

Amniotic Fluid Mesenchymal Stromal Cells Derived from Fetuses with Isolated Cardiac Defects Exhibit Decreased Proliferation and Cardiomyogenic Potential

Abstract
:Simple Summary
Abstract
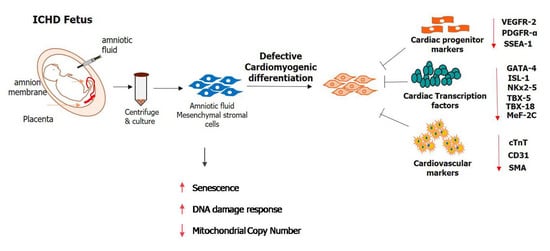
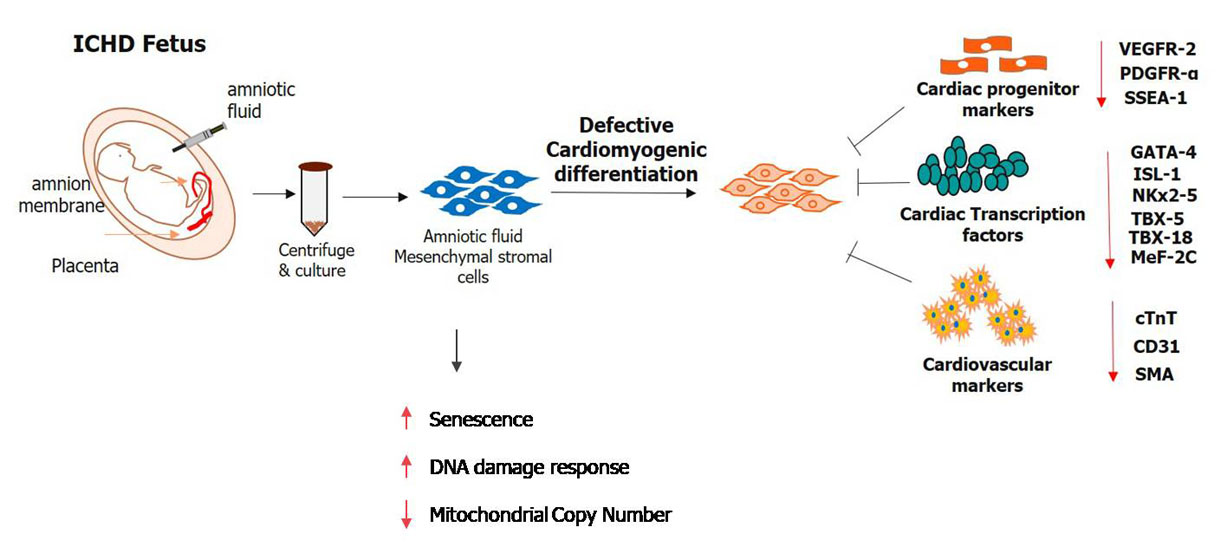
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Isolation and Culture of Normal AF-MSCs and ICHD AF-MSCs
2.3. Immunophenotypic Characterization of Normal AF-MSCs and ICHD AF-MSCs
2.4. In-Vitro Adipogenic, Osteogenic and Chondrogenic Differentiation
2.5. RNA Isolation and Real-Time PCR
2.6. Growth Kinetics
2.7. MTT Assay
2.8. Senescence-Associated (SA) β-Galactosidase Assay
2.9. Expression of Senescence and DNA Damage Associated Genes
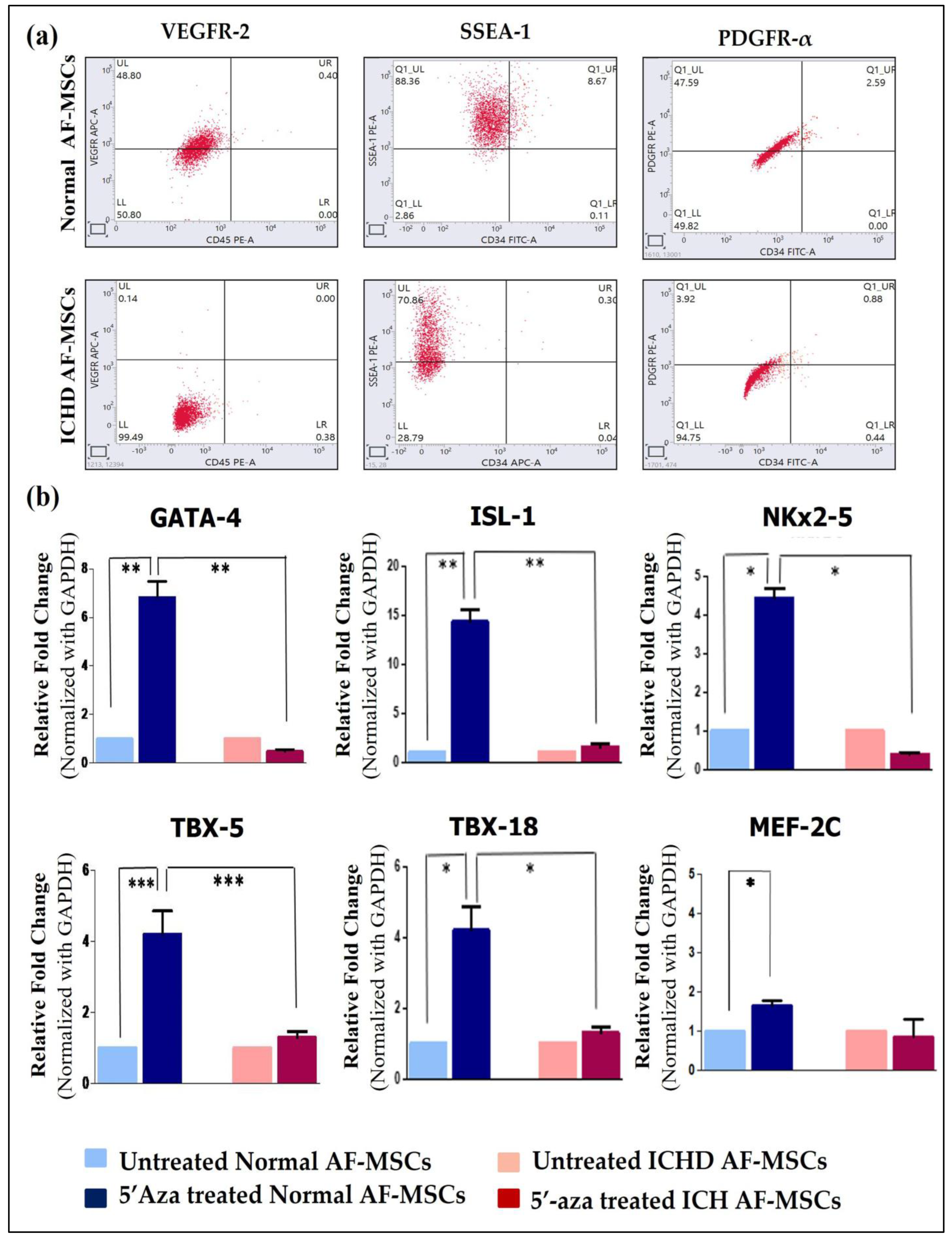
2.10. Characterization of Normal AF-MSCs and ICHD AF-MSCs for the Expression of Cardiac Progenitor Markers
2.11. Analysis of Cardiac Transcription Factors Expression in Normal AF-MSCs and ICHD AF-MSCs
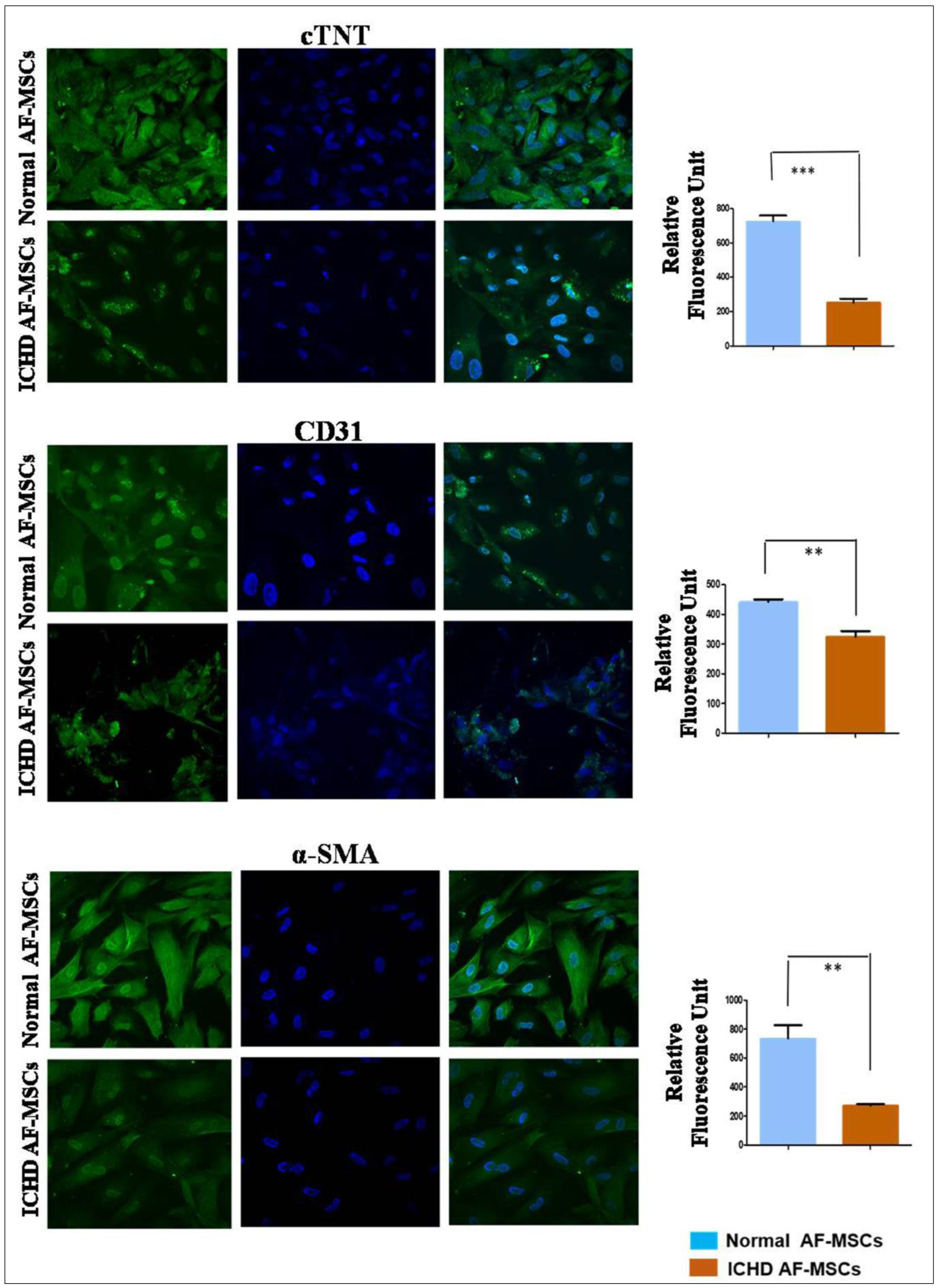
2.12. Cardiovascular Trilineage Differentiation of Normal AF-MSCs and ICHD AF-MSCs
2.13. Endothelial Differentiation
2.14. Smooth Muscle Actin Differentiation
2.15. Statistical Data Analysis
3. Results
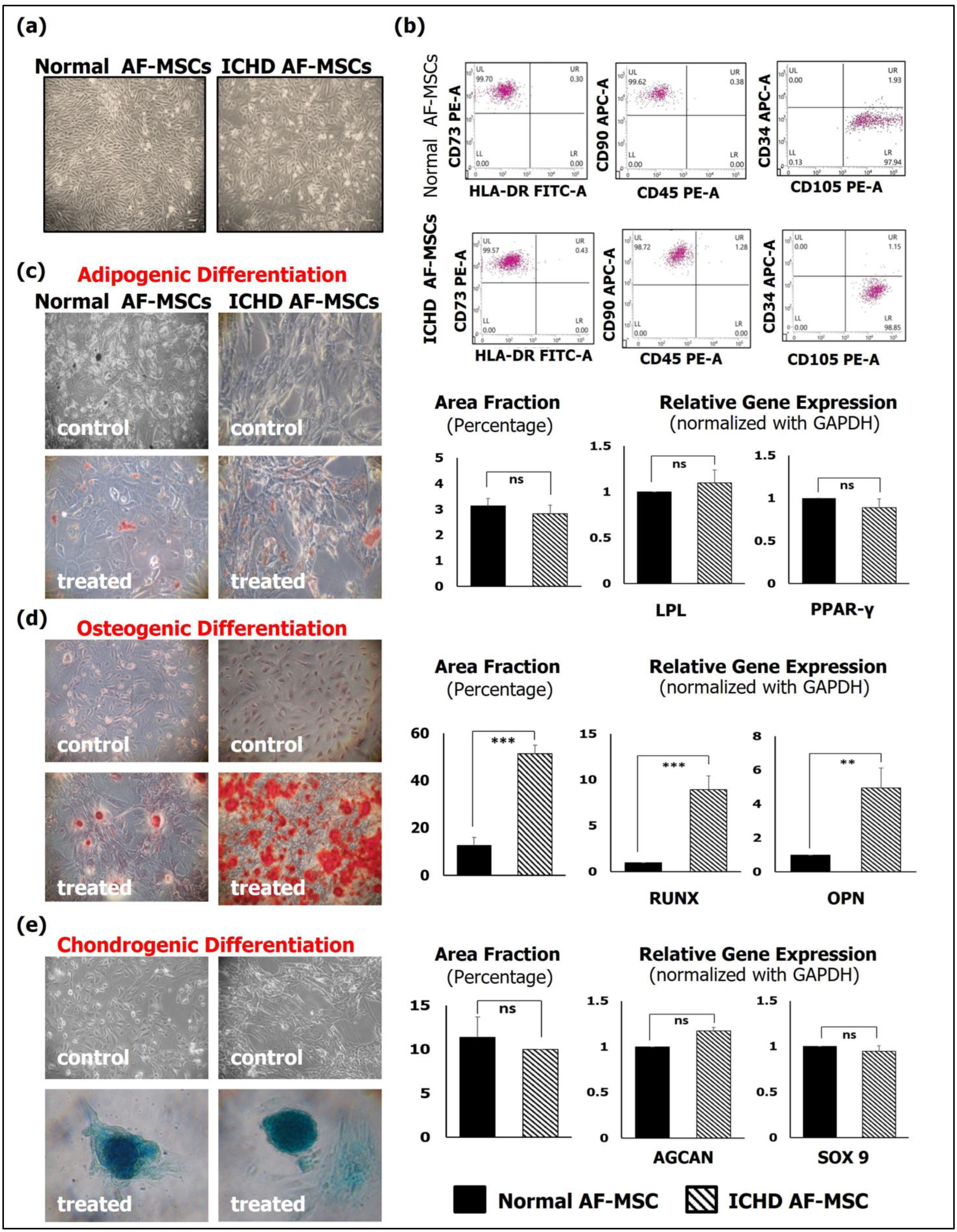
3.1. Morphology, Immunophenotypic Characterization, and Tri-Lineage Differentiation of Normal AF-MSCs and ICHD AF-MSCs
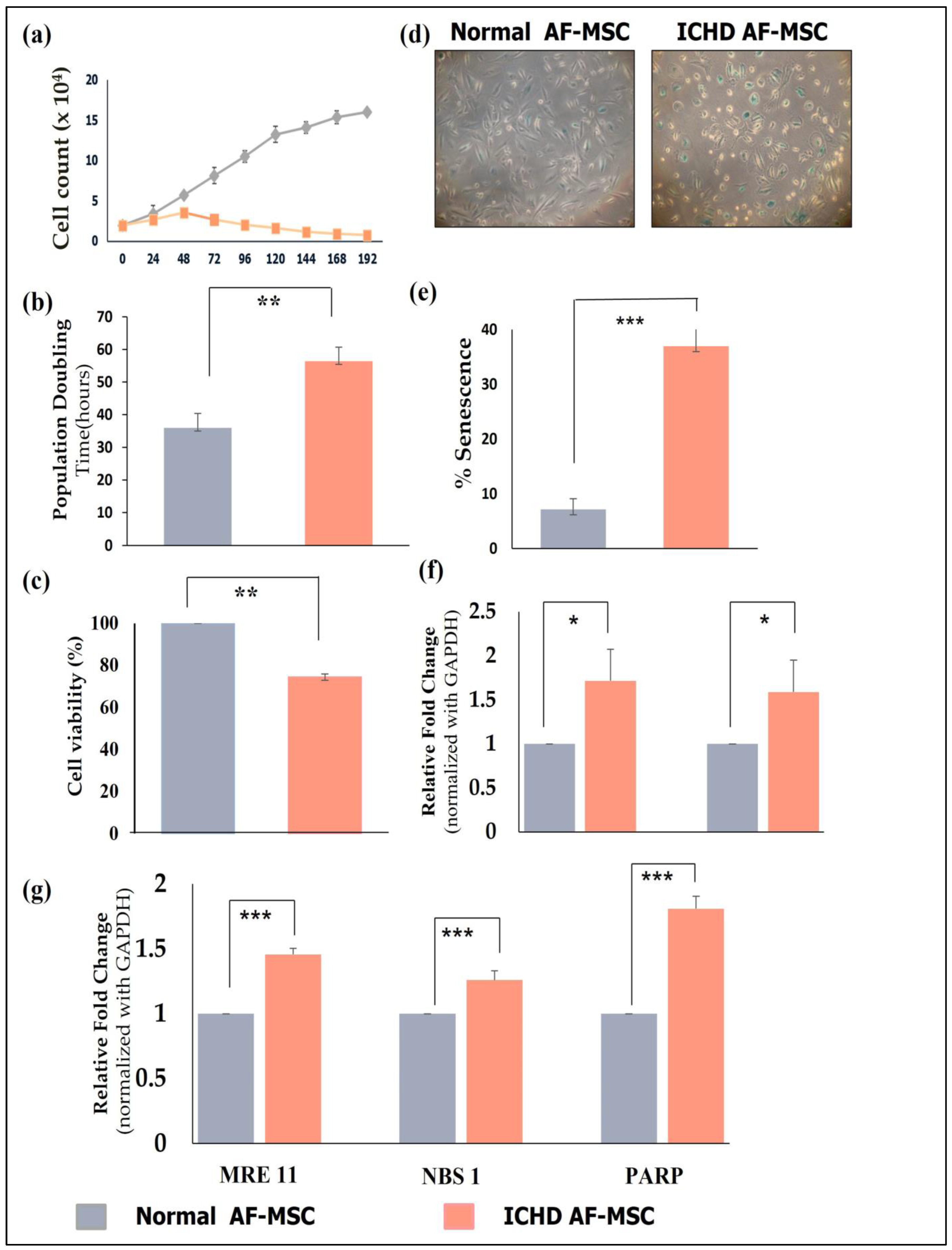
3.2. Growth Kinetics Studies of Normal AF-MSCs and ICHD AF-MSCs
3.3. Expression of Cardiac Progenitor Markers and Transcription Factors in Normal AF-MSCs and ICHD AF-MSCs
3.4. Tri-Lineage Cardiovascular Differentiation Potential of Normal AF-MSCs and ICHD AF-MSCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krantz, I.D.; Smith, R.; Colliton, R.P.; Tinkel, H.; Zackai, E.H.; Piccoli, D.A.; Goldmuntz, E.; Spinner, N.B. Jagged1 Mutations in Patients Ascertained with Isolated Congenital Heart Defects. Am. J. Med. Genet. 1999, 84, 56–60. [Google Scholar] [CrossRef]
