
Nucleolar Protein 56 Deficiency in Zebrafish Leads to Developmental Abnormalities and Anemia via p53 and JAK2-STAT3 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Maintenance
2.2. mRNA and gRNA Synthesis
2.3. Generation of the nop56−/− Homozygous Mutants
2.4. Real-Time Quantitative PCR
2.5. TUNEL Staining
2.6. o-dianisidine Staining
2.7. Wright–Giemsa Staining
2.8. WISH
2.9. RNA-Sequencing
2.10. Inhibitor Treatments
2.11. Statistics and Reproducibility
3. Results
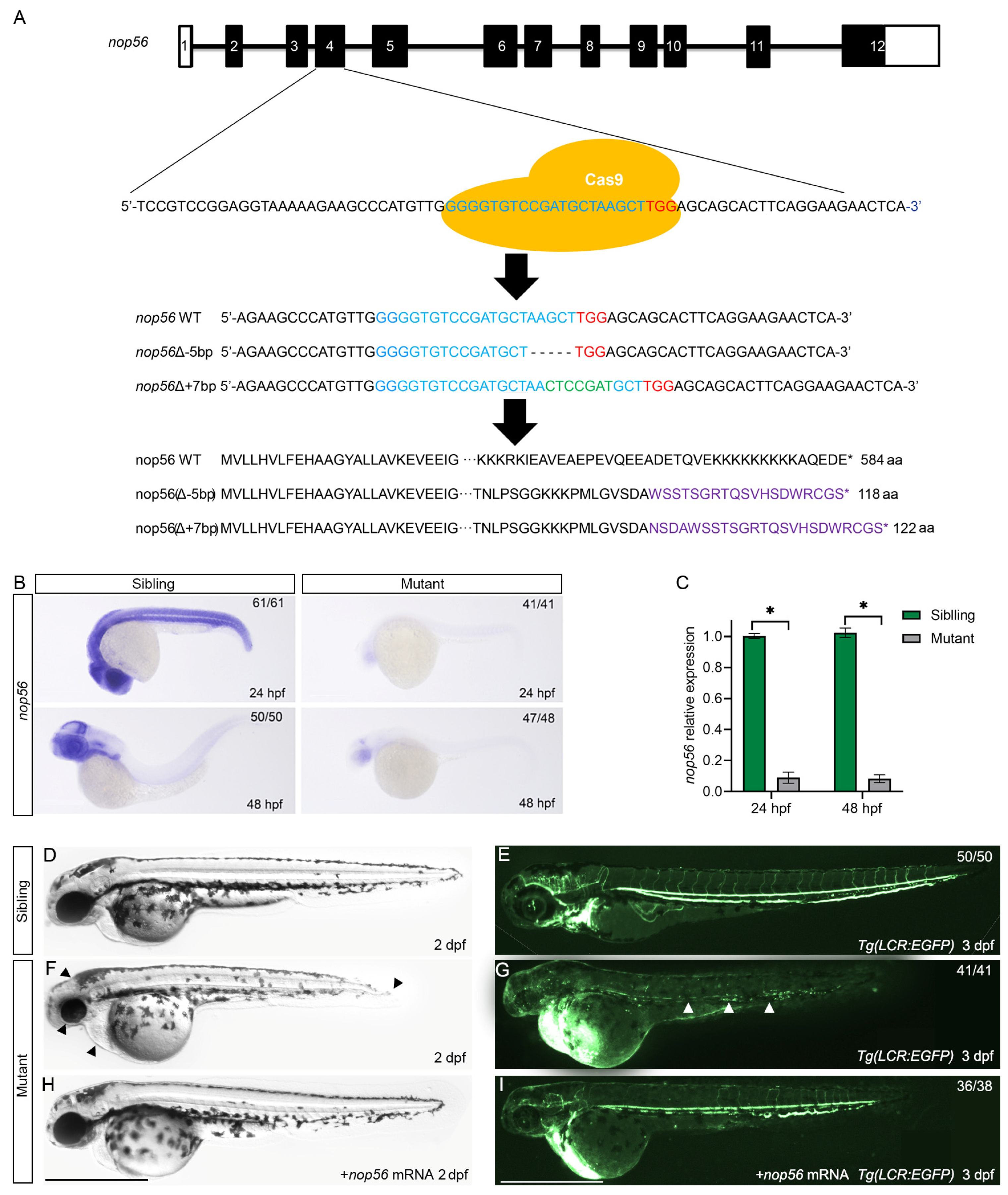
3.1. Loss of nop56 Using CRISPR/Cas9 Leads to Morphological Defects and Anemia in Zebrafish
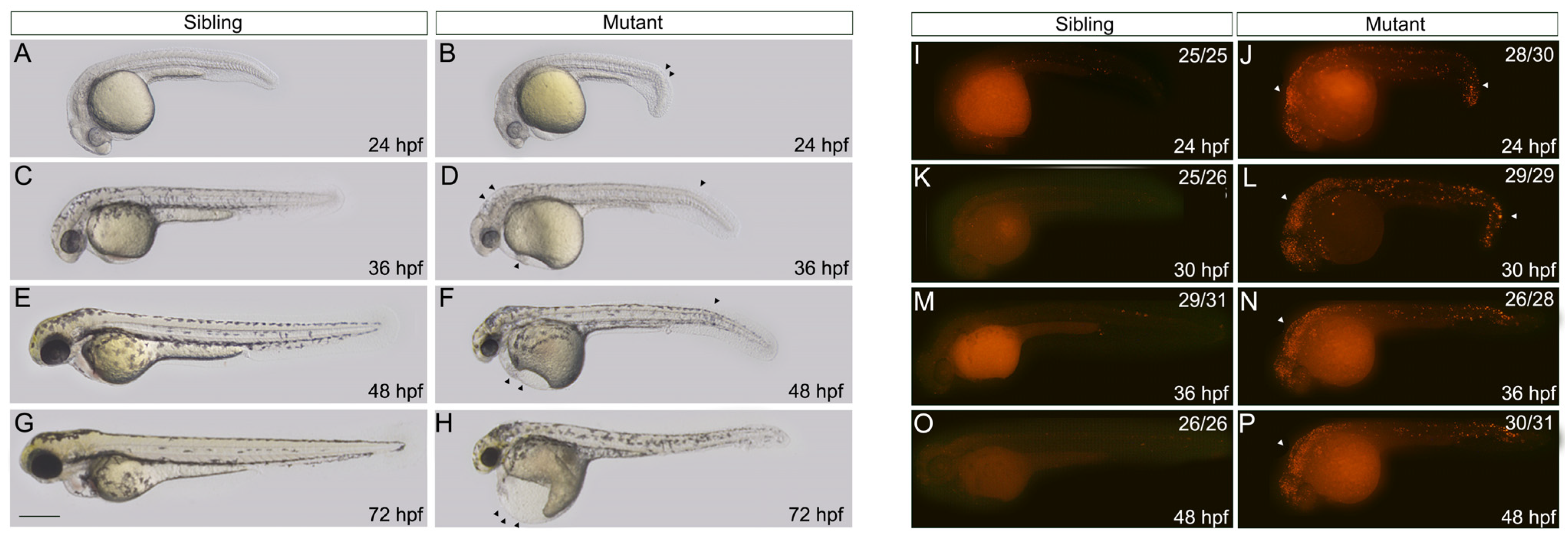
3.2. Morphological Malformation of nop56 Deficiency Is Partially due to Increased Apoptosis
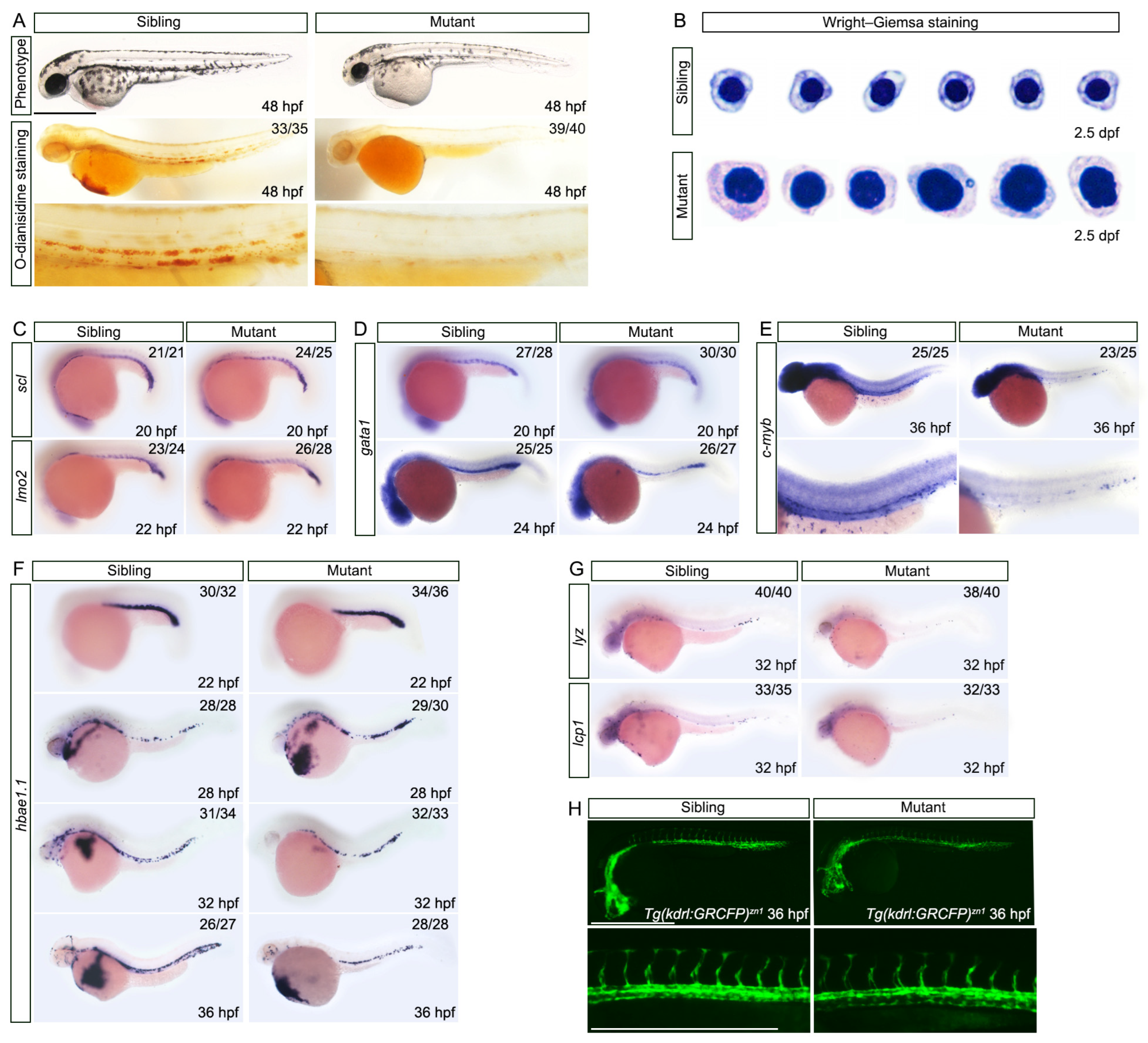
3.3. Maturation of Erythroid Cells and Specification of the Erythroid Lineage in Definitive Hematopoiesis Are Abnormal in nop56 Mutants
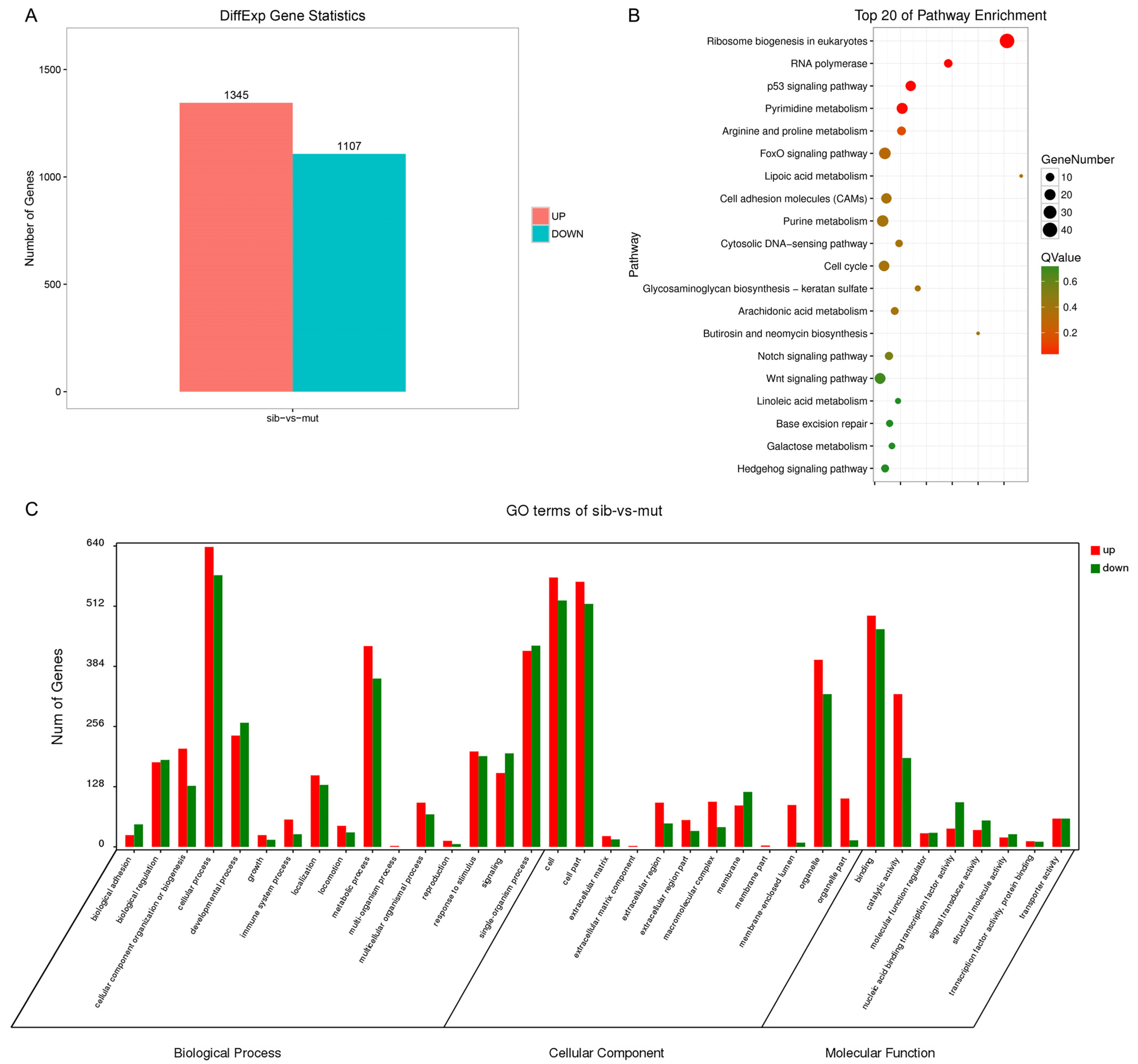
3.4. Transcriptome Analysis of nop56 Mutants
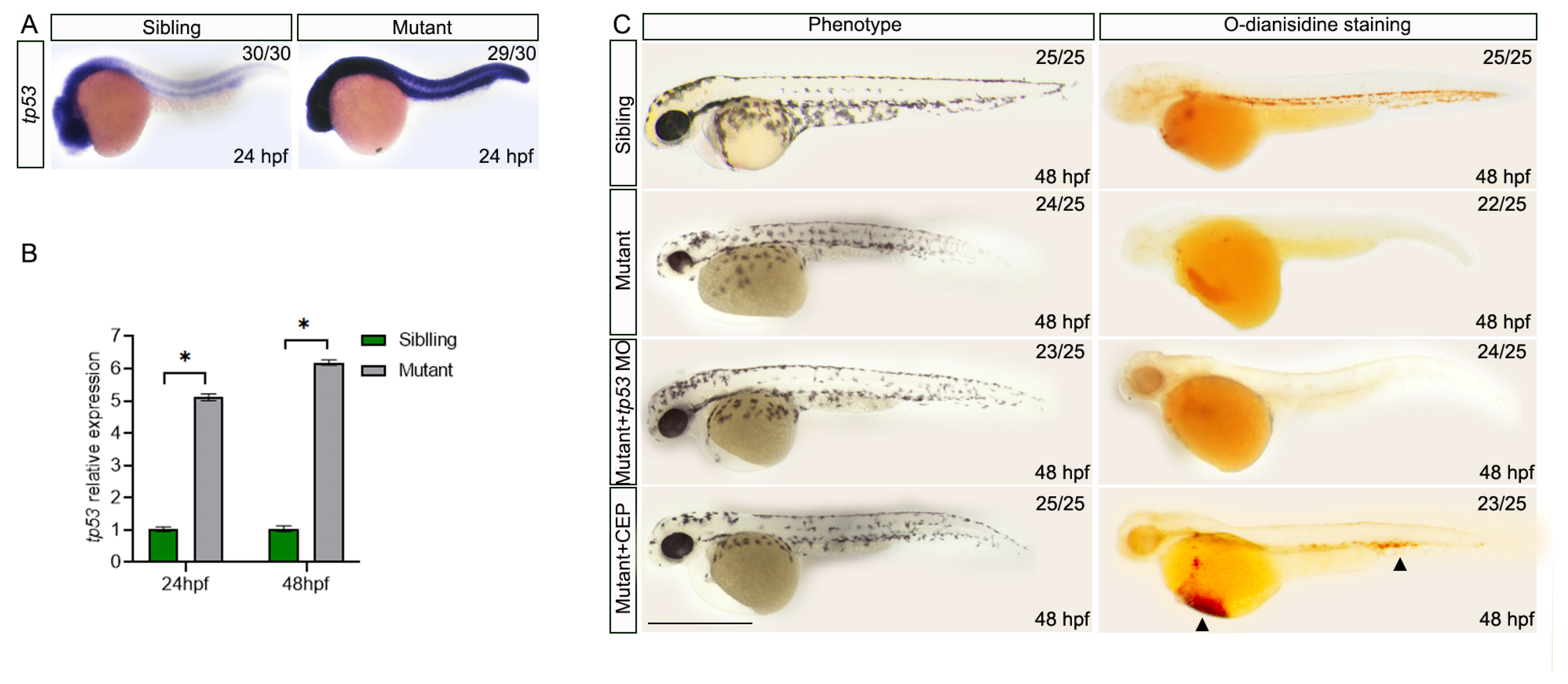
3.5. Knockdown of p53 and Inhibition of jak2 Partially Rescue Malformations and Anemia in nop56 Mutants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Meunier, M.; Park, S. Lower-risk myelodysplastic syndromes: Current treatment options for anemia. EJHaem 2022, 3, 1091–1099. [Google Scholar] [CrossRef]
