
SigPrimedNet: A Signaling-Informed Neural Network for scRNA-seq Annotation of Known and Unknown Cell Types
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
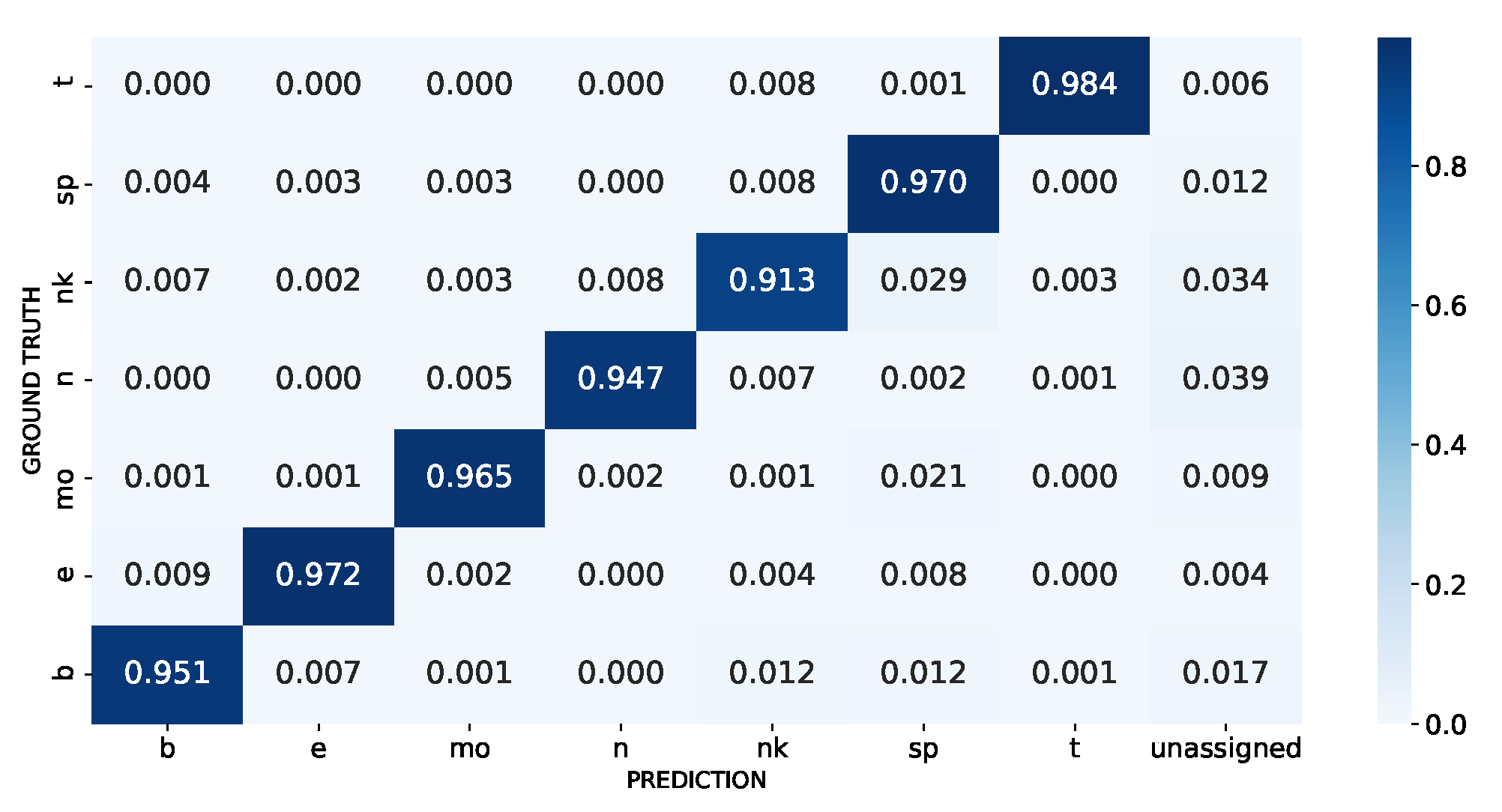
2.1.1. PBMC Dataset
2.1.2. Immune Dataset
2.1.3. Melanoma Dataset
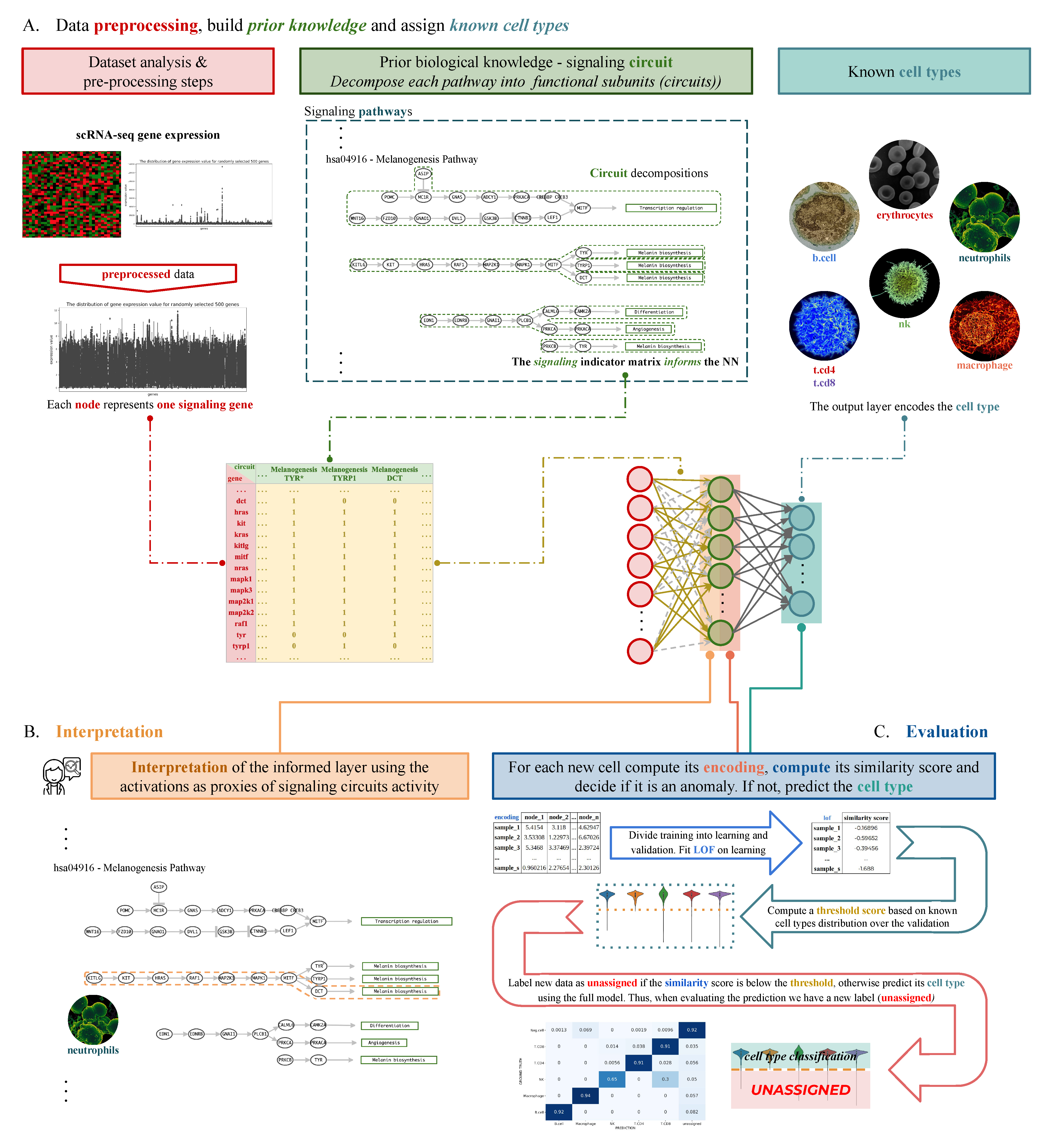
2.2. Analysis Workflow
2.3. Model Design
2.4. Data Preprocessing
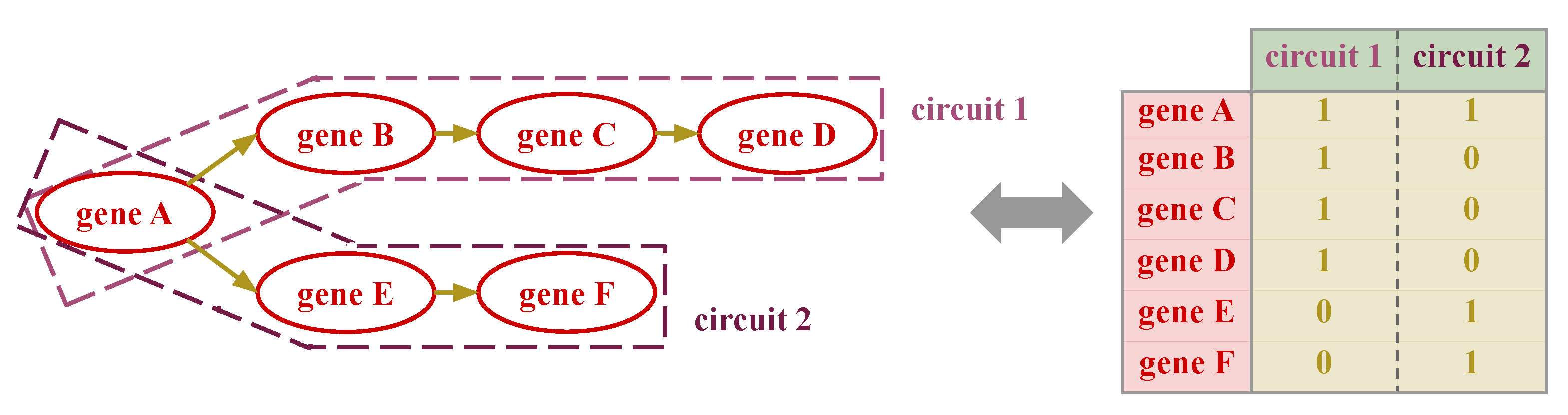
2.5. Signaling-Primed Sparsity-Inducing Layers
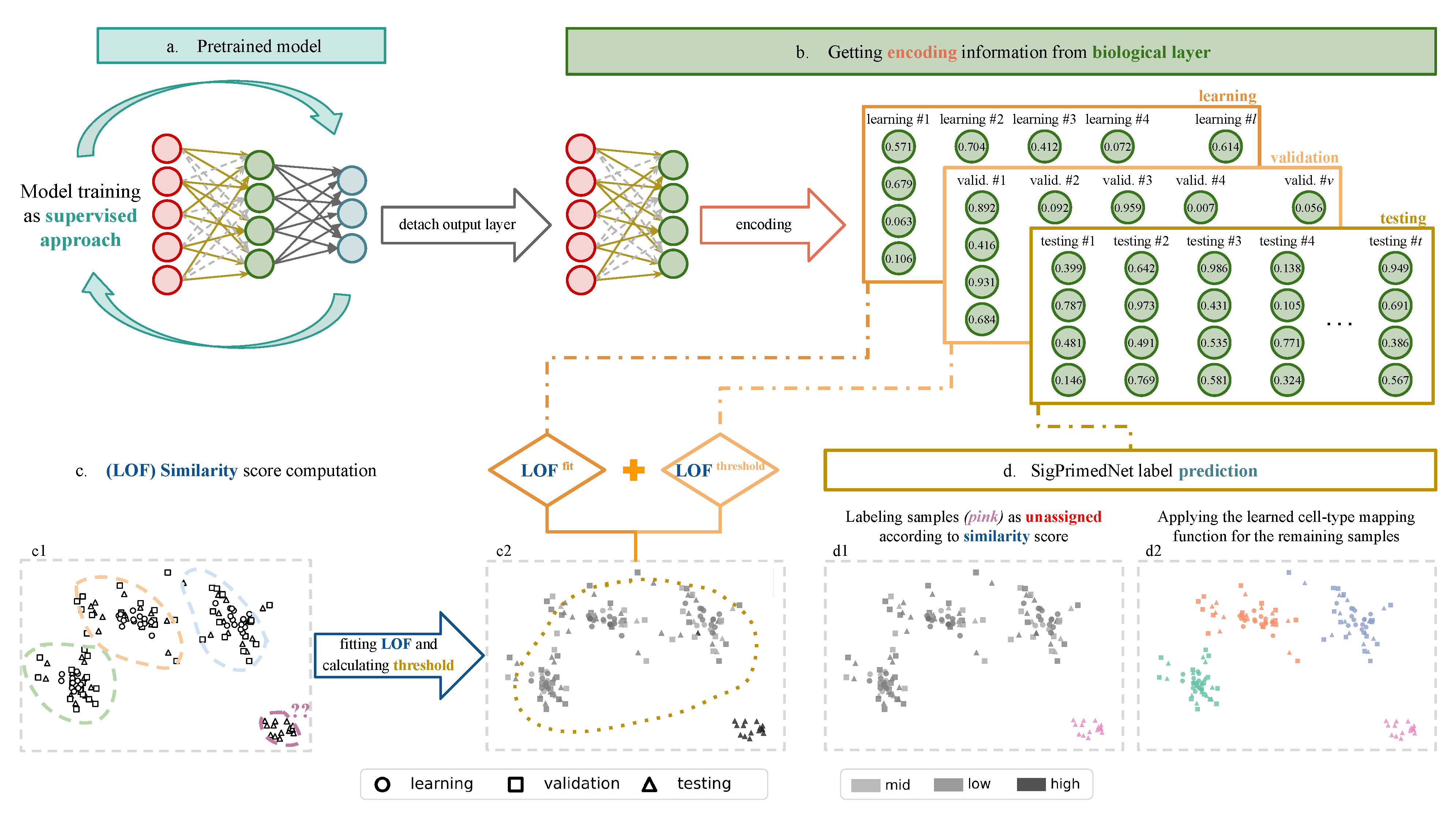
2.6. Network Training and Inference
2.7. Functional Proxies and Representation Learning
3. Results and Discussion
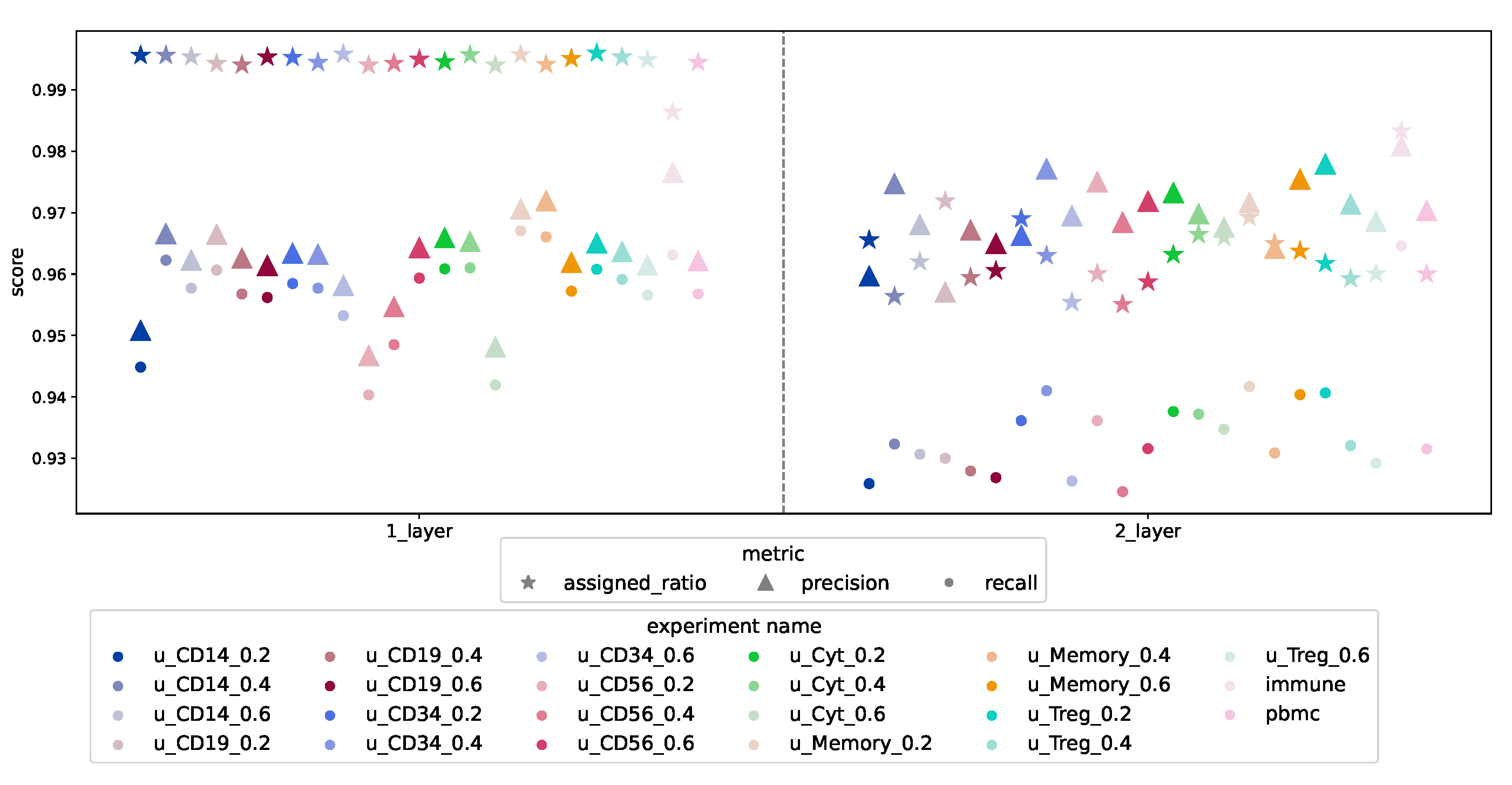
3.1. Model Performance When All Cell Types Are Known
3.1.1. Synthetically Balanced PBMC
3.1.2. Synthetically Unbalanced PBMC
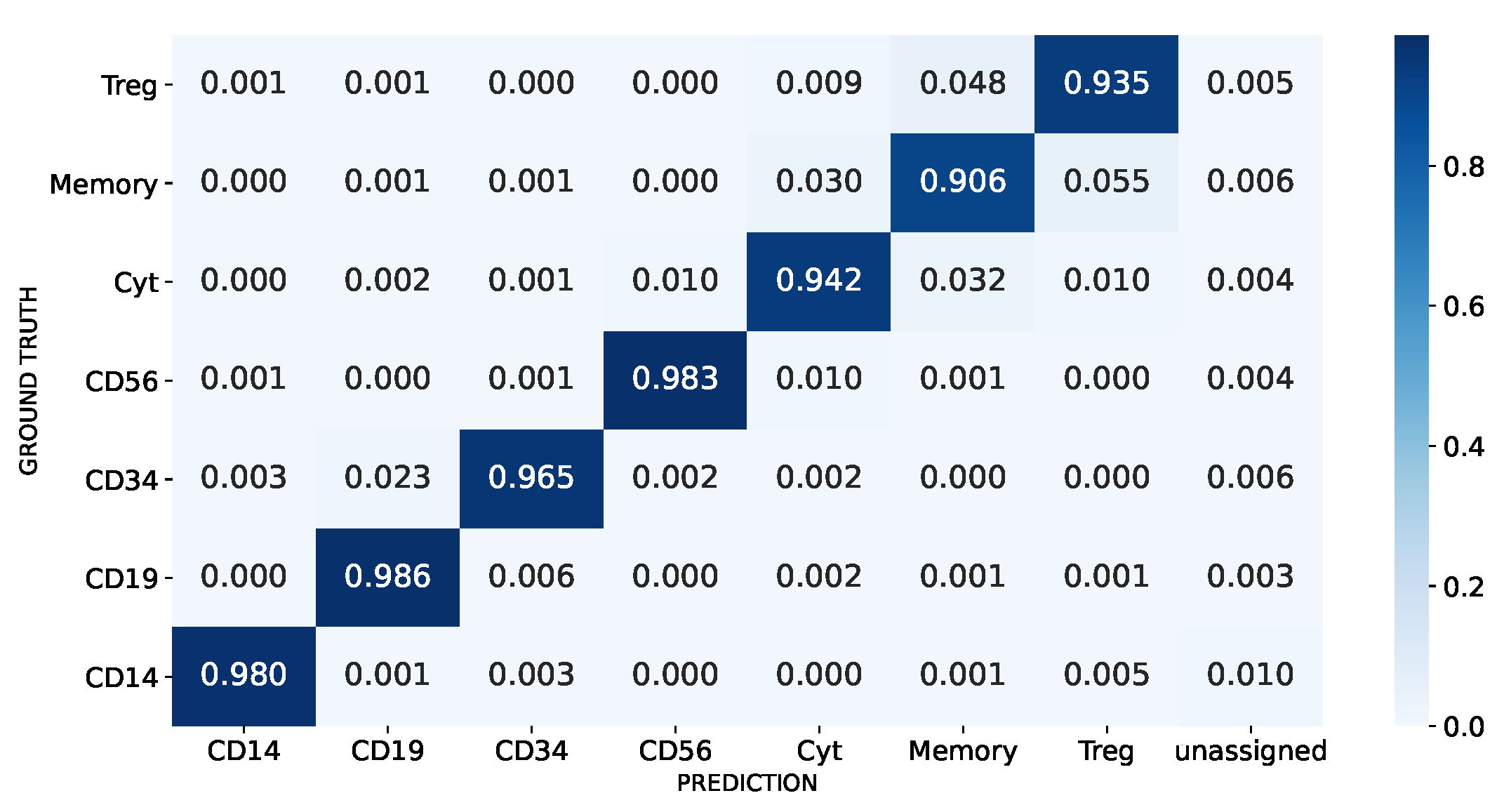
3.1.3. Real-World Unbalanced Scenario
3.1.4. Design Comparison
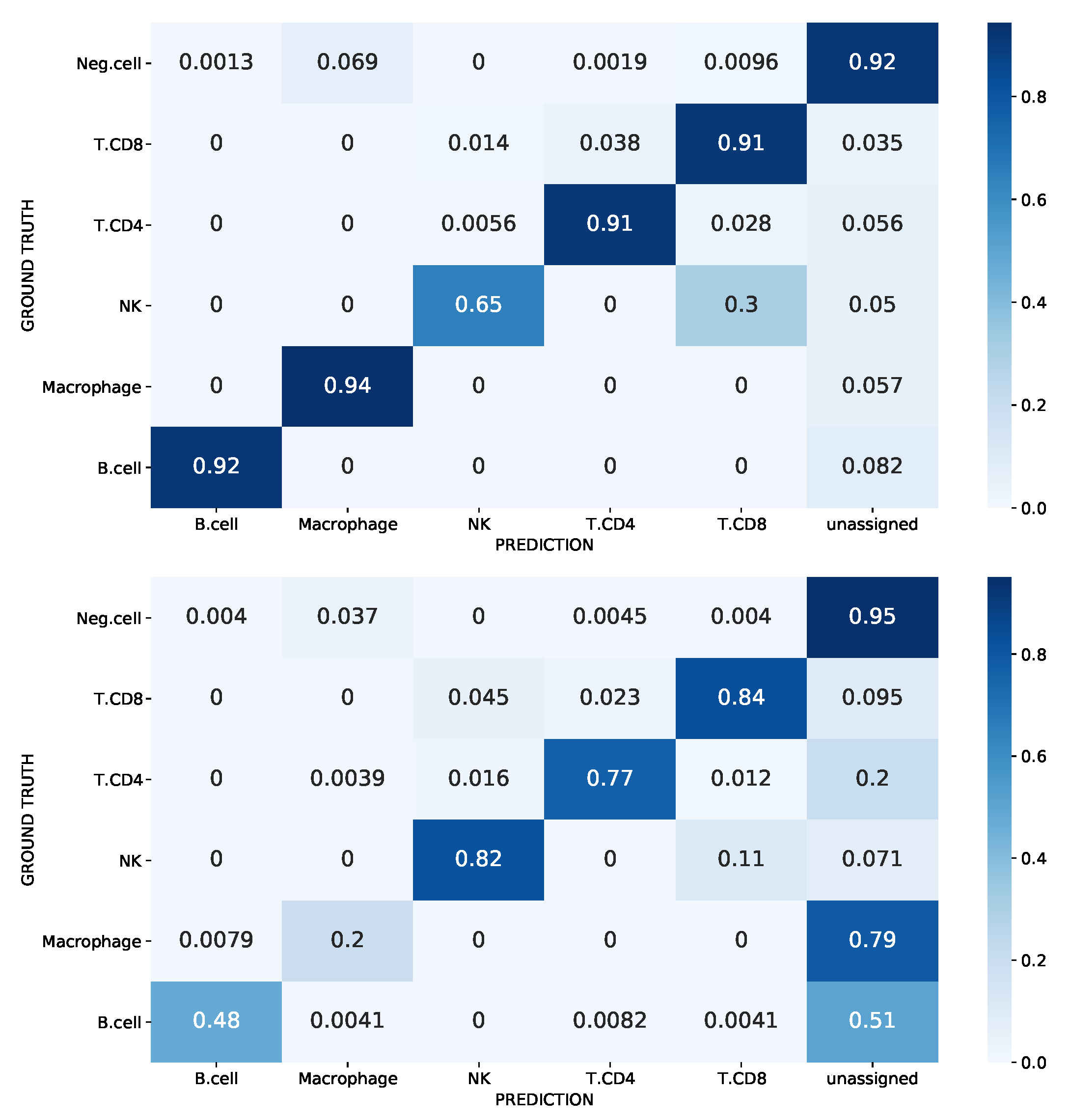
3.2. Unknown Cell-Type Identification
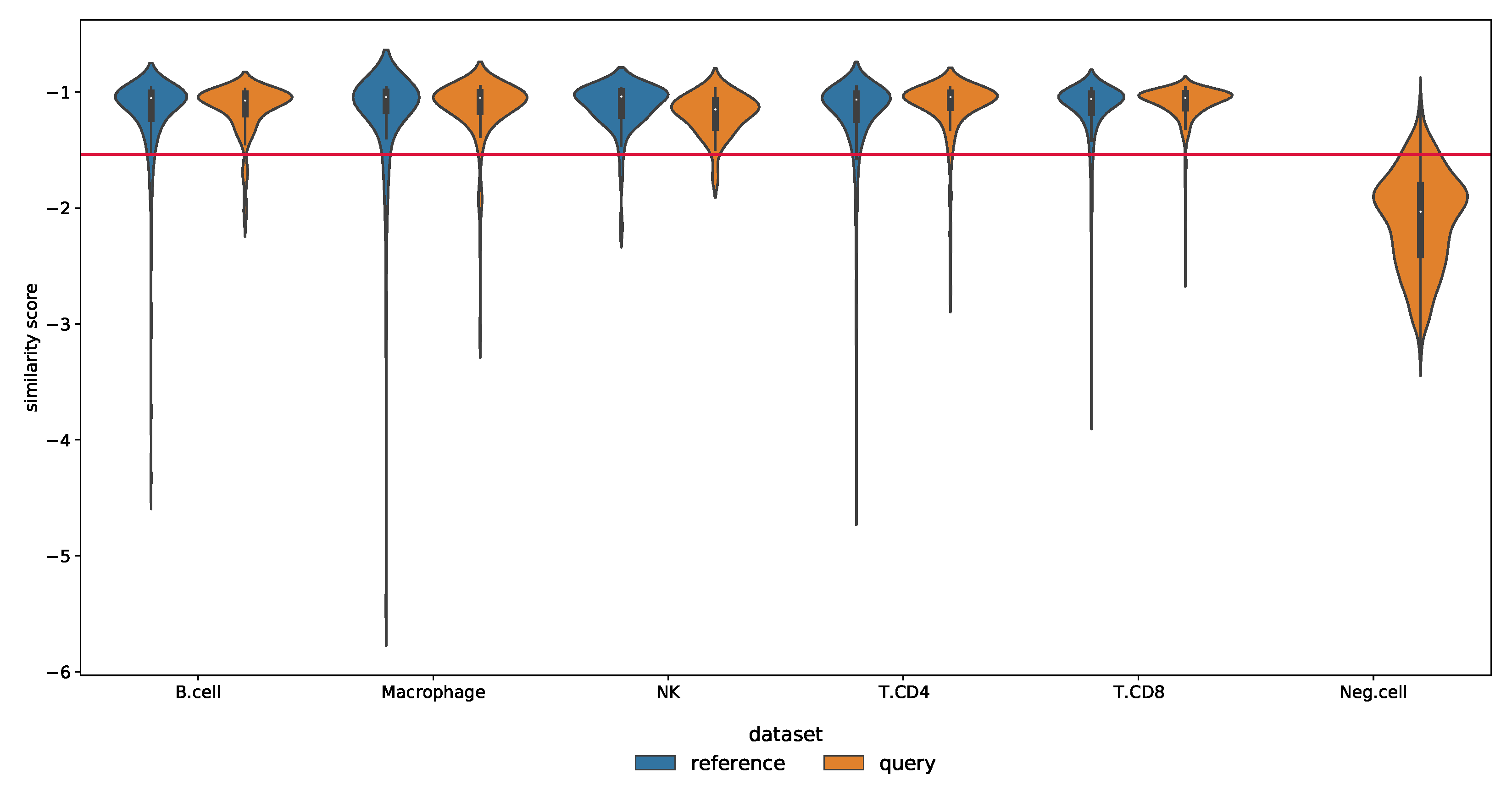
Novelty Detection in the Melanoma Dataset
3.3. SigPrimedNet Provides Biologically Interpretable Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| scRNA-seq | Single-cell RNA sequencing |
| HVGs | Highly variable genes |
| PCA | Principal Component Analysis |
| UMAP | Uniform Manifold Approximation and Projection |
| ANN | Artificial Neural Network |
| UMI | Unique Molecular Identifier |
| KEGG | Kyoto Encyclopedia of Genes and Genomes Database |
| TSNE | t-distributed stochastic neighbor embedding |
| TPM | Transcripts per Million |
| LOF | Local Outlier Factor |
| KNN | K-nearest neighbors |
| PDNN | Pathway-driven Neural Network |
| MACRO | Unweighted average across cell types for any given classification metric |
| WEIGHTED | Support-weighted average across cell types for any given classification metric |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
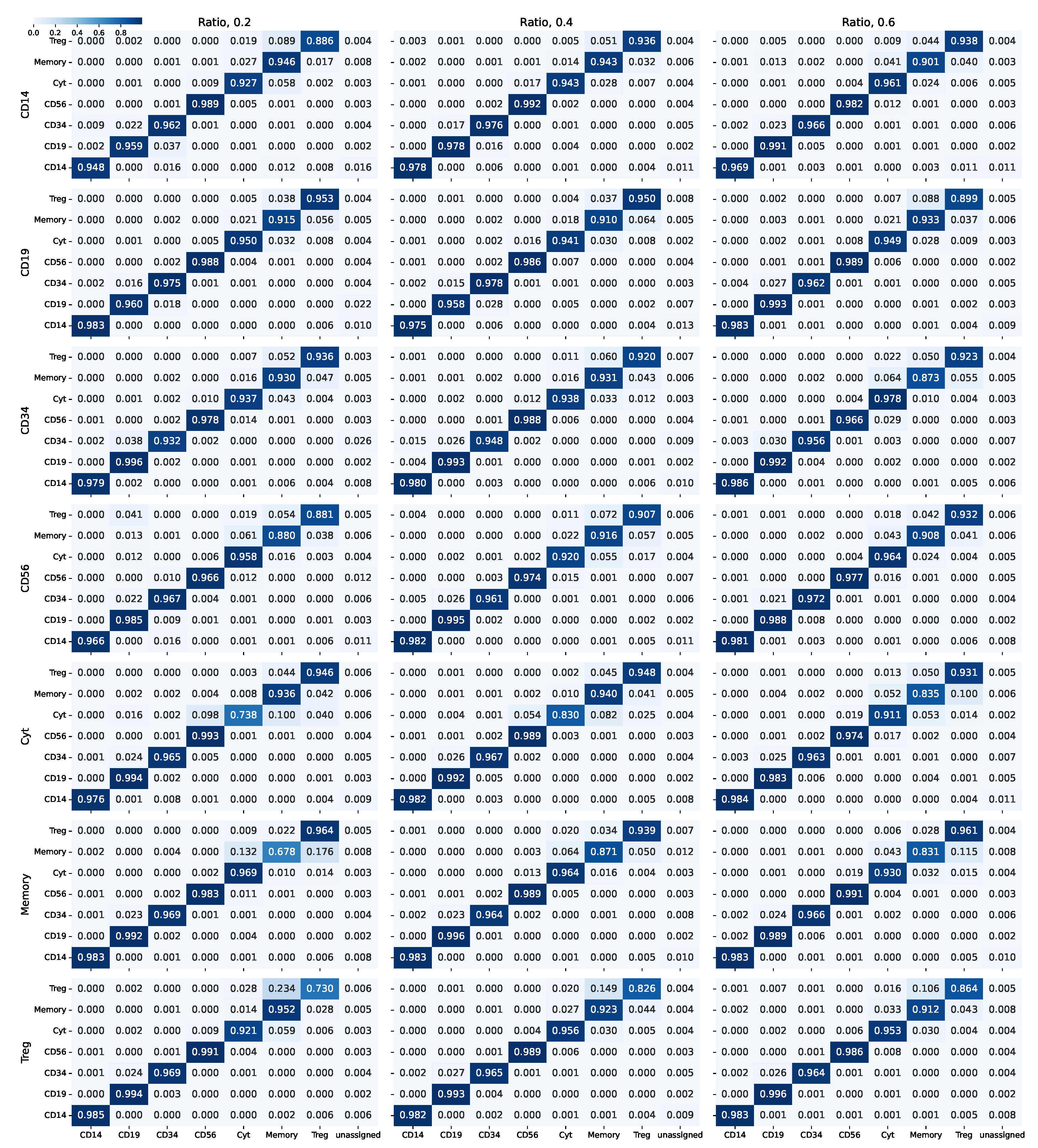{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KeggID | Pathway Name | KeggID | Pathway Name | KeggID | Pathway Name | ||
|---|---|---|---|---|---|---|---|
| hsa03320 | PPAR signaling pathway | hsa04370 | VEGF signaling pathway | hsa04727 | GABAergic synapse | ||
| hsa04010 | MAPK signaling pathway | hsa04380 | Osteoclast differentiation | hsa04728 | Dopaminergic synapse | ||
| hsa04012 | ErbB signaling pathway | hsa04390 | Hippo signaling pathway | hsa04740 | Olfactory transduction | ||
| hsa04014 | Ras signaling pathway | hsa04510 | Focal adhesion | hsa04742 | Taste transduction | ||
| hsa04015 | Rap1 signaling pathway | hsa04520 | Adherens junction | hsa04750 | Inflammatory mediator regulation of TRP channels | ||
