
Establishment of an Integrated CRISPR/Cas9 Plasmid System for Simple and Efficient Genome Editing in Medaka In Vitro and In Vivo
 and
and 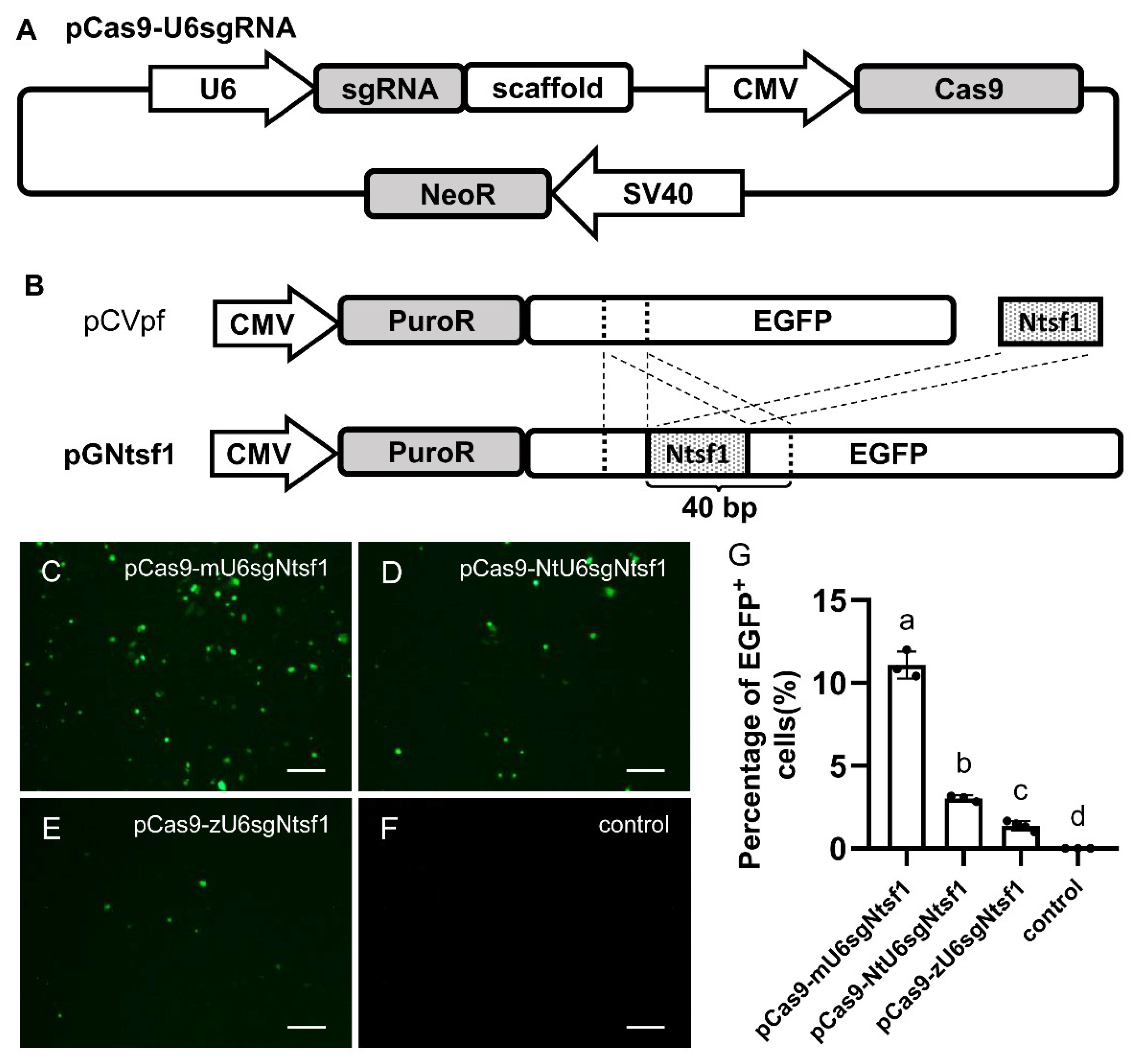{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish
2.2. Plasmids
2.3. Cell Culture and Cell Transfection
2.4. Microinjection
2.5. HMA
2.6. TIDE Analysis
2.7. Statistical Analysis
3. Results
3.1. Construction and Evaluation of an Integrated Plasmid-Based CRISPR/Cas9 System in Medaka Cultured Cells
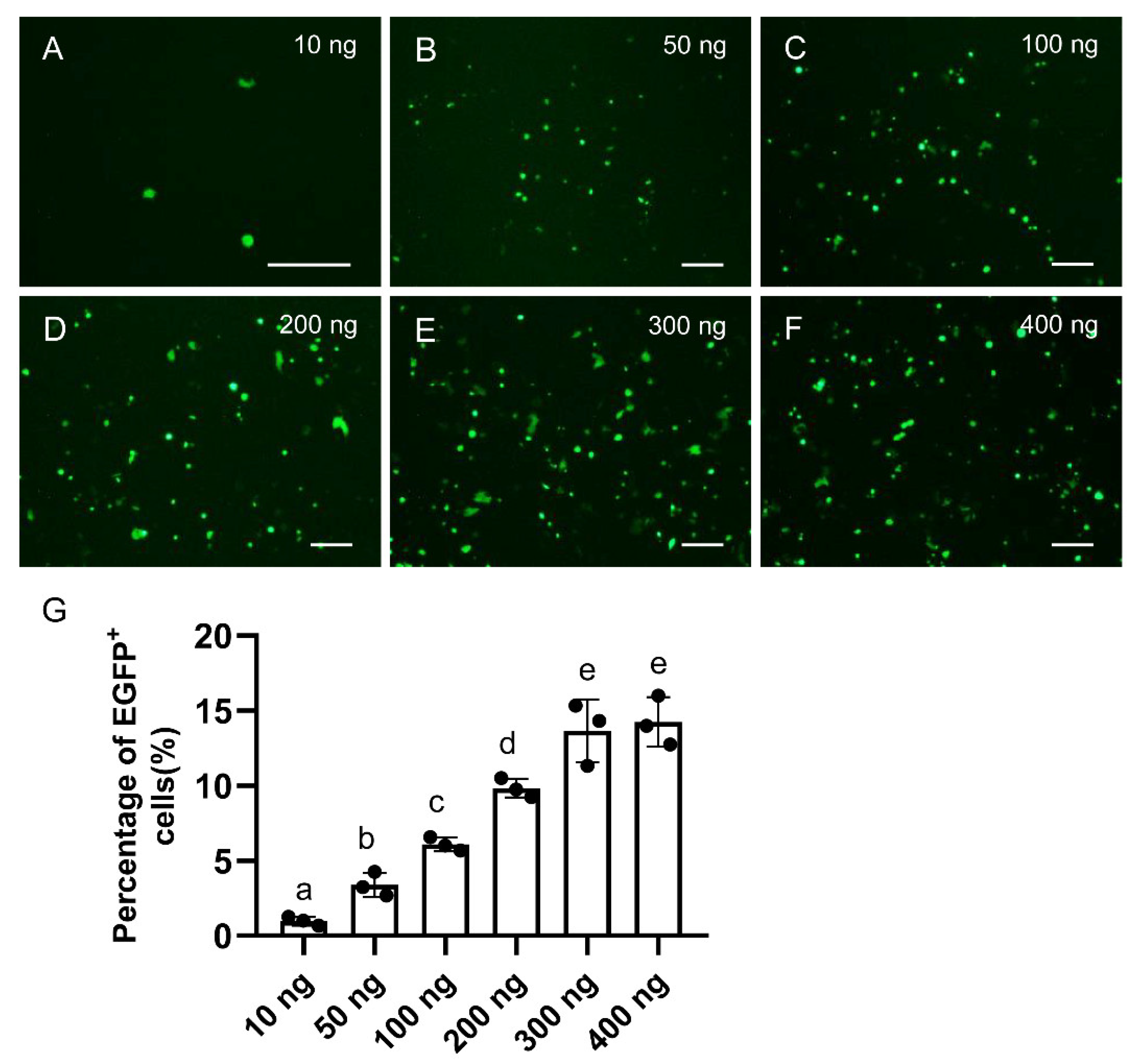
3.2. Dosage Optimization of pCas9-U6sgRNA in Medaka Cultured Cells
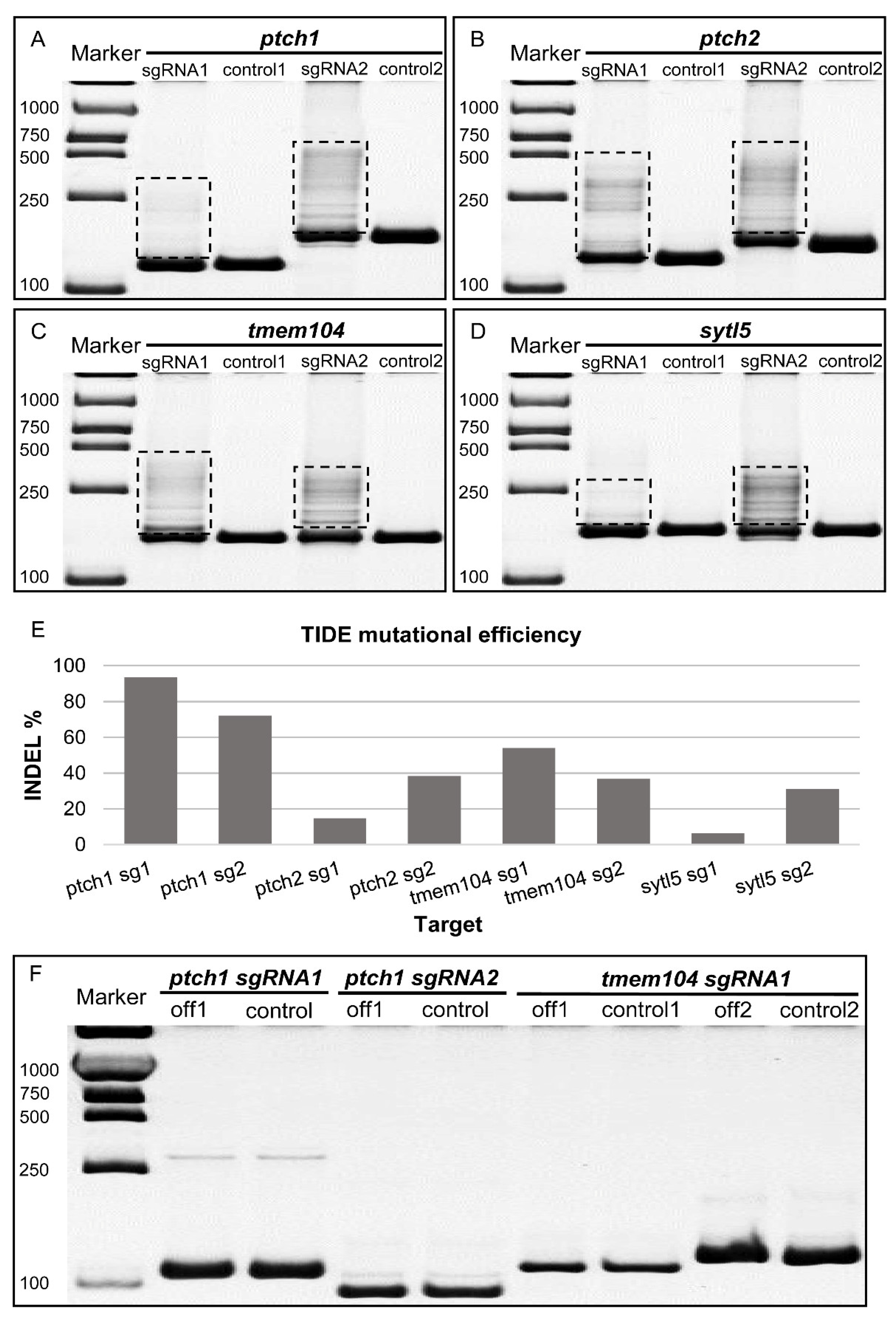
3.3. Endogenous Gene Knock-Out Mediated by pCas9-mU6sgRNA in Medaka Cultured Cells
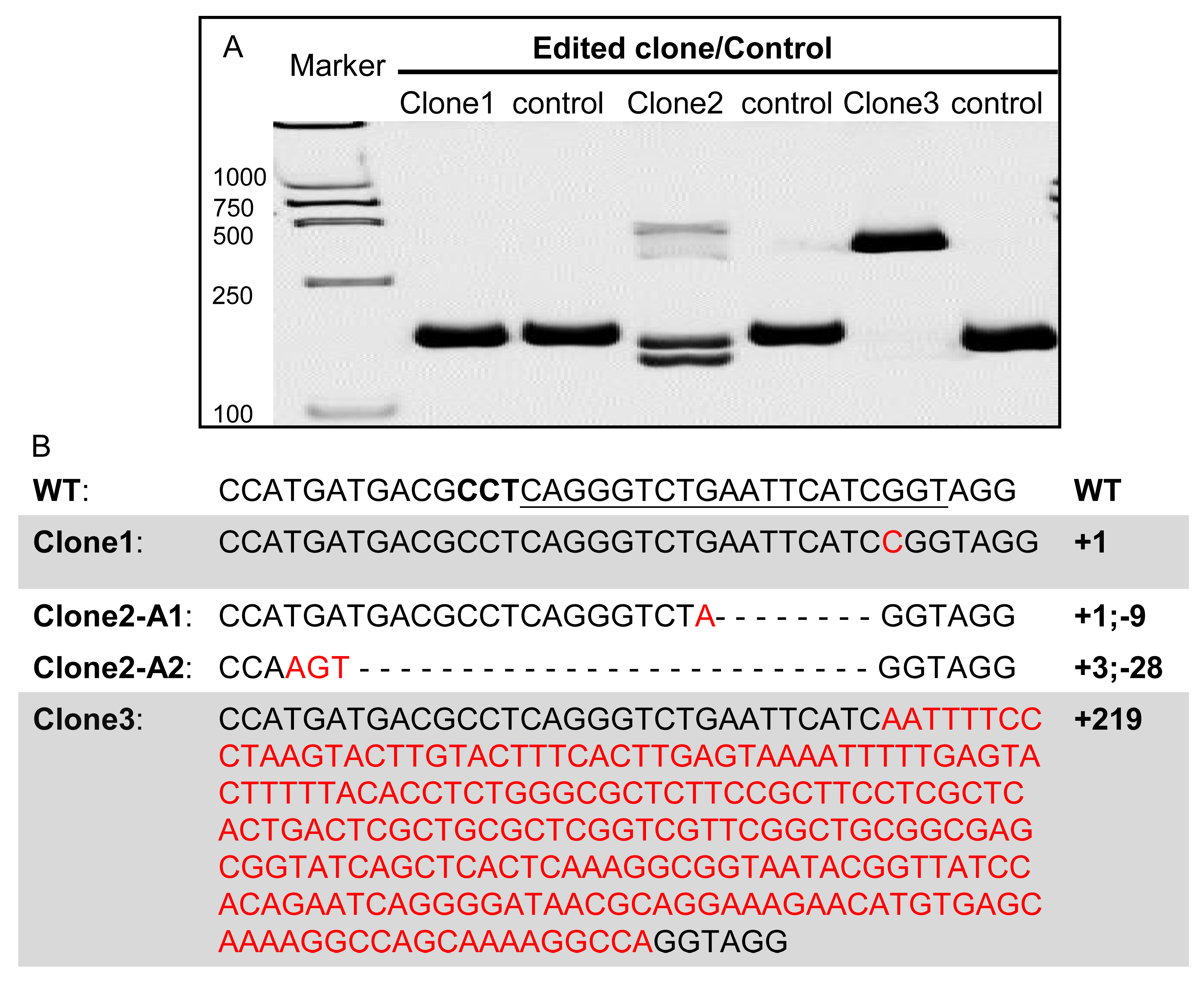
3.4. Mutational Single Cell Clones Generated with pCas9-mU6sgRNA
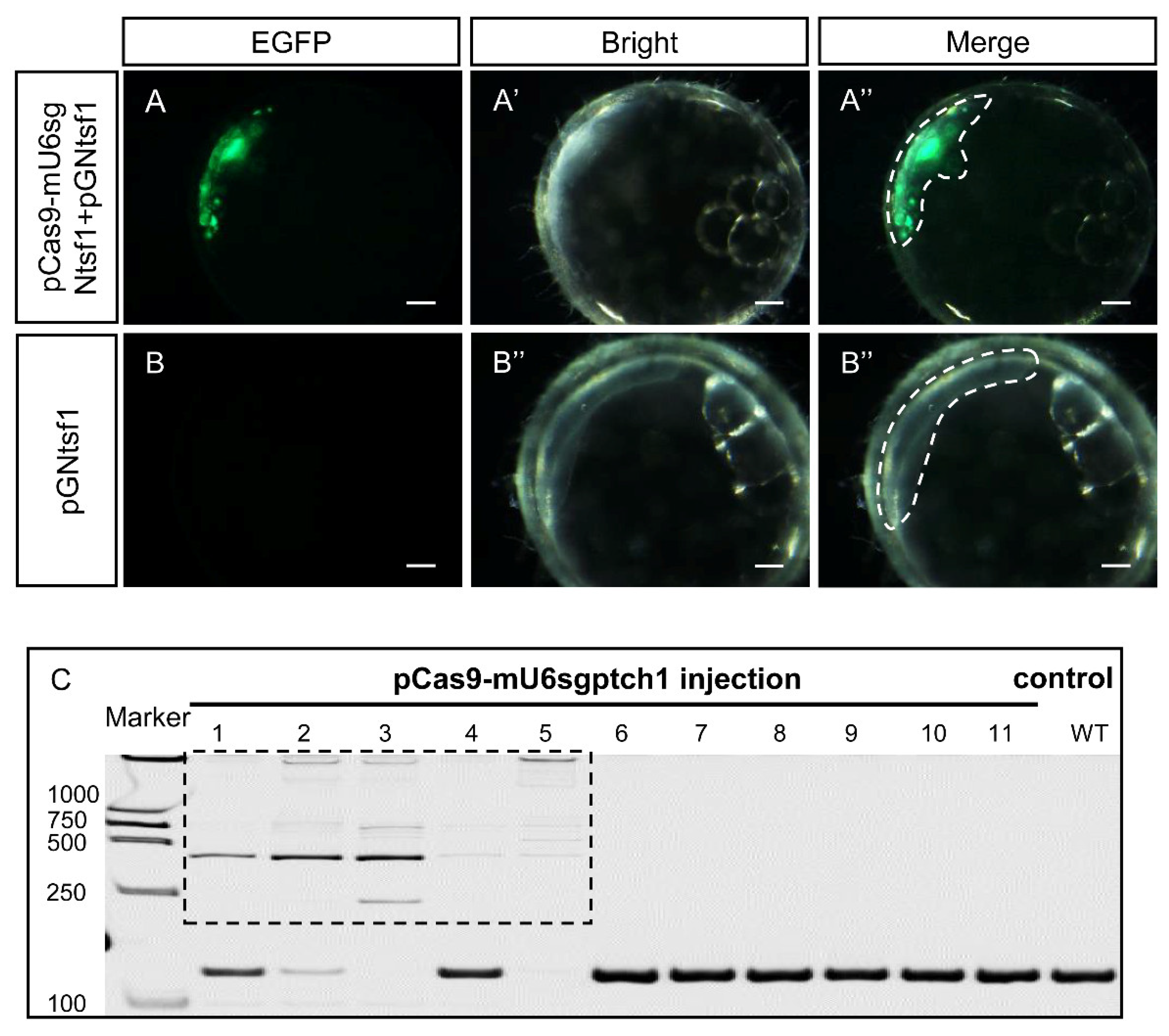
3.5. Gene Knock-Out Mediated by pCas9-mU6sgRNA In Vivo
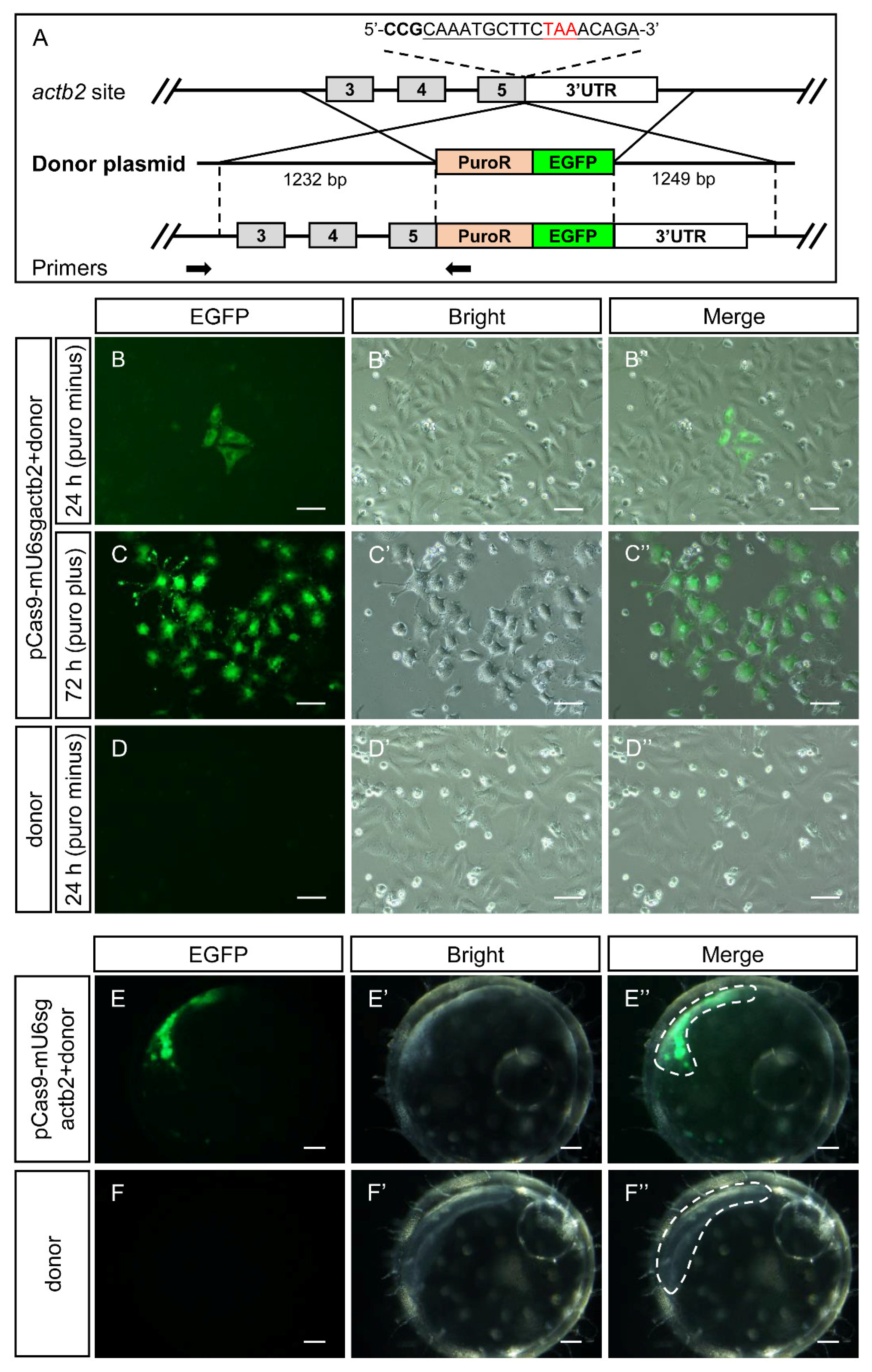
3.6. Gene Knock-In Mediated by pCas9-mU6sgRNA In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–923. [Google Scholar] [CrossRef] [Green Version]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Gratacap, R.L.; Wargelius, A.; Edvardsen, R.B.; Houston, R.D. Potential of Genome Editing to Improve Aquaculture Breeding and Production. Trends Genet. 2019, 35, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Dolskiy, A.A.; Grishchenko, I.V.; Yudkin, D.V. Cell Cultures for Virology: Usability, Advantages, and Prospects. Int. J. Mol. Sci. 2020, 21, 7978. [Google Scholar] [CrossRef]