- Zhang, Y.; Ear, J.; Yang, Z.; Morimoto, K.; Zhang, B.; Lin, S. Defects of protein production in erythroid cells revealed in a zebrafish Diamond-Blackfan anemia model for mutation in RPS19. Cell Death Dis. 2014, 5, e1352. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br. J. Haematol. 2011, 152, 217–228. [Google Scholar] [CrossRef]
- Ear, J.; Hsueh, J.; Nguyen, M.; Zhang, Q.; Sung, V.; Chopra, R.; Sakamoto, K.M.; Lin, S. A Zebrafish Model of 5q-Syndrome Using CRISPR/Cas9 Targeting RPS14 Reveals a p53-Independent and p53-Dependent Mechanism of Erythroid Failure. J. Genet. Genom. = Yi Chuan Xue Bao 2016, 43, 307–318. [Google Scholar] [CrossRef]
- Chen, C.; Lu, M.; Lin, S.; Qin, W. The nuclear gene rpl18 regulates erythroid maturation via JAK2-STAT3 signaling in zebrafish model of Diamond-Blackfan anemia. Cell Death Dis. 2020, 11, 135. [Google Scholar] [CrossRef]
- Youn, M.; Huang, H.; Chen, C.; Kam, S.; Wilkes, M.C.; Chae, H.D.; Sridhar, K.J.; Greenberg, P.L.; Glader, B.; Narla, A.; et al. MMP9 inhibition increases erythropoiesis in RPS14-deficient del(5q) MDS models through suppression of TGF-β pathways. Blood Adv. 2019, 3, 2751–2763. [Google Scholar] [CrossRef]
