombination hotspots. Science 2010, 327, 835. [Google Scholar] [CrossRef]
- Wang, S.; Zickler, D.; Kleckner, N.; Zhang, L. Meiotic crossover patterns: Obligatory crossover, interference and homeostasis in a single process. Cell Cycle 2015, 14, 305–314. [Google Scholar] [CrossRef]
- Zickler, D.; Kleckner, N. Meiotic chromosomes: Integrating structure and function. Annu. Rev. Genet. 1999, 33, 603. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Caballero, C.; Herrán, Y.; Sánchez-Martín, M.; Suja, J.Á.; Barbero, J.L.; Llano, E.; Pendás, A.M. Identification and molecular characterization of the mammalian α-kleisin RAD21L. Cell Cycle 2011, 10, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; Buard, J.; Grey, C.; Fledel-Alon, A.; Ober, C.; Przeworski, M.; Coop, G.; De Massy, B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 2010, 327, 836–840. [Google Scholar] [CrossRef]
- Mihola, O.; Trachtulec, Z.; Vlcek, C.; Schimenti, J.C.; Forejt, J. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 2009, 323, 373–375. [Google Scholar] [CrossRef]
- Pratto, F.; Brick, K.; Khil, P.; Smagulova, F.; Petukhova, G.V.; Camerini-Otero, R.D. Recombination initiation maps of individual human genomes. Science 2014, 346, 1256442. [Google Scholar] [CrossRef] [PubMed]
- Smagulova, F.; Brick, K.; Pu, Y.; Camerini-Otero, R.D.; Petukhova, G.V. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes. Dev. 2016, 30, 266–280. [Google Scholar] [CrossRef]
- Oliver, P.L.; Goodstadt, L.; Bayes, J.J.; Birtle, Z.; Roach, K.C.; Phadnis, N.; Beatson, S.A.; Lunter, G.; Malik, H.S.; Ponting, C.P. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009, 5, e1000753. [Google Scholar] [CrossRef]
- Davies, B.; Hatton, E.; Altemose, N.; Hussin, J.G.; Pratto, F.; Zhang, G.; Hinch, A.G.; Moralli, D.; Biggs, D.; Diaz, R. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 2016, 530, 171–176. [Google Scholar] [CrossRef]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nature Genet. 2008, 40, 1124–1129. [Google Scholar] [CrossRef]
- Duret, L.; Galtier, N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 285–311. [Google Scholar] [CrossRef]
- Janoušek, V.; Munclinger, P.; Wang, L.; Teeter, K.C.; Tucker, P.K. Functional organization of the genome may shape the species boundary in the house mouse. Mol. Biol. Evol. 2015, 32, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.; Schumer, M.; Haba, Y.; Bashkirova, L.; Holland, C.; Rosenthal, G.G.; Przeworski, M. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. Elife 2017, 6, e24133. [Google Scholar] [CrossRef] [PubMed]
- Capilla, L.; Medarde, N.; Alemany-Schmidt, A.; Oliver-Bonet, M.; Ventura, J.; Ruiz-Herrera, A. Genetic recombination variation in wild Robertsonian mice: On the role of chromosomal fusions and Prdm9 allelic background. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140297. [Google Scholar] [CrossRef]
- Faria, R.; Navarro, A. Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends Ecol. Evol. 2010, 25, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Feulner, P.G.; Chain, F.J.; Panchal, M.; Huang, Y.; Eizaguirre, C.; Kalbe, M.; Lenz, T.L.; Samonte, I.E.; Stoll, M.; Bornberg-Bauer, E. Genomics of divergence along a continuum of parapatric population differentiation. PLoS Genet. 2015, 11, e1004966. [Google Scholar] [CrossRef]
- Sodeland, M.; Jorde, P.E.; Lien, S.; Jentoft, S.; Berg, P.R.; Grove, H.; Kent, M.P.; Arnyasi, M.; Olsen, E.M.; Knutsen, H. “Islands of Divergence” in the Atlantic cod genome represent polymorphic chromosomal rearrangements. Genome Biol. Evol. 2016, 8, 1012–1022. [Google Scholar] [CrossRef]
- Ullastres, A.; Farré, M.; Capilla, L.; Ruiz-Herrera, A. Unraveling the effect of genomic structural changes in the rhesus macaque-implications for the adaptive role of inversions. BMC Genom. 2014, 15, 530. [Google Scholar] [CrossRef]