- Zurita, J.; Peso, A.D.; Rojas, R.; Maisanaba, S.; Repetto, G. Integration of fish cell cultures in the toxicological assessment of effluents. Ecotoxicol. Environ. Saf. 2019, 176, 309–320. [Google Scholar] [CrossRef]
- Tan, L.; Schirmer, K. Cell culture-based biosensing techniques for detecting toxicity in water. Curr. Opin. Biotechnol. 2017, 45, 59–68. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Zhang, J.T.; Huang, S.C.; He, X.Y.; Deng, L.X. Double sperm cloning (DSC) is a promising strategy in mammalian genetic engineering and stem cell research. Stem Cell Res. Ther. 2020, 11, 388. [Google Scholar] [CrossRef]
- Saragusty, J.; Walzer, C.; Petit, T.; Stalder, G.; Horowitz, I.; Hermes, R. Cooling and freezing of epididymal sperm in the common hippopotamus (Hippopotamus amphibius). Theriogenology 2010, 74, 1256–1263. [Google Scholar] [CrossRef]
- Liu, S.; Yin, N.; Faiola, F. Prospects and Frontiers of Stem Cell Toxicology. Stem Cells Dev. 2017, 26, 1528–1539. [Google Scholar] [CrossRef]
- Ahmed, U.; Ahmed, R.; Masoud, M.S.; Tariq, M.; Ashfaq, U.A.; Augustine, R.; Hasan, A. Stem cells based in vitro models: Trends and prospects in biomaterials cytotoxicity studies. Biomed. Mater. 2021, 16, 042003. [Google Scholar] [CrossRef]
- Liang, W.; Han, P.; Kim, E.H.; Mak, J.; Zhang, R.; Torrente, A.G.; Goldhaber, J.I.; Marban, E.; Cho, H.C. Canonical Wnt signaling promotes pacemaker cell specification of cardiac mesodermal cells derived from mouse and human embryonic stem cells. Stem Cells 2020, 38, 352–368. [Google Scholar] [CrossRef]