| hsa04020 | Calcium signaling pathway | hsa04530 | Tight junction | hsa04810 | Regulation of actin cytoskeleton | ||
| hsa04022 | cGMP-PKG signaling pathway | hsa04540 | Gap junction | hsa04910 | Insulin signaling pathway | ||
| hsa04024 | cAMP signaling pathway | hsa04550 | Signaling pathways regulating pluripotency of stem cells | hsa04911 | Insulin secretion | ||
| hsa04062 | Chemokine signaling pathway | hsa04610 | Complement and coagulation cascades | hsa04912 | GnRH signaling pathway | ||
| hsa04064 | NF-kappa B signaling pathway | hsa04611 | Platelet activation | hsa04913 | Ovarian steroidogenesis | ||
| hsa04066 | HIF-1 signaling pathway | hsa04612 | Antigen processing and presentation | hsa04914 | Progesterone-mediated oocyte maturation | ||
| hsa04068 | FoxO signaling pathway | hsa04620 | Toll-like receptor signaling pathway | hsa04915 | Estrogen signaling pathway | ||
| hsa04071 | Sphingolipid signaling pathway | hsa04621 | NOD-like receptor signaling pathway | hsa04916 | Melanogenesis | ||
| hsa04072 | Phospholipase D signaling pathway | hsa04622 | RIG-I-like receptor signaling pathway | hsa04917 | Prolactin signaling pathway | ||
| hsa04110 | Cell cycle | hsa04623 | Cytosolic DNA-sensing pathway | hsa04918 | Thyroid hormone synthesis | ||
| hsa04114 | Oocyte meiosis | hsa04630 | Jak-STAT signaling pathway | hsa04919 | Thyroid hormone signaling pathway | ||
| hsa04115 | p53 signaling pathway | hsa04650 | Natural killer cell mediated cytotoxicity | hsa04920 | Adipocytokine signaling pathway | ||
| hsa04150 | mTOR signaling pathway | hsa04660 | T cell receptor signaling pathway | hsa04921 | Oxytocin signaling pathway | ||
| hsa04151 | PI3K-Akt signaling pathway | hsa04662 | B cell receptor signaling pathway | hsa04922 | Glucagon signaling pathway | ||
| hsa04152 | AMPK signaling pathway | hsa04664 | Fc epsilon RI signaling pathway | hsa04923 | Regulation of lipolysis in adipocytes | ||
| hsa04210 | Apoptosis | hsa04666 | Fc gamma R-mediated phagocytosis | hsa04924 | Renin secretion | ||
| hsa04211 | Longevity regulating pathway - mammal | hsa04668 | TNF signaling pathway | hsa04925 | Aldosterone synthesis and secretion | ||
| hsa04213 | Longevity regulating pathway - multiple species | hsa04670 | Leukocyte transendothelial migration | hsa04960 | Aldosterone-regulated sodium reabsorption | ||
| hsa04218 | Cellular senescence | hsa04710 | Circadian rhythm | hsa04961 | Endocrine and other factor-regulated calcium reabsorption | ||
| hsa04261 | Adrenergic signaling in cardiomyocytes | hsa04713 | Circadian entrainment | hsa04962 | Vasopressin-regulated water reabsorption | ||