- Hannan, K.M.; Soo, P.; Wong, M.S.; Lee, J.K.; Hein, N.; Poh, P.; Wysoke, K.D.; Williams, T.D.; Montellese, C.; Smith, L.K.; et al. Nuclear stabilization of p53 requires a functional nucleolar surveillance pathway. Cell Rep. 2022, 41, 111571. [Google Scholar] [CrossRef]
- Wang, B.; Wang, C.; Wan, Y.; Gao, J.; Ma, Y.; Zhang, Y.; Tong, J.; Zhang, Y.; Liu, J.; Chang, L.; et al. Decoding the pathogenesis of Diamond-Blackfan anemia using single-cell RNA-seq. Cell Discov. 2022, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Payne, E.M.; Virgilio, M.; Narla, A.; Sun, H.; Levine, M.; Paw, B.H.; Berliner, N.; Look, A.T.; Ebert, B.L.; Khanna-Gupta, A. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012, 120, 2214–2224. [Google Scholar] [CrossRef]
- Doulatov, S.; Vo, L.T.; Macari, E.R.; Wahlster, L.; Kinney, M.A.; Taylor, A.M.; Barragan, J.; Gupta, M.; McGrath, K.; Lee, H.Y.; et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med. 2017, 9, eaah5645. [Google Scholar] [CrossRef]
- Lawrimore, J.; Kolbin, D.; Stanton, J.; Khan, M.; de Larminat, S.C.; Lawrimore, C.; Yeh, E.; Bloom, K. The rDNA is biomolecular condensate formed by polymer–polymer phase separation and is sequestered in the nucleolus by transcription and R-loops. Nucleic Acids Res. 2021, 49, 4586–4598. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Ardal, B.K.; Hollensen, A.K.; Damgaard, C.K.; Jensen, T.H. Box C/D snoRNP Autoregulation by a cis-Acting snoRNA in the NOP56 Pre-mRNA. Mol. Cell 2018, 72, 99–111.e115. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Watkins, N.J.; Ségault, V.; Charpentier, B.; Nottrott, S.; Fabrizio, P.; Bachi, A.; Wilm, M.; Rosbash, M.; Branlant, C.; Lührmann, R. A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell 2000, 103, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Li, Y.; Liu, C.J.; Xiang, Y.; Li, C.; Ye, Y.; Zhang, Z.; Hawke, D.H.; Park, P.K.; Diao, L.; et al. A Pan-cancer Analysis of the Expression and Clinical Relevance of Small Nucleolar RNAs in Human Cancer. Cell Rep. 2017, 21, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt’s lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- García-Murias, M.; Quintáns, B.; Arias, M.; Seixas, A.I.; Cacheiro, P.; Tarrío, R.; Pardo, J.; Millán, M.J.; Arias-Rivas, S.; Blanco-Arias, P.; et al. ‘Costa da Morte’ ataxia is spinocerebellar ataxia 36: Clinical and genetic characterization. Brain A J. Neurol. 2012, 135, 1423–1435. [Google Scholar] [CrossRef]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef]
- Quelle-Regaldie, A.; Folgueira, M.; Yáñez, J.; Sobrido-Cameán, D.; Alba-González, A.; Barreiro-Iglesias, A.; Sobrido, M.J.; Sánchez, L. A nop56 Zebrafish Loss-of-Function Model Exhibits a Severe Neurodegenerative Phenotype. Biomedicines 2022, 10, 1814. [Google Scholar] [CrossRef] [PubMed]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Chen, C.; Huang, H.; Yan, R.; Lin, S.; Qin, W. Loss of rps9 in Zebrafish Leads to p53-Dependent Anemia. G3 Genes Genomes Genet. 