- Dumas, D.; Britton-Davidian, J. Chromosomal rearrangements and evolution of recombination: Comparison of chiasma distribution patterns in standard and Robertsonian populations of the house mouse. Genetics 2002, 162, 1355–1366. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. γ-H2AX-a novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Murakami, H.; Keeney, S. Regulating the formation of DNA double-strand breaks in meiosis. Genes Dev. 2008, 22, 286–292. [Google Scholar] [CrossRef]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Lange, J.; Keeney, S. Genome destabilization by homologous recombination in the germ line. Nat. Rev. Mol. Cell Biol. 2010, 11, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Álvarez-González, L.; Marín-Gual, L.; Garcia, F.; Florit-Sabater, B.; Capilla, L.; Sanchéz-Guillén, R.A.; Sarrate, Z. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nat. Commun. 2021, 12, 2981. [Google Scholar] [CrossRef] [PubMed]
- Britton-Davidian, J.; Catalan, J.; da Graça Ramalhinho, M.; Ganem, G.; Auffray, J.-C.; Capela, R.; Biscoito, M.; Searle, J.B.; da Luz Mathias, M. Rapid chromosomal evolution in island mice. Nature 2000, 403, 158. [Google Scholar] [CrossRef]
- Dutrillaux, B. Chromosomal evolution in primates: Tentative phylogeny from Microcebus murinus (Prosimian) to man. Human Genet. 1979, 48, 251–314. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Sawyer, J.R.; Dunham, K. The striking resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science 1980, 208, 1145–1148. [Google Scholar] [CrossRef]
- Yunis, J.J.; Prakash, O. The origin of man: A chromosomal pictorial legacy. Science 1982, 215, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.L.; Tuzun, E.; Morrison, V.A.; Hayden, K.E.; Ventura, M.; McGrath, S.D.; Rocchi, M.; Eichler, E.E. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005, 15, 1344–1356. [Google Scholar] [CrossRef]
- Fan, Y.; Linardopoulou, E.; Friedman, C.; Williams, E.; Trask, B.J. Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes. Genome Res. 2002, 12, 1651–1662. [Google Scholar] [CrossRef]
- Pevzner, P.; Tesler, G. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 7672–7677. [Google Scholar] [CrossRef]
- Dennis, M.Y.; Harshman, L.; Nelson, B.J.; Penn, O.; Cantsilieris, S.; Huddleston, J.; Antonacci, F.; Penewit, K.; Denman, L.; Raja, A. The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol. 2017, 1, 0069. [Google Scholar] [CrossRef]
- Waters, P.D.; Patel, H.R.; Ruiz-Herrera, A.; Álvarez-González, L.; Lister, N.C.; Simakov, O.; Ezaz, T.; Kaur, P.; Frere, C.; Grützner, F.; et al. Microchromosomes are building blocks of bird, reptile, and mammal chromosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2112494118. [Google Scholar] [CrossRef]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Crow, K.D.; Wagner, G.P. What is the role of genome duplication in the evolution of complexity and diversity? Mol. Biol. Evol. 2005, 23, 887–892. [Google Scholar] [CrossRef]
- Kellis, M.; Birren, B.W.; Lander, E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 2004, 428, 617–624. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef]
- Mottes, F.; Villa, C.; Osella, M.; Caselle, M. The impact of whole genome duplications on the human gene regulatory networks. PLoS Comput. Biol. 2021, 17, e1009638. [Google Scholar] [CrossRef]
- Ohno, S. The enormous diversity in genome sizes of fish as a reflection of nature’s extensive experiments with gene duplication. Trans. Am. Fish. Soc. 1970, 99, 120–130. [Google Scholar] [CrossRef]
- D’Antonio, M.; Ciccarelli, F.D. Integrated analysis of recurrent properties of cancer genes to identify novel drivers. Genome Biol. 2013, 14, R52. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Arora, J.; Isambert, H. Identification of ohnolog genes originating from whole genome duplication in early vertebrates, based on synteny comparison across multiple genomes. PLoS Comput. Biol. 2015, 11, e1004394. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA 2010, 107, 9270–9274. [Google Scholar] [CrossRef]