- Lindner, B.; Martin, E.; Steininger, M.; Bundalo, A.; Lenter, M.; Zuber, J.; Schuler, M. A genome-wide CRISPR/Cas9 screen to identify phagocytosis modulators in monocytic THP-1 cells. Sci. Rep. 2021, 11, 12973. [Google Scholar] [CrossRef]
- Hoshijima, K.; Jurynec, M.J.; Klatt Shaw, D.; Jacobi, A.M.; Behlke, M.A.; Grunwald, D.J. Highly Efficient CRISPR-Cas9-Based Methods for Generating Deletion Mutations and F0 Embryos that Lack Gene Function in Zebrafish. Dev. Cell 2019, 51, 645–657e4. [Google Scholar] [CrossRef]
- Seleit, A.; Aulehla, A.; Paix, A. Endogenous protein tagging in medaka using a simplified CRISPR/Cas9 knock-in approach. eLife 2021, 10, e75050. [Google Scholar] [CrossRef]
- Li, M.; Yang, H.; Zhao, J.; Fang, L.; Shi, H.; Li, M.; Sun, Y.; Zhang, X.; Jiang, D.; Zhou, L.; et al. Efficient and heritable gene targeting in tilapia by CRISPR/Cas9. Genetics 2014, 197, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Feng, R.; Ma, H.; Dong, R.; Liu, Z.; Jiang, W.; Tao, W.; Wang, D. Retinoic acid triggers meiosis initiation via stra8-dependent pathway in Southern catfish, Silurus meridionalis. Gen. Comp. Endocrinol. 2016, 232, 191–198. [Google Scholar] [CrossRef]
- Chen, G.; Xiong, L.; Wang, Y.; He, L.; Huang, R.; Liao, L.; Zhu, Z.; Wang, Y. ITGB1b-Deficient Rare Minnows Delay Grass Carp Reovirus (GCRV) Entry and Attenuate GCRV-Triggered Apoptosis. Int. J. Mol. Sci. 2018, 19, 3175. [Google Scholar] [CrossRef] [Green Version]
- Wargelius, A.; Leininger, S.; Skaftnesmo, K.O.; Kleppe, L.; Andersson, E.; Taranger, G.L.; Schulz, R.W.; Edvardsen, R.B. Dnd knockout ablates germ cells and demonstrates germ cell independent sex differentiation in Atlantic salmon. Sci. Rep. 2016, 6, 21284. [Google Scholar] [CrossRef] [Green Version]