- Fantasia, I.; Kasapoglu, D.; Kasapoglu, T.; Syngelaki, A.; Akolekar, R.; Nicolaides, K.H. Fetal Major Cardiac Defects and Placental Dysfunction at 11–13 Weeks’ Gestation. Ultrasound Obstet. Gynecol. 2018, 51, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Llurba, E.; Syngelaki, A.; Sánchez, O.; Carreras, E.; Cabero, L.; Nicolaides, K.H. Maternal Serum Placental Growth Factor at 11-13 Weeks’ Gestation and Fetal Cardiac Defects. Ultrasound Obstet. Gynecol. 2013, 42, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Haveman, I.; Fleurke-Rozema, J.H.; Mulder, E.J.H.; Benders, M.; du Marchie Sarvaas, G.; ter Heide, H.; de Heus, R.H.; Bilardo, C.M. Growth Patterns in Fetuses with Isolated Cardiac Defects. Prenat. Diagn. 2018, 38, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; McBride, K.L.; Garg, V.; Zhao, M.T. Decoding Genetics of Congenital Heart Disease Using Patient-Derived Induced Pluripotent Stem Cells (IPSCs). Front. Cell. Dev. Biol. 2021, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Lin, L.; Yu, X.; Song, Y.; Yang, Q.; He, L.; Pan, B.; Jiang, H. Congenital Heart Defects in Patients with Isolated Microtia: Evaluation Using Colour Doppler Echocardiographic Image. Cardiol. Young 2021, 31, 260–263. [Google Scholar] [CrossRef]
- Yun, S.W. Congenital Heart Disease in the Newborn Requiring Early Intervention. Korean J. Pediatr. 2011, 54, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell. Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Oikonomou, E.K.; Moris, D.; Schizas, D.; Economopoulos, K.P.; Mylonas, K.S. Stem Cell Therapy for Congenital Heart Disease: A Systematic Review. Circulation 2017, 136, 2373–2385. [Google Scholar] [CrossRef]
- Wehman, B.; Kaushal, S. The Emergence of Stem Cell Therapy for Patients with Congenital Heart Disease. Circ. Res. 2015, 116, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Ambastha, C.; Bittle, G.J.; Morales, D.; Parchment, N.; Saha, P.; Mishra, R.; Sharma, S.; Vasilenko, A.; Gunasekaran, M.; Al-Suqi, M.T.; et al. Regenerative Medicine Therapy for Single Ventricle Congenital Heart Disease. Transl. Pediatr. 