- Acharya, D.; Ghosh, T.C. Global analysis of human duplicated genes reveals the relative importance of whole-genome duplicates originated in the early vertebrate evolution. BMC Genom. 2016, 17, 71. [Google Scholar] [CrossRef]
- Huminiecki, L.; Heldin, C.H. 2R and remodeling of vertebrate signal transduction engine. BMC Biol. 2010, 8, 146. [Google Scholar] [CrossRef]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genom. 2014, 15, 82. [Google Scholar] [CrossRef]
- Weckselblatt, B.; Rudd, M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef] [PubMed]
- De Pagter, M.S.; Van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; Van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; Van Der Veken, L.T.; Cuppen, E. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. Am. J. Human. Genet. 2015, 96, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A piece of the macroevolution scenario. Mol. Cytogenet. 2020, 13, 3. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Cooper, D.N. Structural divergence between the human and chimpanzee genomes. Human. Genet. 2007, 120, 759–778. [Google Scholar] [CrossRef]
- Zhao, H.; Bourque, G. Recovering genome rearrangements in the mammalian phylogeny. Genome Res. 2009, 19, 934–942. [Google Scholar] [CrossRef]
- Farré, M.; Bosch, M.; López-Giráldez, F.; Ponsà, M.; Ruiz-Herrera, A. Assessing the role of tandem repeats in shaping the genomic architecture of great apes. PLoS ONE 2011, 6, e27239. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Sandig, C.; Goidts, V.; Hameister, H. Breakpoint analysis of the pericentric inversion between chimpanzee chromosome 10 and the homologous chromosome 12 in humans. Cytogenet. Genome Res. 2005, 108, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Harris, R.A.; Mootnick, A.R.; Milosavljevic, A.; Martin, D.I.; Rocchi, M.; Capozzi, O.; Archidiacono, N.; Konkel, M.K.; Walker, J.A. Centromere remodeling in Hoolock leuconedys (Hylobatidae) by a new transposable element unique to the gibbons. Genome Biol. Evol. 2012, 4, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: The emerging players in cells, cellular pathways and cancer. Chromosome Res. 2015, 23, 479–493. [Google Scholar] [CrossRef]
- Larracuente, A.M. The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: Dynamic evolution of a target of meiotic drive. BMC Evol. Biol. 2014, 14, 233. [Google Scholar] [CrossRef]
- Nazaryan-Petersen, L.; Bertelsen, B.; Bak, M.; Jønson, L.; Tommerup, N.; Hancks, D.C.; Tümer, Z. Germline chromothripsis driven by L1-mediated retrotransposition and Alu/Alu homologous recombination. Human. Mutat. 2016, 37, 385–395. [Google Scholar] [CrossRef]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef]
- Carbone, L.; Alan Harris, R.; Gnerre, S.; Veeramah, K.R.; Lorente-Galdos, B.; Huddleston, J.; Meyer, T.J.; Herrero, J.; Roos, C.; Aken, B. Gibbon genome and the fast karyotype evolution of small apes. Nature 2014, 513, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Held, U.; Nevonen, K.A.; Klawitter, S.; Pirzer, T.; Carbone, L.; Schumann, G.G. The flow of the gibbon LAVA element is facilitated by the LINE-1 retrotransposition machinery. Genome Biol. Evol. 2016, 8, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the role of satellite DNA in genome architecture and plasticity—An evolutionary and clinical affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef]
- Marín-Gual, L.; González-Rodelas, L.; Pujol, G.; Vara, C.; Martín-Ruiz, M.; Berríos, S.; Fernández-Donoso, R.; Pask, A.; Renfree, M.B.; Page, J.; et al. Strategies for meiotic sex chromosome dynamics and telomeric elongation in Marsupials. PLoS Genet. 2022, 18, e1010040. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Liu, L.; Bailey, S.M.; Okuka, M.; Muñoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere lengthening early in development. Nat. Cell Biol. 2007, 9, 1436–1441. [Google Scholar] [CrossRef]