- Datsomor, A.K.; Zic, N.; Li, K.; Olsen, R.E.; Jin, Y.; Vik, J.O.; Edvardsen, R.B.; Grammes, F.; Wargelius, A.; Winge, P. CRISPR/Cas9-mediated ablation of elovl2 in Atlantic salmon (Salmo salar L.) inhibits elongation of polyunsaturated fatty acids and induces Srebp-1 and target genes. Sci. Rep. 2019, 9, 7533. [Google Scholar] [CrossRef] [Green Version]
- Edvardsen, R.B.; Leininger, S.; Kleppe, L.; Skaftnesmo, K.O.; Wargelius, A. Targeted mutagenesis in Atlantic salmon (Salmo salar L.) using the CRISPR/Cas9 system induces complete knockout individuals in the F0 generation. PLoS ONE 2014, 9, e108622. [Google Scholar] [CrossRef]
- Liu, Q.; Yuan, Y.; Zhu, F.; Hong, Y.; Ge, R. Efficient genome editing using CRISPR/Cas9 ribonucleoprotein approach in cultured Medaka fish cells. Biol. Open 2018, 7, bio035170. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Fan, Y.; Zhou, Y.; Liu, W.; Jiang, N.; Zhang, J.; Zeng, L. Efficient resistance to grass carp reovirus infection in JAM-A knockout cells using CRISPR/Cas9. Fish Shellfish Immunol. 2018, 76, 206–215. [Google Scholar] [CrossRef]
- Escobar-Aguirre, S.; Arancibia, D.; Escorza, A.; Bravo, C.; Andres, M.E.; Zamorano, P.; Martinez, V. Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines. Cells 2019, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Gratacap, R.L.; Regan, T.; Dehler, C.E.; Martin, S.A.M.; Boudinot, P.; Collet, B.; Houston, R.D. Efficient CRISPR/Cas9 genome editing in a salmonid fish cell line using a lentivirus delivery system. BMC Biotechnol. 2020, 20, 35. [Google Scholar] [CrossRef]