2018, 7, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and Clinical Applications of Mesenchymal Stem Cells State-of-the-Art Review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Chen, W.; Liu, W.; Du, G.Q.; Jiang, S.L.; Tian, W.C.; Sun, L.; Li, R.K.; Tian, H. The Effect of Age on the Efficacy of Human Mesenchymal Stem Cell Transplantation after a Myocardial Infarction. Rejuvenation Res. 2010, 13, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, A.; Sen, D. Mesenchymal Stem Cells in Cardiac Regeneration: A Detailed Progress Report of the Last 6 Years (2010–2015). Stem Cell. Res. Ther. 2016, 7, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, Y.; Yang, L.; Huang, M.; Zhang, X.; Wang, X.; Guan, X.; Yang, P.; Wang, Y.; Meng, L.; et al. Analysis of Biomarkers for Congenital Heart Disease Based on Maternal Amniotic Fluid Metabolomics. Front. Cardiovasc. Med. 2021, 8, 671191. [Google Scholar] [CrossRef]
- Qiu, C.; Ge, Z.; Cui, W.; Yu, L.; Li, J. Human Amniotic Epithelial Stem Cells: A Promising Seed Cell for Clinical Applications. Int. J. Mol. Sci. 2020, 21, 7730. [Google Scholar] [CrossRef]
- Maguire, C.T.; Sunderland, R.; Demarest, B.; Gorsi, B.; Jackson, J.; Lopez-Izquierdo, A.; Tristani-Firouzi, M.; Yost, H.J.; Condic, M.L. Deriving Cardiomyocytes from Human Amniocytes. Biorxiv. Org. 2018, 475624. [Google Scholar]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Jain, M.; Minocha, E.; Tripathy, N.K.; Singh, N.; Chaturvedi, C.P.; Nityanand, S. Comparison of the Cardiomyogenic Potency of Human Amniotic Fluid and Bone Marrow Mesenchymal Stem Cells. Int. J. Stem Cells 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Utama, B.; Afriwardi, A.; Santoso, B.; Ali, H. Isolation of Amniotic Fluid Mesenchymal Stem Cells Obtained from Cesarean Sections. Open. Access. Maced. J. Med. Sci. 2020, 8, 245–249. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Somal, A.; Bhat, I.A.; Indu, B.; Pandey, S.; Panda, B.S.K.; Thakur, N.; Sarkar, M.; Chandra, V.; Saikumar, G.; Sharma, G.T. A Comparative Study of Growth Kinetics, in Vitro Differentiation Potential and Molecular Characterization of Fetal Adnexa Derived Caprine Mesenchymal Stem Cells. PLoS ONE 2016, 11, e0156821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessio, N.; Pipino, C.; Mandatori, D.; di Tomo, P.; Ferone, A.; Marchiso, M.; Melone, M.A.B.; Peluso, G.; Pandolfi, A.; Galderisi, U. Mesenchymal Stromal Cells from Amniotic Fluid Are Less Prone to Senescence Compared to Those Obtained from Bone Marrow: An in Vitro Study. J. Cell Physiol. 2018, 233, 8996–9006. [Google Scholar] [CrossRef]
- Davydova, D.A.; Vorotelyak, E.A.; Zinovieva, R.D.; Kabaeva, N.V.; Terskikh, V.V.; Vasiliev, A.V. Cell Phenotypes in Human Amniotic Fluid. Acta Nat. 2009, 1, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.S.; Lee, J.L.; Chang, Y.J.; Hwang, S.M. Isolation of human multipotent mesenchymal stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Human. Reprod. 2004, 19, 1450–1456. [Google Scholar] [CrossRef]
- Mazzoni, E.; Mazziotta, C.; Iaquinta, M.R.; Lanzillotti, C.; Fortini, F.; D’Agostino, A.; Martini, F. Enhanced osteogenic differentiation of human bone marrow-derived mesenchymal stem cells by a hybrid hydroxylapatite/collagen scaffold. Front. Cell. Dev. Biol. 2021, 8, 610570. [Google Scholar] [CrossRef]
- James, A.W. Review of signaling pathways governing MSC osteogenic and adipogenic differentiation. Scientifica 2013, 2013, 684736. [Google Scholar] [CrossRef]
- McKee, M.D.; Nanci, A. Osteopontin at mineralized tissue interfaces in bone, teeth, and osseointegrated implants: Ultrastructural distribution and implications for mineralized tissue formation, turnover, and repair. Microsc. Res. Tech. 1996, 33, 141–164. [Google Scholar] [CrossRef]
