
Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy
 , and
, and
Abstract
:Simple Summary
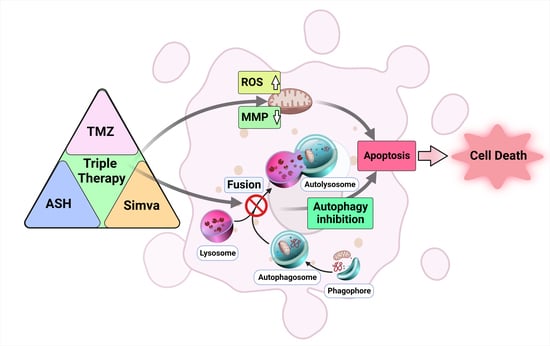
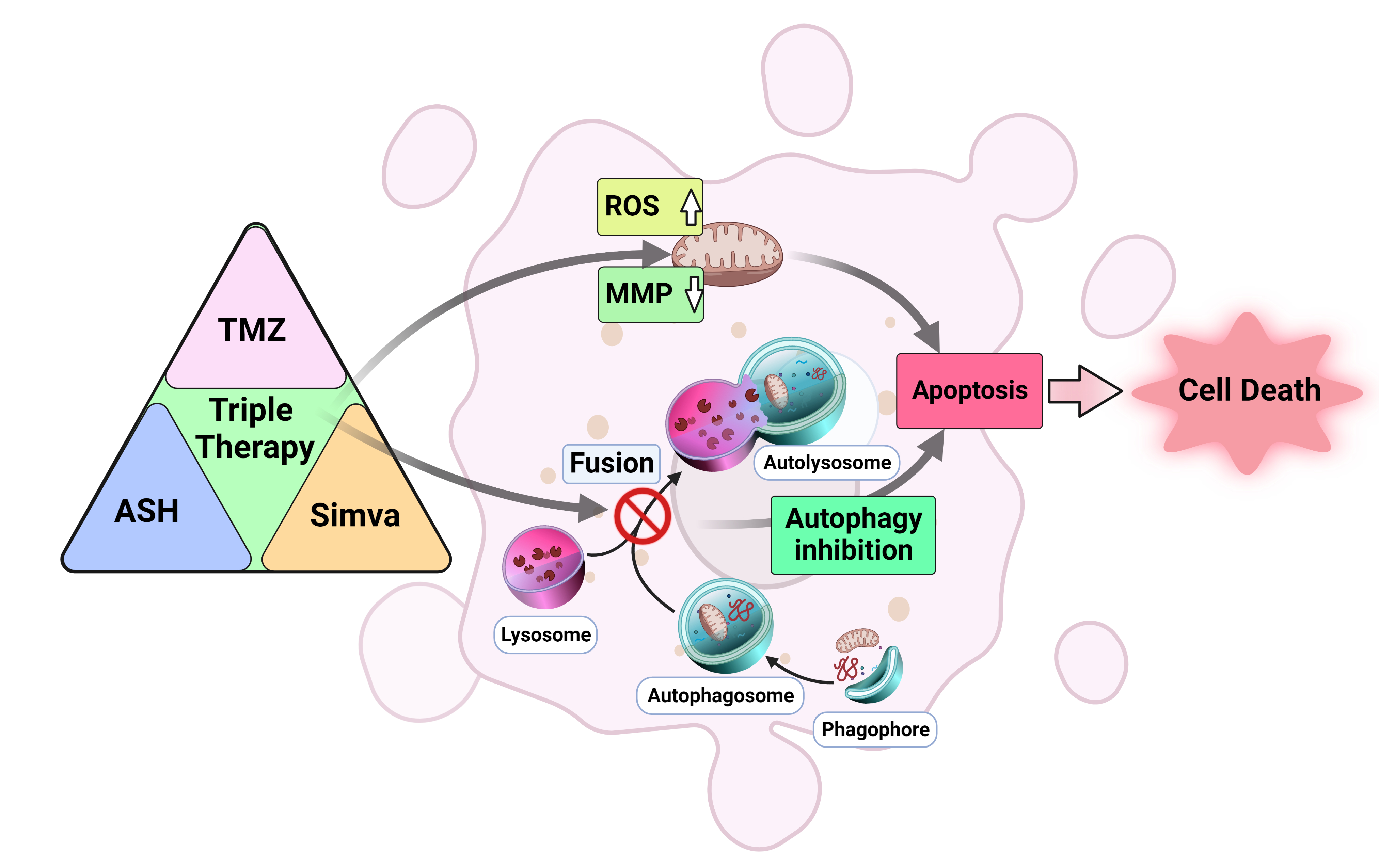