2019, 9, 4149–4157. [Google Scholar] [CrossRef]
- Qian, F.; Zhen, F.; Xu, J.; Huang, M.; Li, W.; Wen, Z. Distinct functions for different scl isoforms in zebrafish primitive and definitive hematopoiesis. PLoS Biol. 2007, 5, e132. [Google Scholar] [CrossRef] [PubMed]
- Porcher, C.; Liao, E.C.; Fujiwara, Y.; Zon, L.I.; Orkin, S.H. Specification of hematopoietic and vascular development by the bHLH transcription factor SCL without direct DNA binding. Development 1999, 126, 4603–4615. [Google Scholar] [CrossRef]
- Davidson, A.J.; Ernst, P.; Wang, Y.; Dekens, M.P.; Kingsley, P.D.; Palis, J.; Korsmeyer, S.J.; Daley, G.Q.; Zon, L.I. cdx4 mutants fail to specify blood progenitors and can be rescued by multiple hox genes. Nature 2003, 425, 300–306. [Google Scholar] [CrossRef]
- Stanulovic, V.S.; Cauchy, P.; Assi, S.A.; Hoogenkamp, M. LMO2 is required for TAL1 DNA binding activity and initiation of definitive haematopoiesis at the haemangioblast stage. Nucleic Acids Res. 2017, 45, 9874–9888. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, H.; Li, L.; Qin, F.X.; Wen, Z. cMyb regulates hematopoietic stem/progenitor cell mobilization during zebrafish hematopoiesis. Blood 2011, 118, 4093–4101. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Pellagatti, A.; Wainscoat, J.S. Haploinsufficiency of ribosomal proteins and p53 activation in anemia: Diamond-Blackfan anemia and the 5q- syndrome. Adv. Biol. Regul. 2012, 52, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008, 112, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Foley, H.A.; Ofori-Acquah, S.F.; Yoshimura, A.; Critz, S.; Baliga, B.S.; Pace, B.S. Stat3 beta inhibits gamma-globin gene expression in erythroid cells. J. Biol. Chem. 2002, 277, 16211–16219. [Google Scholar] [CrossRef]
- Yao, X.; Kodeboyina, S.; Liu, L.; Dzandu, J.; Sangerman, J.; Ofori-Acquah, S.F.; Pace, B.S. Role of STAT3 and GATA-1 interactions in gamma-globin gene expression. Exp. Hematol. 2009, 37, 889–900. [Google Scholar] [CrossRef]
- Stump, K.L.; Lu, L.D.; Dobrzanski, P.; Serdikoff, C.; Gingrich, D.E.; Dugan, B.J.; Angeles, T.S.; Albom, M.S.; Ator, M.A.; Dorsey, B.D.; et al. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R68. [Google Scholar] [CrossRef]
- Yadav, G.V.; Chakraborty, A.; Uechi, T.; Kenmochi, N. Ribosomal protein deficiency causes Tp53-independent erythropoiesis failure in zebrafish. Int. J. Biochem. Cell Biol. 2014, 49, 1–7. [Google Scholar] [CrossRef]