- Liu, H.Y.; Liu, M.C.; Wang, M.F.; Chen, W.H.; Tsai, C.Y.; Wu, K.H.; Deng, W.P. Potential osteoporosis recovery by deep sea water through bone regeneration in SAMP8 mice. Evid. Based Complement. Altern. Med. 2013, 2013, 161976. [Google Scholar] [CrossRef] [Green Version]
- Perin, L.; Sedrakyan, S.; Da Sacco, S.; De Filippo, R. Characterization of human amniotic fluid stem cells and their pluripotential capability. Methods Cell Biol. 2008, 86, 85–99. [Google Scholar]
- Atala, A. Amniotic Fluid-Derived Pluripotential Cells. Essent. Stem Cell Biol. Second. Ed. 2009, 145–150. [Google Scholar]
- Loukogeorgakis, S.P.; De Coppi, P. Concise review: Amniotic fluid stem cells: The known, the unknown, and potential regenerative medicine applications. Stem Cells. 2017, 35, 1663–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieternella, S. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood 2003, 102, 1548–1549. [Google Scholar]
- Zheng, Y.B.; Gao, Z.L.; Xie, C.; Zhu, H.P.; Peng, L.; Chen, J.H.; Chong, Y.T. Characterization and Hepatogenic Differentiation of Mesenchymal Stem Cells from Human Amniotic Fluid and Human Bone Marrow: A Comparative Study. Cell Biol. Int. 2008, 32, 1439–1448. [Google Scholar] [CrossRef]
- Baxter, M.A.; Wynn, R.F.; Jowitt, S.N.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004, 22, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The Essence of Senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Baldassarre, A.; D’Amico, M.A.; Izzicupo, P.; Gaggi, G.; Guarnieri, S.; Mariggiò, M.A.; Antonucci, I.; Corneo, B.; Sirabella, D.; Stuppia, L.; et al. Cardiomyocytes Derived from Human CardiopoieticAmniotic Fluids. Sci. Rep. 2018, 8, 12028. [Google Scholar] [CrossRef]
- Chong, J.J.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell. Stem Cell 2011, 9, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.J.H.; Reinecke, H.; Iwata, M.; Torok-Storb, B.; Stempien-Otero, A.; Murry, C.E. Progenitor Cells Identified by PDGFR-Alpha Expression in the Developing and Diseased Human Heart. Stem Cells Dev. 2013, 22, 1932–1943. [Google Scholar] [CrossRef] [Green Version]