| hsa04270 | Vascular smooth muscle contraction | hsa04720 | Long-term potentiation | hsa04970 | Salivary secretion | ||
| hsa04310 | Wnt signaling pathway | hsa04722 | Neurotrophin signaling pathway | hsa04971 | Gastric acid secretion | ||
| hsa04330 | Notch signaling pathway | hsa04723 | Retrograde endocannabinoid signaling | hsa04972 | Pancreatic secretion | ||
| hsa04340 | Hedgehog signaling pathway | hsa04724 | Glutamatergic synapse | hsa04973 | Carbohydrate digestion and absorption | ||
| hsa04350 | TGF-beta signaling pathway | hsa04725 | Cholinergic synapse | hsa04976 | Bile secretion | ||
| hsa04360 | Axon guidance | hsa04726 | Serotonergic synapse | hsa05100 | Bacterial invasion of epithelial cells |
| CT | KeggID | Circuit Name | CT | KeggID | Circuit Name | |
|---|---|---|---|---|---|---|
| B cells | hsa03320 | PPAR signaling pathway: DBI | NKs | hsa04115 | p53 signaling pathway: TP73 | |
| hsa04670 | Leukocyte transendothelial migration: CDH5 | hsa04151 | PI3K-Akt signaling pathway: EIF4B | |||
| hsa04666 | Fc gamma R-mediated phagocytosis: PLA2G4B | hsa04390 | Hippo signaling pathway: SERPINE1 | |||
| hsa04115 | p53 signaling pathway: CD82 | hsa04152 | AMPK signaling pathway: CCNA2 | |||
| hsa04340 | Hedgehog signaling pathway: GLI1 SUFU | hsa04151 | PI3K-Akt signaling pathway: CDKN1B | |||
| hsa04390 | Hippo signaling pathway: SERPINE1 | hsa04520 | Adherens junction: LEF1 CTNNB1 | |||
| hsa04064 | NF-kappa B signaling pathway: PLCG2 | hsa04210 | Apoptosis: BID | |||
| hsa04724 | Glutamatergic synapse: ADRBK1 | hsa04064 | NF-kappa B signaling pathway: PLCG2 | |||
| hsa04115 | p53 signaling pathway: TP73 | hsa04340 | Hedgehog signaling pathway: GLI1 SUFU | |||
| hsa04620 | Toll-like receptor signaling pathway: CCL5 | hsa04620 | Toll-like receptor signaling pathway: CCL5 | |||
| Erythrocytes | hsa04724 | Glutamatergic synapse: MAPK1 | HSPCs | hsa04340 | Hedgehog signaling pathway: GLI1 SUFU | |
| hsa05100 | Bacterial invasion of epithelial cells: ACTB | hsa04390 | Hippo signaling pathway: SERPINE1 | |||
| hsa04115 | p53 signaling pathway: CD82 | hsa04724 | Glutamatergic synapse: ADRBK1 | |||
| hsa04340 | Hedgehog signaling pathway: GLI1 SUFU | hsa04151 | PI3K-Akt signaling pathway: EIF4B | |||
| hsa03320 | PPAR signaling pathway: FADS2 | hsa04919 | Thyroid hormone signaling pathway: SLC9A1 | |||
| hsa04110 | Cell cycle: ORC3 ORC5 ORC4 ORC2 ORC1 ORC6 MCM7 MCM6 MCM5 MCM4 MCM3 MCM2 | hsa04520 | Adherens junction: LEF1 CTNNB1 | |||
| hsa04670 | Leukocyte transendothelial migration: ACTB CTNNA1 CTNNB1 | hsa04210 | Apoptosis: BID | |||