- Orea-Soufi, A.; Paik, J.; Bragança, J.; Donlon, T.A.; Willcox, B.J.; Link, W. FOXO transcription factors as therapeutic targets in human diseases. Trends Pharmacol. Sci. 2022, 43, 1070–1084. [Google Scholar] [CrossRef]
- Liang, R.; Campreciós, G.; Kou, Y.; McGrath, K.; Nowak, R.; Catherman, S.; Bigarella, C.L.; Rimmelé, P.; Zhang, X.; Gnanapragasam, M.N.; et al. A Systems Approach Identifies Essential FOXO3 Functions at Key Steps of Terminal Erythropoiesis. PLoS Genet. 2015, 11, e1005526. [Google Scholar] [CrossRef]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef]
- Peng, K.; Li, Y.; Long, L.; Li, D.; Jia, Q.; Wang, Y.; Shen, Q.; Tang, Y.; Wen, L.; Kung, H.F.; et al. Knockdown of FoxO3a induces increased neuronal apoptosis during embryonic development in zebrafish. Neurosci. Lett. 2010, 484, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Wang, S.; Zhang, Q.; Zheng, J.; Yang, Y.; Qi, H.; Qu, H.; Zhang, Z.; Liu, F.; et al. Knockdown of transcription factor forkhead box O3 (FOXO3) suppresses erythroid differentiation in human cells and zebrafish. Biochem. Biophys. Res. Commun. 2015, 460, 923–930. [Google Scholar] [CrossRef]
- Bu, Y.; Su, F.; Wang, X.; Gao, H.; Lei, L.; Chang, N.; Wu, Q.; Hu, K.; Zhu, X.; Chang, Z.; et al. Protein tyrosine phosphatase PTPN9 regulates erythroid cell development through STAT3 dephosphorylation in zebrafish. J. Cell Sci. 2014, 127, 2761–2770. [Google Scholar] [CrossRef]