- Hamar, J.; Kultz, D. An efficient vector-based CRISPR/Cas9 system in an Oreochromis mossambicus cell line using endogenous promoters. Sci. Rep. 2021, 11, 7854. [Google Scholar] [CrossRef]
- Zoppo, M.; Okoniewski, N.; Pantelyushin, S.; Vom Berg, J.; Schirmer, K. A ribonucleoprotein transfection strategy for CRISPR/Cas9-mediated gene editing and single cell cloning in rainbow trout cells. Cell Biosci. 2021, 11, 103. [Google Scholar] [CrossRef]
- Dehler, C.E.; Boudinot, P.; Martin, S.A.; Collet, B. Development of an Efficient Genome Editing Method by CRISPR/Cas9 in a Fish Cell Line. Mar. Biotechnol. 2016, 18, 449–452. [Google Scholar] [CrossRef] [Green Version]
- Stromsnes, T.A.H.; Schmidke, S.E.; Azad, M.; Singstad, O.; Gronsberg, I.M.; Dalmo, R.A.; Okoli, A.S. CRISPR/Cas9-Mediated Gene Editing in Salmonids Cells and Efficient Establishment of Edited Clonal Cell Lines. Int. J. Mol. Sci. 2022, 23, 16218. [Google Scholar] [CrossRef]
- Liu, Z.H.; Zhang, Y.G.; Wang, D.S. Studies on feminization, sex determination, and differentiation of the Southern catfish, Silurus meridionalis—A review. Fish Physiol. Biochem. 2010, 36, 223–235. [Google Scholar] [CrossRef]
- Boonanuntanasarn, S.; Panyim, S.; Yoshizaki, G. Characterization and organization of the U6 snRNA gene in zebrafish and usage of their promoters to express short hairpin RNA. Mar. Genom. 2008, 1, 115–121. [Google Scholar] [CrossRef]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Boonanuntanasarn, S.; Panyim, S.; Yoshizaki, G. Usage of putative zebrafish U6 promoters to express shRNA in Nile tilapia and shrimp cell extracts. Transgenic Res. 2009, 18, 323–325. [Google Scholar] [CrossRef]