| hsa04810 | Regulation of actin cytoskeleton: MYL12B MYH9 ACTB | hsa04740 | Olfactory transduction: PDE2A | |||
| hsa04014 | Ras signaling pathway: PLCE1 | hsa04064 | NF-kappa B signaling pathway: TNFSF13B | |||
| hsa03320 | PPAR signaling pathway: DBI | hsa04014 | Ras signaling pathway: PAK4 | |||
| Monocytes | hsa04110 | Cell cycle: CDC6 ORC3 ORC5 ORC4 ORC2 ORC1 ORC6 | T cells | hsa04110 | Cell cycle: CDC6 ORC3 ORC5 ORC4 ORC2 ORC1 ORC6 | |
| hsa04740 | Olfactory transduction: PDE2A | hsa04919 | Thyroid hormone signaling pathway: THRA Triiodothyronine | |||
| hsa04022 | cGMP-PKG signaling pathway: ITPR1 | hsa04724 | Glutamatergic synapse: MAPK1 | |||
| hsa04670 | Leukocyte transendothelial migration: CDH5 | hsa04713 | Circadian entrainment: PRKCA | |||
| hsa04914 | Progesterone-mediated oocyte maturation: CDK1 | hsa04666 | Fc gamma R-mediated phagocytosis: ARF6 | |||
| hsa04210 | Apoptosis: BCL2L1 | hsa04014 | Ras signaling pathway: PLCE1 | |||
| hsa05100 | Bacterial invasion of epithelial cells: ACTB | hsa04650 | Natural killer cell mediated cytotoxicity: TNFRSF10D | |||
| hsa04915 | Estrogen signaling pathway: ESR1 Estradiol-17beta | hsa04024 | cAMP signaling pathway: LIPE | |||
| hsa04668 | TNF signaling pathway: DNM1L | hsa04110 | Cell cycle: TFDP1 E2F4 | |||
| hsa04020 | Calcium signaling pathway: Sphingosine 1-phosphate | hsa04919 | Thyroid hormone signaling pathway: SLC9A1 | |||
| Neutrophyls | hsa04668 | TNF signaling pathway: DNM1L | hsa04919 | Thyroid hormone signaling pathway: NOTCH1 | ||
| hsa04915 | Estrogen signaling pathway: ESR1 Estradiol-17beta | hsa04020 | Calcium signaling pathway: Sphingosine 1-phosphate | |||
| hsa04916 | Melanogenesis: DCT | hsa04660 | T cell receptor signaling pathway: CD40LG | |||
| hsa04970 | Salivary secretion: KCNN4 | hsa04915 | Estrogen signaling pathway: CREB3 | |||
| hsa04014 | Ras signaling pathway: RHOA | hsa04340 | Hedgehog signaling pathway: SMO |
Appendix B
References
- Alavi, A.; Ruffalo, M.; Parvangada, A.; Huang, Z.; Bar-Joseph, Z. A Web Server for Comparative Analysis of Single-Cell RNA-seq Data. Nat. Commun. 2018, 9, 4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlJanahi, A.A.; Danielsen, M.; Dunbar, C.E. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol. Ther. Methods Clin. Dev. 2018, 10, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. Scmap: Projection of Single-Cell RNA-seq Data across Data Sets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kołodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for Technical Noise in Single-Cell RNA-seq Experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics 2019, 35, 2865–2867. [Google Scholar] [CrossRef] [Green Version]