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Cell Lines, Culture, and Treatment
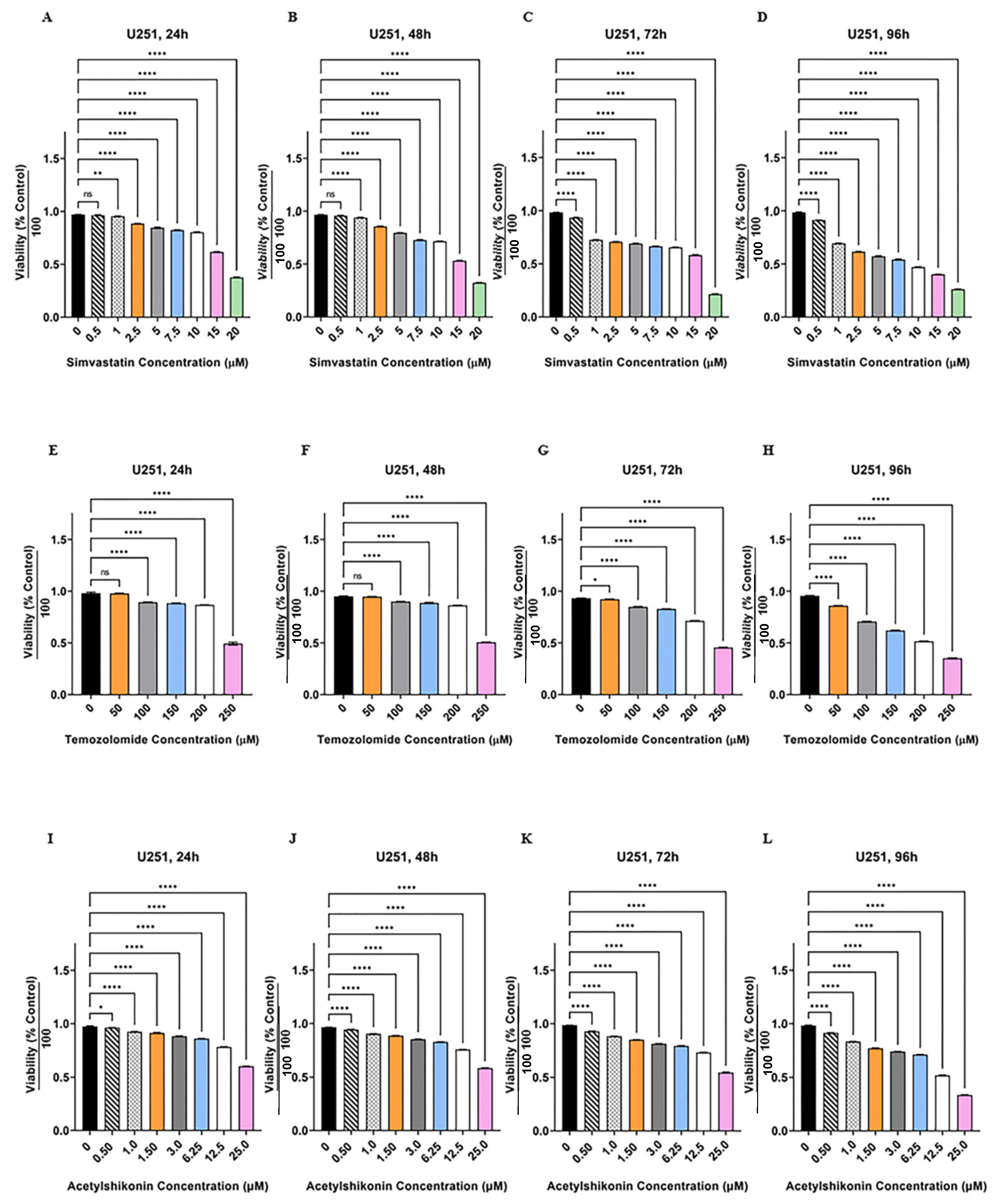
2.3. Cytotoxicity Assay
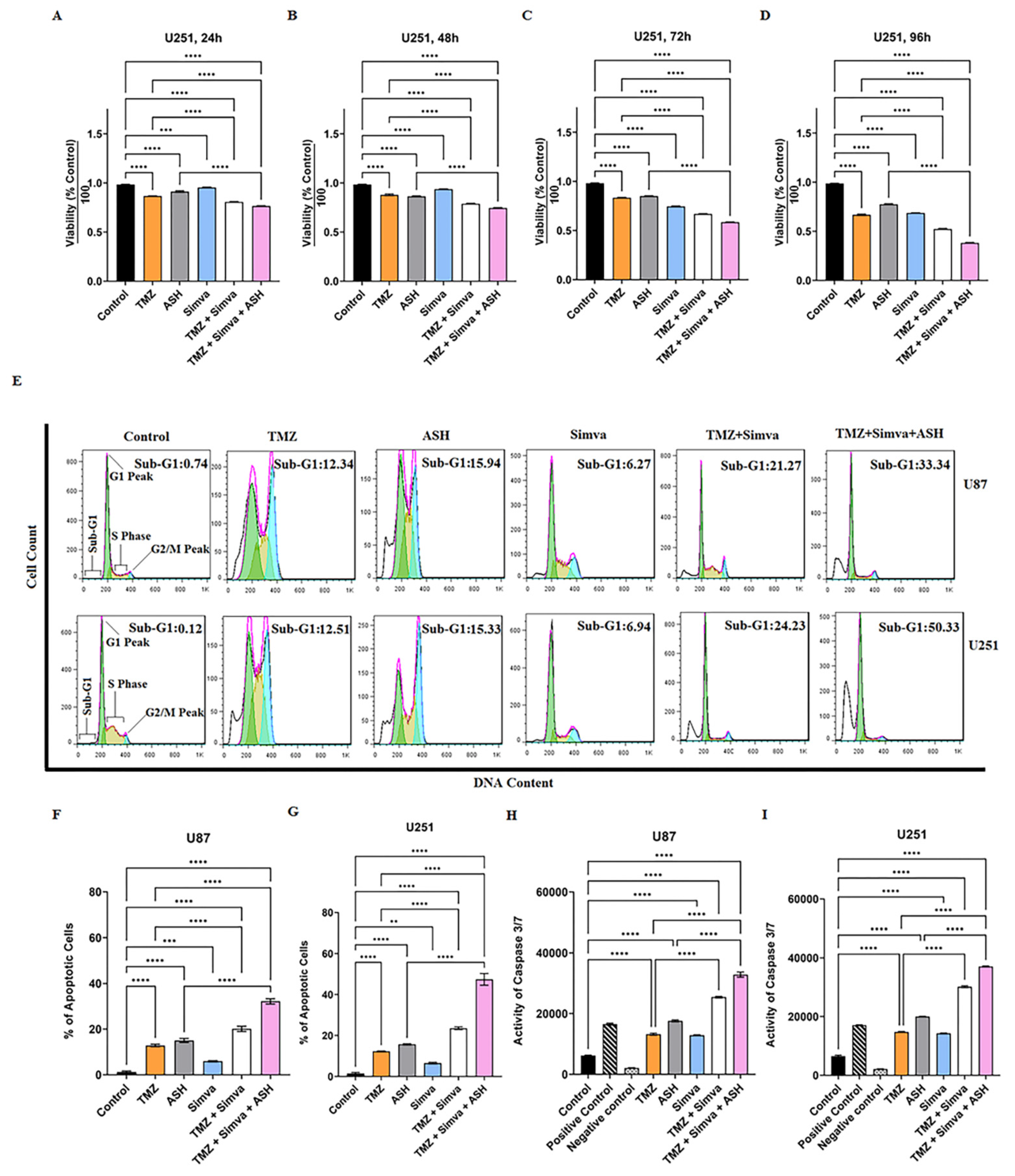
2.4. Apoptosis Assay Using Flow Cytometry