- Stankunas, K.; Ma, G.K.; Kuhnert, F.J.; Kuo, C.J.; Chang, C.P. VEGF Signaling Has Distinct Spatiotemporal Roles during Heart Valve Development. Dev. Biol. 2010, 347, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivelä, R.; Hemanthakumar, K.A.; Vaparanta, K.; Robciuc, M.; Izumiya, Y.; Kidoya, H.; Takakura, N.; Peng, X.; Sawyer, D.B.; Elenius, K.; et al. Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation 2019, 139, 2570–2584. [Google Scholar] [CrossRef]
- Shibuya, M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 2006, 39, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Finan, A.; Sopko, N.; Dong, F.; Turturice, B.; Kiedrowski, M.; Penn, M.S. Bone Marrow SSEA1+ Cells Support the Myocardium in Cardiac Pressure Overload. PLoS ONE 2013, 8, e68528. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Hiriart, E.; van Vliet, P.P.; Faure, E.; Norris, R.A.; Farhat, B.; Jagla, B.; Lefrancois, J.; Sugi, Y.; Moore-Morris, T.; et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 2019, 10, 1929. [Google Scholar] [CrossRef] [Green Version]
- Kohli, S.; Ahuja, S.; Rani, V. Transcription factors in heart: Promising therapeutic targets in cardiac hypertrophy. Curr. Cardiol. Rev. 2011, 7, 262–271. [Google Scholar] [CrossRef] [Green Version]
- McCulley, D.J.; Black, B.L. Transcription Factor Pathways and Congenital Heart Disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar] [PubMed] [Green Version]
- Hori, Y.; Tanimoto, Y.; Takahashi, S.; Furukawa, T.; Koshiba-Takeuchi, K.; Takeuchi, J.K. Important Cardiac Transcription Factor Genes Are Accompanied by Bidirectional Long Non-Coding RNAs. BMC Genom. 2018, 19, 967. [Google Scholar] [CrossRef]
- Govindsamy, A.; Naidoo, S.; Cerf, M.E. Cardiac Development and Transcription Factors: Insulin Signaling, Insulin Resistance, and Intrauterine Nutritional Programming of Cardiovascular Disease. J. Nutr. Metab. 2018, 2018, 8547976. [Google Scholar] [CrossRef] [Green Version]