- Tsuyuzaki, K.; Sato, H.; Sato, K.; Nikaido, I. Benchmarking Principal Component Analysis for Large-Scale Single-Cell RNA-sequencing. Genome Biol. 2020, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- Pierson, E.; Yau, C. ZIFA: Dimensionality Reduction for Zero-Inflated Single-Cell Gene Expression Analysis. Genome Biol. 2015, 16, 241. [Google Scholar] [CrossRef] [Green Version]
- van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep Generative Modeling for Single-Cell Transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Naghipourfar, M.; Luecken, M.D.; Khajavi, M.; Büttner, M.; Wagenstetter, M.; Avsec, Ž.; Gayoso, A.; Yosef, N.; Interlandi, M.; et al. Mapping Single-Cell Data to Reference Atlases by Transfer Learning. Nat. Biotechnol. 2022, 40, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.B.; Nathan, A.; Weinand, K.; Zhang, F.; Millard, N.; Rumker, L.; Moody, D.B.; Korsunsky, I.; Raychaudhuri, S. Efficient and Precise Single-Cell Reference Atlas Mapping with Symphony. Nat. Commun. 2021, 12, 5890. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Rybakov, S.; Hrovatin, K.; Hediyeh-zadeh, S.; Talavera-López, C.; Misharin, A.V.; Theis, F.J. Biologically Informed Deep Learning to Infer Gene Program Activity in Single Cells. Nat. Cell Biol. 2023, 25, 337–350. [Google Scholar] [CrossRef]
- Li, C.; Liu, B.; Kang, B.; Liu, Z.; Liu, Y.; Chen, C.; Ren, X.; Zhang, Z. SciBet as a Portable and Fast Single Cell Type Identifier. Nat. Commun. 2020, 11, 1818. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Lin, X.; Li, Z.; Wu, H. A Comprehensive Comparison of Supervised and Unsupervised Methods for Cell Type Identification in Single-Cell RNA-seq. Brief. Bioinform. 2022, 23, bbab567. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, M.; Gu, Z.; Pan, G. Overfitting Remedy by Sparsifying Regularization on Fully-Connected Layers of CNNs. Neurocomputing 2019, 328, 69–74. [Google Scholar] [CrossRef]
- Elmarakeby, H.A.; Hwang, J.; Arafeh, R.; Crowdis, J.; Gang, S.; Liu, D.; AlDubayan, S.H.; Salari, K.; Kregel, S.; Richter, C.; et al. Biologically Informed Deep Neural Network for Prostate Cancer Discovery. Nature 2021, 598, 348–352. [Google Scholar] [CrossRef]
- Ma, J.; Yu, M.K.; Fong, S.; Ono, K.; Sage, E.; Demchak, B.; Sharan, R.; Ideker, T. Using Deep Learning to Model the Hierarchical Structure and Function of a Cell. Nat. Methods 2018, 15, 290–298. [Google Scholar] [CrossRef]
- Lin, C.; Jain, S.; Kim, H.; Bar-Joseph, Z. Using Neural Networks for Reducing the Dimensions of Single-Cell RNA-Seq Data. Nucleic Acids Res. 2017, 45, e156. [Google Scholar] [CrossRef] [Green Version]