- Kim, N.Y.; Baek, J.Y.; Choi, H.S.; Chung, I.S.; Shin, S.; Lee, J.I.; Choi, J.Y.; Yang, J.M. Short-hairpin RNA-mediated gene expression interference in Trichoplusia ni cells. J. Microbiol. Biotechnol. 2012, 22, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Henning, D.; Redd, R. Structure, organization, and transcription of Drosophila U6 small nuclear RNA genes. J. Biol. Chem. 1986, 262, 1187–1193. [Google Scholar] [CrossRef]
- Zhao, C.L.; Zhang, Z.M.; Qu, X.M.; Bai, X.M.; Liu, X.Y.; Tao, W.J.; Zhou, L.Y.; Wang, D.S.; Wei, J. Desert hedgehog mediates the proliferation of medaka spermatogonia through Smoothened signaling. Reproduction 2022, 163, 209–218. [Google Scholar] [CrossRef]
- Xie, Q.P.; He, X.; Sui, Y.N.; Chen, L.L.; Sun, L.N.; Wang, D.S. Haploinsufficiency of SF-1 Causes Female to Male Sex Reversal in Nile Tilapia, Oreochromis niloticus. Endocrinology 2016, 157, 2500–2514. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Hong, N.; Lu, W.; Zeng, H.; Song, J.; Hong, Y. Fusion gene vectors allowing for simultaneous drug selection, cell labeling, and reporter assay in vitro and in vivo. Anal. Chem. 2012, 84, 987–993. [Google Scholar] [CrossRef]
- Zhao, H.; Li, M.; Purwanti, Y.I.; Liu, R.; Chen, T.; Li, Z.; Hong, N.; Guan, G.; Yin, A.; Xiao, L.; et al. Mitf is a transcriptional activator of medaka germ genes in culture. Biochimie 2012, 94, 759–767. [Google Scholar] [CrossRef]