- Smemo, S.; Campos, L.C.; Moskowitz, I.P.; Krieger, J.E.; Pereira, A.C.; Nobrega, M.A. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 2012, 21, 3255–3263. [Google Scholar] [CrossRef] [Green Version]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes. Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ai, F.; Zheng, J.; Peng, B. Associations of GATA4 genetic mutations with the risk of congenital heart disease: A meta-analysis. Medicine 2017, 96, e6857. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Shi, G.P. CD31: Beyond a marker for endothelial cells. Cardiovasc. Res. 2012, 94, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Catalogue Number | Dilution | Source |
|---|---|---|---|
| CD73 (PE) | 344,003 | 1:100 | BioLegend, San Diego, CA, USA |
| CD90 (APC) | 328,113 | 1:100 | BioLegend, San Diego, CA, USA |
| CD105 (FITC) | 323,203 | 1:100 | BioLegend, San Diego, CA, USA |
| CD34 (FITC) | 343,603 | 1:100 | BioLegend, San Diego, CA, USA |
| CD45 (PE) | 304,007 | 1:100 | BioLegend, San Diego, CA, USA |
| HLA-DR (APC) | 307,605 | 1:100 | BioLegend, San Diego, CA, USA |
| PDGFR-α (PE) | 323,505 | 1:100 | BioLegend, San Diego, CA, USA |
| SSEA-1 (PE) | 330,405 | 1:100 | BioLegend, San Diego, CA, USA |
| VEGFR-2 (APC) | 359,915 | 1:100 | BioLegend, San Diego, CA, USA |
| cTNT | ab45932 | 1:200 | Abcam, Cambridge, UK |
| anti-rabbit secondary antibodies (FITC) | ab6717 | 1:200 | Abcam, Cambridge, UK |
| CD31 | ab24590 | 1:100 | Abcam, Cambridge, UK |
| α-SMA | ab5694 | 1: 50 | Abcam, Cambridge, UK |
| Gene Name | Target Gene-Primer Sequence |
|---|---|
| Lipoprotein Lipase | Forward-5′ TCCAAACCAGAAAACGGAAG3′ Reverse-5′ ACAGCCAGTCCACCACAATG3′ |
| PPAR-ϒ | Forward-5′ TCAGGGCTGCCAGTTTCG 3′ Reverse-5′GCTTTTGGCATACTCTGTGATCTC 3′ |
| Osteopontin (OPN) | Forward-5′ CCTGCCAGCAACCGAAGT 3′ Reverse-5′ CCTCGGCCATCATATGTGTCT 3′ |
| RUNX | Forward-5′ TCGAATGGCAGCACGCTAT 3′ Reverse-5′ CATCAGCGTCAACACCATCAT 3′ |
| SOX 9 | Forward-5′ AGCGACGTCATCTCCAACATC 3′ Reverse-5′ GTTGGGCGGCAGGTACTG 3′ |