- Gundogdu, P.; Loucera, C.; Alamo-Alvarez, I.; Dopazo, J.; Nepomuceno, I. Integrating Pathway Knowledge with Deep Neural Networks to Reduce the Dimensionality in Single-Cell RNA-seq Data. BioData Min. 2022, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.R.; Cubuk, C.; Amadoz, A.; Salavert, F.; Carbonell-Caballero, J.; Dopazo, J. High Throughput Estimation of Functional Cell Activities Reveals Disease Mechanisms and Predicts Relevant Clinical Outcomes. Oncotarget 2016, 8, 5160–5178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Liu, M.; Zhang, Y.; Wang, B.; Zhu, C.; Wang, C.; Li, Q.; Huo, Y.; Guo, J.; Xu, C.; et al. Single-Cell Transcriptomic Landscape of Human Blood Cells. Natl. Sci. Rev. 2021, 8, nwaa180. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, T.; Vähärautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting Absolute Numbers of Molecules Using Unique Molecular Identifiers. Nat. Methods 2012, 9, 72–74. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Nair, V.; Hinton, G.E. Rectified Linear Units Improve Restricted Boltzmann Machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Madison, WI, USA, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA Abundance Using RNA-seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. = Theor. Den Biowiss. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Glorot, X.; Bengio, Y. Understanding the Difficulty of Training Deep Feedforward Neural Networks. In Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics, JMLR Workshop and Conference Proceedings, Sardinia, Italy, 13–15 May 2010; pp. 249–256. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2017, arXiv:cs/1412.6980. [Google Scholar] [CrossRef]
- Breunig, M.M.; Kriegel, H.P.; Ng, R.T.; Sander, J. LOF: Identifying Density-Based Local Outliers. ACM Sigmod Rec. 2000, 29, 93–104. [Google Scholar] [CrossRef]
- Hein, M.; Andriushchenko, M.; Bitterwolf, J. Why Relu Networks Yield High-Confidence Predictions Far Away from the Training Data and How to Mitigate the Problem. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 15–20 June 2019; pp. 41–50. [Google Scholar]
- Bengio, Y.; Courville, A.; Vincent, P. Representation Learning: A Review and New Perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpf, P.S.; Du, X.; Imanishi, H.; Kunisaki, Y.; Semba, Y.; Noble, T.; Smith, R.C.G.; Rose-Zerili, M.; West, J.J.; Oreffo, R.O.C.; et al. Transfer Learning Efficiently Maps Bone Marrow Cell Types from Mouse to Human Using Single-Cell RNA Sequencing. Commun. Biol. 2020, 3, 736. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human Haematopoietic Stem Cell Lineage Commitment Is a Continuous Process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detmer, K.; Walker, A.N.; Jenkins, T.M.; Steele, T.A.; Dannawi, H. Erythroid Differentiation in Vitro Is Blocked by Cyclopamine, an Inhibitor of Hedgehog Signaling. Blood Cells Mol. Dis. 2000, 26, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, F.; He, D.; Ji, X.; Gao, J.; Liu, W.; Wang, Y.; Liu, Q.; Xin, T. Immune-Related Gene SERPINE1 Is a Novel Biomarker for Diffuse Lower-Grade Gliomas via Large-Scale Analysis. Front. Oncol. 2021, 11, 646060. [Google Scholar] [CrossRef]
- Yahata, T.; Ibrahim, A.A.; Muguruma, Y.; Eren, M.; Shaffer, A.M.; Watanabe, N.; Kaneko, S.; Nakabayashi, T.; Dan, T.; Hirayama, N.; et al. TGF-β–Induced Intracellular PAI-1 Is Responsible for Retaining Hematopoietic Stem Cells in the Niche. Blood 2017, 130, 2283–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, K.; Yoshioka, K.; Kano, K.; Kurano, M.; Saigusa, D.; Aoki, J.; Yatomi, Y.; Takuwa, N.; Okamoto, Y.; Proia, R.L.; et al. Sphingosine Kinase-2 Prevents Macrophage Cholesterol Accumulation and Atherosclerosis by Stimulating Autophagic Lipid Degradation. Sci. Rep. 2019, 9, 18329. [Google Scholar] [CrossRef] [Green Version]
- Galkina, S.I.; Fedorova, N.V.; Ksenofontov, A.L.; Stadnichuk, V.I.; Baratova, L.A.; Sud’Ina, G.F. Neutrophils as a Source of Branched-Chain, Aromatic and Positively Charged Free Amino Acids. Cell Adhes. Migr. 2019, 13, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Rijken, F.; Bruijnzeel, P.L.B. The Pathogenesis of Photoaging: The Role of Neutrophils and Neutrophil-Derived Enzymes. J. Investig. Dermatol. Symp. Proc. 2009, 14, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Semenov, A.N.; Shirshin, E.A.; Muravyov, A.V.; Priezzhev, A.V. The Effects of Different Signaling Pathways in Adenylyl Cyclase Stimulation on Red Blood Cells Deformability. Front. Physiol. 2019, 10, 923. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, S.; Natalini, A.; Folgori, A.; Capone, S.; Nicosia, A.; Santoni, A.; Di Rosa, F. Antigen-Specific CD8 T Cells in Cell Cycle Circulate in the Blood after Vaccination. Scand. J. Immunol. 2019, 89, e12735. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. arXiv 2015, arXiv:1603.04467. [Google Scholar]









| PBMC | Immune | Melanoma | |||
|---|---|---|---|---|---|
| Cell Type | # of Samples | Cell Type | # of Samples | Cell Type | # of Samples |
| CD14+ | 2500 | B cells | 1465 | B.cell | 818 |
| CD19+ | 2500 | Erythrocytes | 1747 | Macrophage | 420 |
| CD34+ | 2500 | HSPCs | 3742 | NK | 92 |