- Hong, Y.H.; Winklerl, C.; Schartl, M. Pluripotency and differentiation of embryonic stem cell lines. Mech. Dev. 1996, 60, 33–44. [Google Scholar] [CrossRef]
- Hong, Y.H.; Winkler, C.; Schartl, M. Production of medakafish chimeras from a stable embryonic stem cell line. Proc. Natl. Acad. Sci. USA 1998, 95, 3679–3684. [Google Scholar] [CrossRef] [Green Version]
- Porazinski, S.R.; Wang, H.; Furutani-Seiki, M. Microinjection of medaka embryos for use as a model genetic organism. J. Vis. Exp. 2010, 46, e1937. [Google Scholar]
- Chen, J.; Zhang, X.; Wang, T.; Li, Z.; Guan, G.; Hong, Y. Efficient detection, quantification and enrichment of subtle allelic alterations. DNA Res. 2012, 19, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Z.; Wang, Y.; Luo, Y.; Da, F.; Tao, W.; Zhou, L.; Wang, D.; Wei, J. Both Gfrα1a and Gfrα1b Are Involved in the Self-renewal and Maintenance of Spermatogonial Stem Cells in Medaka. Stem Cells Dev. 2018, 27, 1658–1670. [Google Scholar] [CrossRef]
- Jing, W.; Xiaohuan, H.; Zhenhua, F.; Zhuo, Y.; Fan, D.; Wenjing, T.; Linyan, Z.; Deshou, W. Promoter activity and regulation of the Pou5f1 homolog from a teleost, Nile tilapia. Gene 2018, 642, 277–283. [Google Scholar] [CrossRef]
- Yan, Y.; Du, J.; Chen, T.; Yi, M.; Li, M.; Wang, S.; Li, C.M.; Hong, Y. Establishment of medakafish as a model for stem cell-based gene therapy: Efficient gene delivery and potential chromosomal integration by baculoviral vectors. Exp. Cell Res. 2009, 315, 2322–2331. [Google Scholar] [CrossRef]