| AGCAN | Forward-5′ GGAAGGCTGCTATGGAGACAAG 3′ Reverse-5′ GGTGTCTCGGATGCCATACG 3′ |
| GATA-4 | Forward-5′TCCAAACCAGAAAACGGAAG3′ Reverse-5′CTGTGCCCGTAGTGAGATGA3′ |
| NKx 2-5 | Forward -5′AGTTTGTGGCGGCGATTAT3′ Reverse-5′AGCTCAGTCCCAGTTCCA3′ |
| ISL-1 | Forward-5′GCCTTGCAGAGTGACATAGAT3′ Reverse-5′CTGGAAGTTGAGAGGACATTGA3′ |
| TBX-5 | Forward-5′AACCACAAGATCACGCAATTAAAG3′ Reverse-5′GTCATCACTGCCCCGAAATC3′ |
| TBX-18 | Forward-5′CGGTGGAGGCGCTGATC3′ Reverse-5′CAGTTTTCGCCGCTTCT3′ |
| MeF-2C | Forward-5′CACCAGGCAGCAAGAATACGA3′ Reverse-5′CTCAGCCGACTGGGAGTTATTT3′ |
| TP53 | Forward-5′GTCCCAAGCAATGGATGATTTG3′ Reverse-5′GCATTCTGGGAGCTTCATCT3′ |
| CDKN1A | Forward -5′TGGAGACTCTCAGGGTCGAAAA3′ Reverse-5′CGGCGTTTGGAGTGGTAGAA3′ |
| MRE11 | Forward-5′GTGGACAAGGAGGAGAAAGATG3′ Reverse-5′TGTCTTCGAGGGCATCAATATG3′ |
| NBS1 | Forward-5′GTCAGGACGGCAGGAAAGAA3′ Reverse-5′TCAACCTAGCTTCCCCACCT3′ |
| PARP | Forward-5′AGTGCCAACTACTGCCATAC3′ Reverse-5′AGCGTGCTTCAGTTCATACA3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, M.; Singh, N.; Fatima, R.; Nachanekar, A.; Pradhan, M.; Nityanand, S.; Chaturvedi, C.P. Amniotic Fluid Mesenchymal Stromal Cells Derived from Fetuses with Isolated Cardiac Defects Exhibit Decreased Proliferation and Cardiomyogenic Potential. Biology 2023, 12, 552. https://doi.org/10.3390/biology12040552
Jain M, Singh N, Fatima R, Nachanekar A, Pradhan M, Nityanand S, Chaturvedi CP. Amniotic Fluid Mesenchymal Stromal Cells Derived from Fetuses with Isolated Cardiac Defects Exhibit Decreased Proliferation and Cardiomyogenic Potential. Biology. 2023; 12(4):552. https://doi.org/10.3390/biology12040552
Chicago/Turabian StyleJain, Manali, Neeta Singh, Raunaq Fatima, Aditya Nachanekar, Mandakini Pradhan, Soniya Nityanand, and Chandra Prakash Chaturvedi. 2023. "Amniotic Fluid Mesenchymal Stromal Cells Derived from Fetuses with Isolated Cardiac Defects Exhibit Decreased Proliferation and Cardiomyogenic Potential" Biology 12, no. 4: 552. https://doi.org/10.3390/biology12040552