2.5. Immunoblotting
2.6. Caspase3/7 Activation Assay
2.7. Reactive Oxygen Assay
2.8. Determination of Mitochondrial Membrane Potential (MMP)
2.9. Acridine Orange Acidic Vacuole Assay
2.10. Statistical Analysis
3. Results
3.1. TMZ/Simva/ASH Combination Treatment Induces More Cell Death Compared to Single Treatment in Human GBM Cells
3.2. TMZ, Simva, ASH, TMZ/Simva and TMZ/Simva/ASH Treatments Induce Caspase-Dependent Apoptosis in GBM Cells
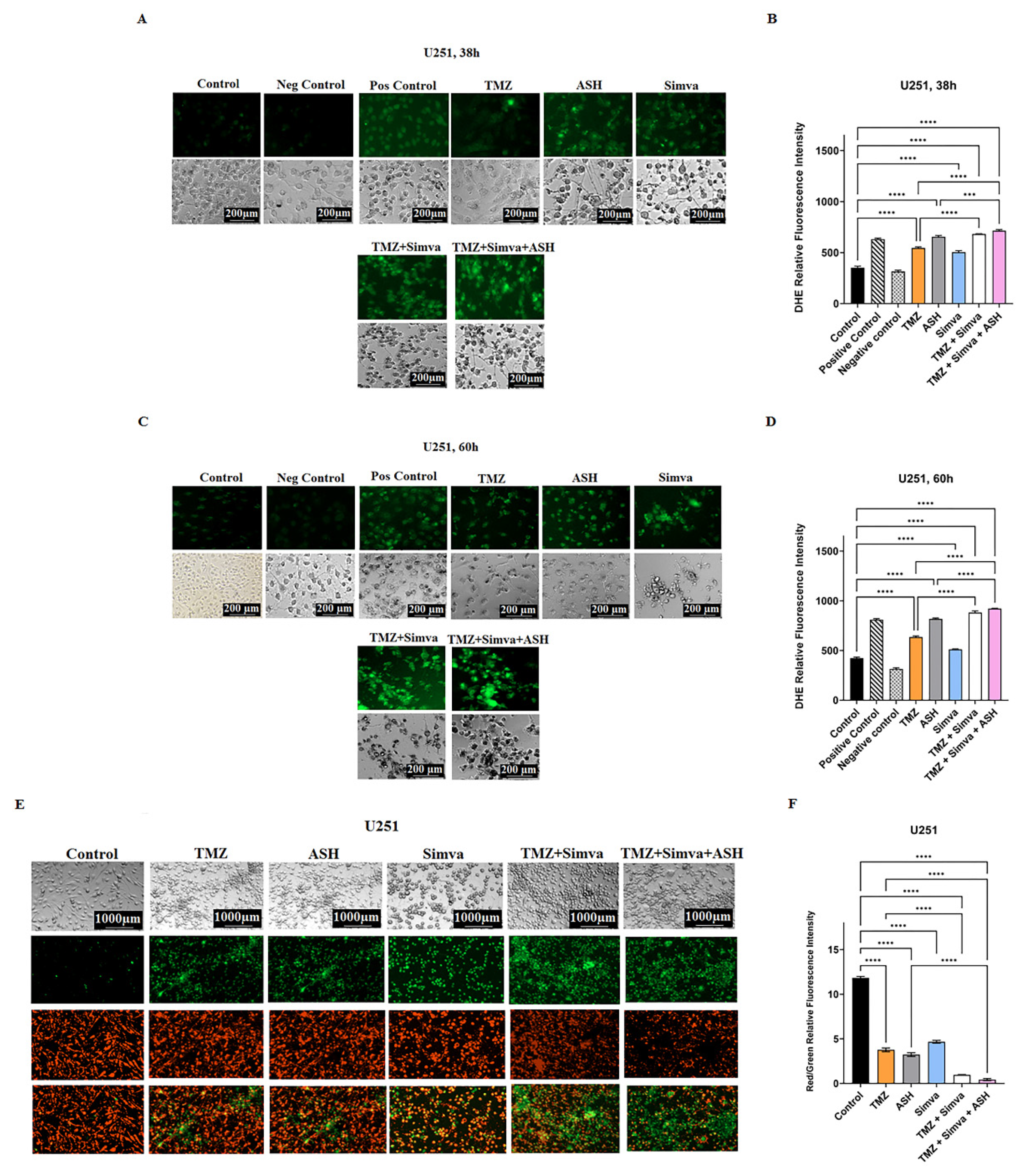
3.3. TMZ, Simva, ASH, TMZ/Simva and TMZ/Simva/ASH Treatments Increase Reactive Oxygen Species and Decrease Mitochondrial Membrane Potential in GBM Cells