- Reig-Viader, R.; Brieño-Enríquez, M.A.; Khoriauli, L.; Toran, N.; Cabero, L.; Giulotto, E.; Garcia-Caldés, M.; Ruiz-Herrera, A. Telomeric repeat-containing RNA and telomerase in human fetal oocytes. Hum. Reprod. 2013, 28, 414–422. [Google Scholar] [CrossRef]
- Reig-Viader, R.; Garcia-Caldés, M.; Ruiz-Herrera, A. Telomere homeostasis in mammalian germ cells: A review. Chromosoma 2016, 125, 337–351. [Google Scholar] [CrossRef]
- Reig-Viader, R.; Vila-Cejudo, M.; Vitelli, V.; Buscà, R.; Sabaté, M.; Giulotto, E.; Caldés, M.G.; Ruiz-Herrera, A. Telomeric repeat-containing RNA (TERRA) and telomerase are components of telomeres during mammalian gametogenesis. Biol. Reprod. 2014, 90, 103. [Google Scholar] [CrossRef]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef]
- Ingles, E.D.; Deakin, J.E. Telomeres, species differences, and unusual telomeres in vertebrates: Presenting challenges and opportunities to understanding telomere dynamics. Aims Genet. 2016, 3, 001–024. [Google Scholar] [CrossRef]
- Hoang, S.M.; O’Sullivan, R.J. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends Cancer 2020, 6, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Linking Telomere Regulation to Stem Cell Pluripotency. Trends Genet. 2017, 33, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Canfield, P.J.; Hartley, W.J.; Reddacliff, G.L. Spontaneous proliferations in Australian marsupials--a survey and review. 2. Dasyurids and bandicoots. J. Comp. Pathol. 1990, 103, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, Q.; Liu, Y.; Jiang, Y.; Yang, X.; Liu, R.; Zhang, H. Fertility problems in males carrying an inversion of chromosome 10. Open Med. 2021, 16, 316–321. [Google Scholar] [CrossRef]
- Fan, H.; Liu, Z.; Zhan, P.; Jia, G. Pericentric inversion of chromosome 6 and male fertility problems. Open Med. 2022, 17, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Takahashi, M.; Nonaka, C.; Enomoto, T.; Takakuwa, K. The analysis of chromosomal abnormalities in patients with recurrent pregnancy loss, focusing on the prognosis of patients with inversion of chromosome (9). Reprod. Med. Biol. 2019, 18, 296–301. [Google Scholar] [CrossRef]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature 2022, 612, 495–502. [Google Scholar] [CrossRef]
- Gross, M.; Hanenberg, H.; Lobitz, S.; Friedl, R.; Herterich, S.; Dietrich, R.; Gruhn, B.; Schindler, D.; Hoehn, H. Reverse mosaicism in Fanconi anemia: Natural gene therapy via molecular self-correction. Cytogenet. Genome Res. 2002, 98, 126–135. [Google Scholar] [CrossRef]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–331. [Google Scholar] [CrossRef]
- Marshall, C.R.; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; Pinto, D.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2017, 49, 27–35. [Google Scholar] [CrossRef] [PubMed]
- van Belzen, I.A.E.M.; Cai, C.; van Tuil, M.; Badloe, S.; Strengman, E.; Janse, A.; Verwiel, E.T.; van der Leest, D.F.M.; Kester, L.; Molenaar, J.J.; et al. Systematic discovery of gene fusions in pediatric cancer by integrating RNA-seq and WGS. bioRxiv 2008. [Google Scholar] [CrossRef]
- Schuy, J.; Grochowski, C.M.; Carvalho, C.M.B.; Lindstrand, A. Complex genomic rearrangements: An underestimated cause of rare diseases. Trends Genet. 2022, 38, 1134–1146. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comaills, V.; Castellano-Pozo, M. Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution. Biology 2023, 12, 671. https://doi.org/10.3390/biology12050671
Comaills V, Castellano-Pozo M. Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution. Biology. 2023; 12(5):671. https://doi.org/10.3390/biology12050671
Chicago/Turabian StyleComaills, Valentine, and Maikel Castellano-Pozo. 2023. "Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution" Biology 12, no. 5: 671. https://doi.org/10.3390/biology12050671