- Ferry, A.E.; Baliga, S.B.; Monteiro, C.; Pace, B.S. Globin gene silencing in primary erythroid cultures. An inhibitory role for interleukin-6. J. Biol. Chem. 1997, 272, 20030–20037. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Zhang, Q.; Zhang, Z.; Wang, Y.; Zhang, W.; Zhou, Y.; Wan, Y.; Cheng, T.; Zhu, X.; Fang, X.; et al. Transcriptome analysis of the zebrafish model of Diamond-Blackfan anemia from RPS19 deficiency via p53-dependent and -independent pathways. PLoS ONE 2013, 8, e71782. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, F.; Lu, X.; Wu, B.; Yang, Y.; Qin, W. Nucleolar Protein 56 Deficiency in Zebrafish Leads to Developmental Abnormalities and Anemia via p53 and JAK2-STAT3 Signaling. Biology 2023, 12, 538. https://doi.org/10.3390/biology12040538
Liang F, Lu X, Wu B, Yang Y, Qin W. Nucleolar Protein 56 Deficiency in Zebrafish Leads to Developmental Abnormalities and Anemia via p53 and JAK2-STAT3 Signaling. Biology. 2023; 12(4):538. https://doi.org/10.3390/biology12040538
Chicago/Turabian StyleLiang, Fang, Xiaochan Lu, Biyu Wu, Yexin Yang, and Wei Qin. 2023. "Nucleolar Protein 56 Deficiency in Zebrafish Leads to Developmental Abnormalities and Anemia via p53 and JAK2-STAT3 Signaling" Biology 12, no. 4: 538. https://doi.org/10.3390/biology12040538