- Gratacap, R.L.; Jin, Y.H.; Mantsopoulou, M.; Houston, R.D. Efficient Genome Editing in Multiple Salmonid Cell Lines Using Ribonucleoprotein Complexes. Mar. Biotechnol. 2020, 22, 717–724. [Google Scholar] [CrossRef]
- Collet, B.; Collins, C.; Lester, K. Engineered cell lines for fish health research. Dev. Comp. Immunol. 2018, 80, 34–40. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, T.; Yu, Z.; Wang, H.; Liu, B.; Wu, C.; Teng, C.B. Inhibiting cyprinid herpesvirus-3 replication with CRISPR/Cas9. Biotechnol. Lett. 2016, 38, 573–578. [Google Scholar] [CrossRef]
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.G.; Zhang, J.; Wu, H. Pol III Promoters to Express Small RNAs: Delineation of Transcription Initiation. Mol. Ther. Nucleic Acids 2014, 3, e161. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wang, J.; Li, J.; Liu, X.; Liu, L.; Zhao, C.; Tao, W.; Wang, D.; Wei, J. Establishment of an Integrated CRISPR/Cas9 Plasmid System for Simple and Efficient Genome Editing in Medaka In Vitro and In Vivo. Biology 2023, 12, 336. https://doi.org/10.3390/biology12020336
Zhang Z, Wang J, Li J, Liu X, Liu L, Zhao C, Tao W, Wang D, Wei J. Establishment of an Integrated CRISPR/Cas9 Plasmid System for Simple and Efficient Genome Editing in Medaka In Vitro and In Vivo. Biology. 2023; 12(2):336. https://doi.org/10.3390/biology12020336
Chicago/Turabian StyleZhang, Zeming, Jie Wang, Jianeng Li, Xiang Liu, Lei Liu, Changle Zhao, Wenjing Tao, Deshou Wang, and Jing Wei. 2023. "Establishment of an Integrated CRISPR/Cas9 Plasmid System for Simple and Efficient Genome Editing in Medaka In Vitro and In Vivo" Biology 12, no. 2: 336. https://doi.org/10.3390/biology12020336