| CD56+ | 2500 | Monocytes | 954 | T.CD4+ | 856 |
| CD8+ Cytotoxic | 2500 | Neutrophils | 485 | T.CD8+ | 1759 |
| CD4+/CD45RO+ Memory | 2500 | NK | 546 | Negative cells | 2228 |
| Treg | 2500 | T cells | 517 | ||
| Dataset | Hyperparameter | Hyperparameter Value |
|---|---|---|
| PBMC | epochs | 100 |
| batch_size | 10 | |
| Immune | kernel_initializer | glorot_uniform + sig-informed |
| bias_initializer | zeros | |
| Melanoma | activation | relu (hidden layers)/softmax (last layer) |
| optimizer | Adam |
| DESIGN | |||
|---|---|---|---|
| Dataset | Experiment | 1-Layer | 2-Layer |
| PBMC | RepeatedStratifiedKFold (10 k-fold with 50 iterations) | mean, 3.20 min std, 0.77 min total execution time is 13.28 h | mean, 3.26 min std, 0.87 min total execution time is 13.52 h |
| Immune | RepeatedStratifiedKFold (10 k-fold with 30 iterations) | mean, 2.82 min std, 0.69 min total execution time is 14.07 h | mean, 4.31 min std, 1.30 min total execution time is 21.48 h |
| train_test_split (50% test size with 100 iterations) | mean, 1.94 min std, 0.92 min total execution time is 3.2 h | mean, 1.79 min std, 0.51 min total execution time is 2.95 h | |
| Melanoma | training with reference dataset (one iteration) | total execution time is 1.96 min | total execution time is 3.7 min |
| MACRO | WEIGHTED | |||||||
|---|---|---|---|---|---|---|---|---|
| Design | F1 | Precision | Recall | F1 | Precision | Recall | Accuracy | Balanced Accuracy |
| SigPrimedNet (1L) | 0.838 | 0.823 | 0.884 | 0.926 | 0.945 | 0.919 | 0.919 | 0.884 |
| SigPrimedNet (2L) | 0.743 | 0.785 | 0.796 | 0.878 | 0.927 | 0.846 | 0.846 | 0.796 |
| PDNN | 0.861 | 0.922 | 0.844 | 0.933 | 0.938 | 0.936 | 0.936 | 0.844 |
| PDNN (*) | 0.499 | 0.454 | 0.753 | 0.241 | 0.224 | 0.326 | 0.326 | 0.753 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gundogdu, P.; Alamo, I.; Nepomuceno-Chamorro, I.A.; Dopazo, J.; Loucera, C. SigPrimedNet: A Signaling-Informed Neural Network for scRNA-seq Annotation of Known and Unknown Cell Types. Biology 2023, 12, 579. https://doi.org/10.3390/biology12040579
Gundogdu P, Alamo I, Nepomuceno-Chamorro IA, Dopazo J, Loucera C. SigPrimedNet: A Signaling-Informed Neural Network for scRNA-seq Annotation of Known and Unknown Cell Types. Biology. 2023; 12(4):579. https://doi.org/10.3390/biology12040579
Chicago/Turabian StyleGundogdu, Pelin, Inmaculada Alamo, Isabel A. Nepomuceno-Chamorro, Joaquin Dopazo, and Carlos Loucera. 2023. "SigPrimedNet: A Signaling-Informed Neural Network for scRNA-seq Annotation of Known and Unknown Cell Types" Biology 12, no. 4: 579. https://doi.org/10.3390/biology12040579