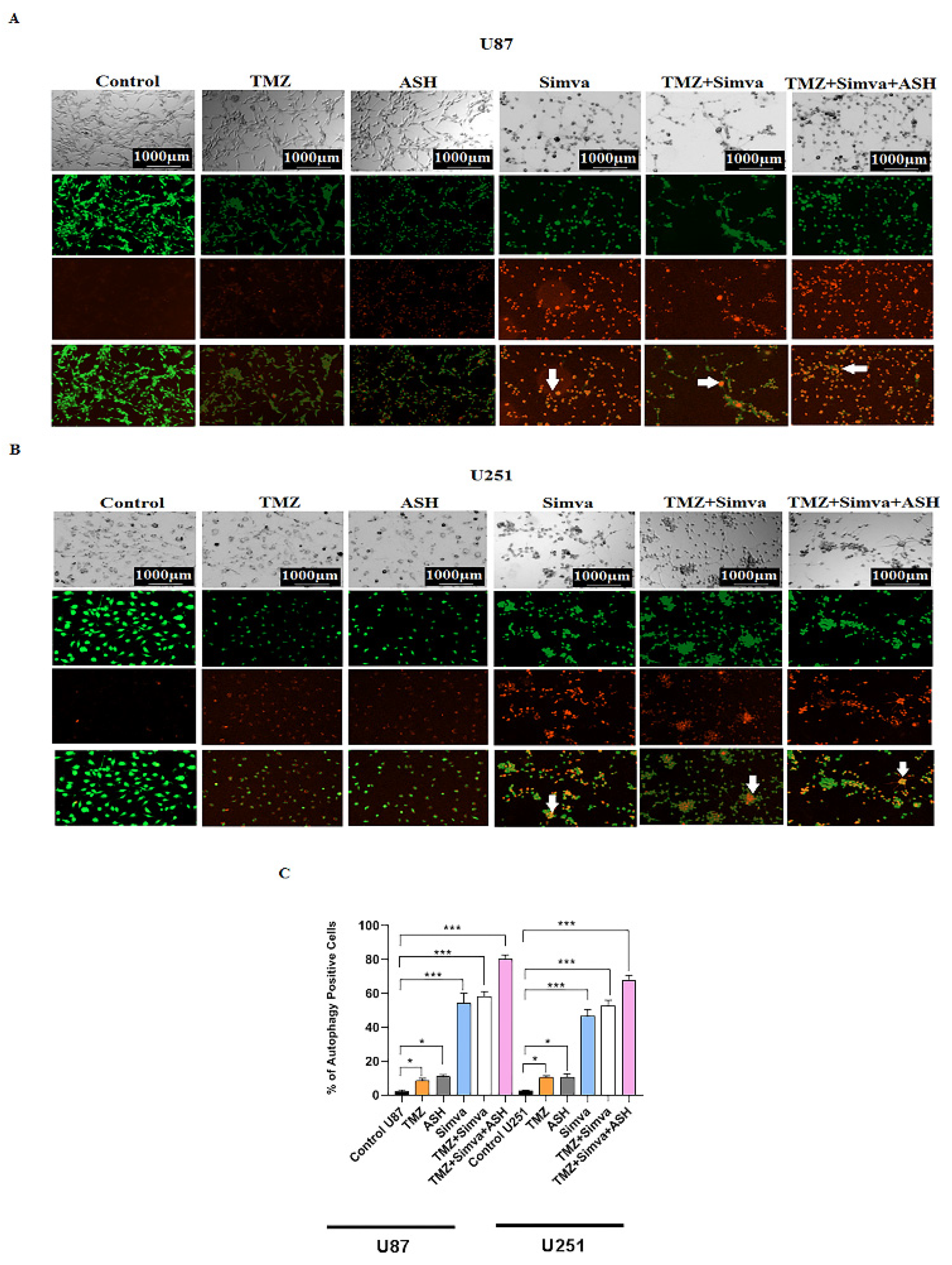
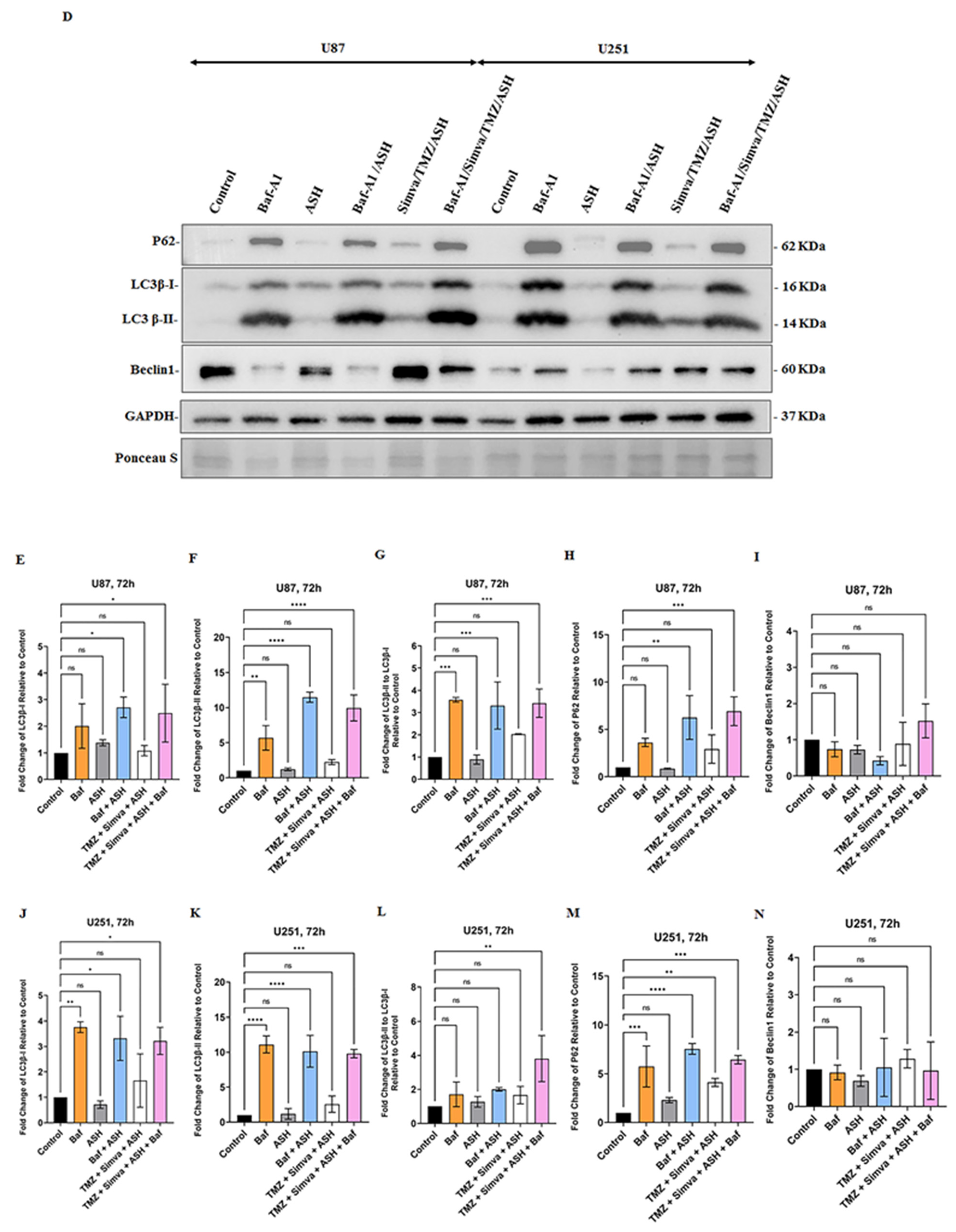
3.4. The Impact of TMZ, Simva, ASH, TMZ/Simva and TMZ/Simva/ASH Treatments on Autophagy in GBM Cells
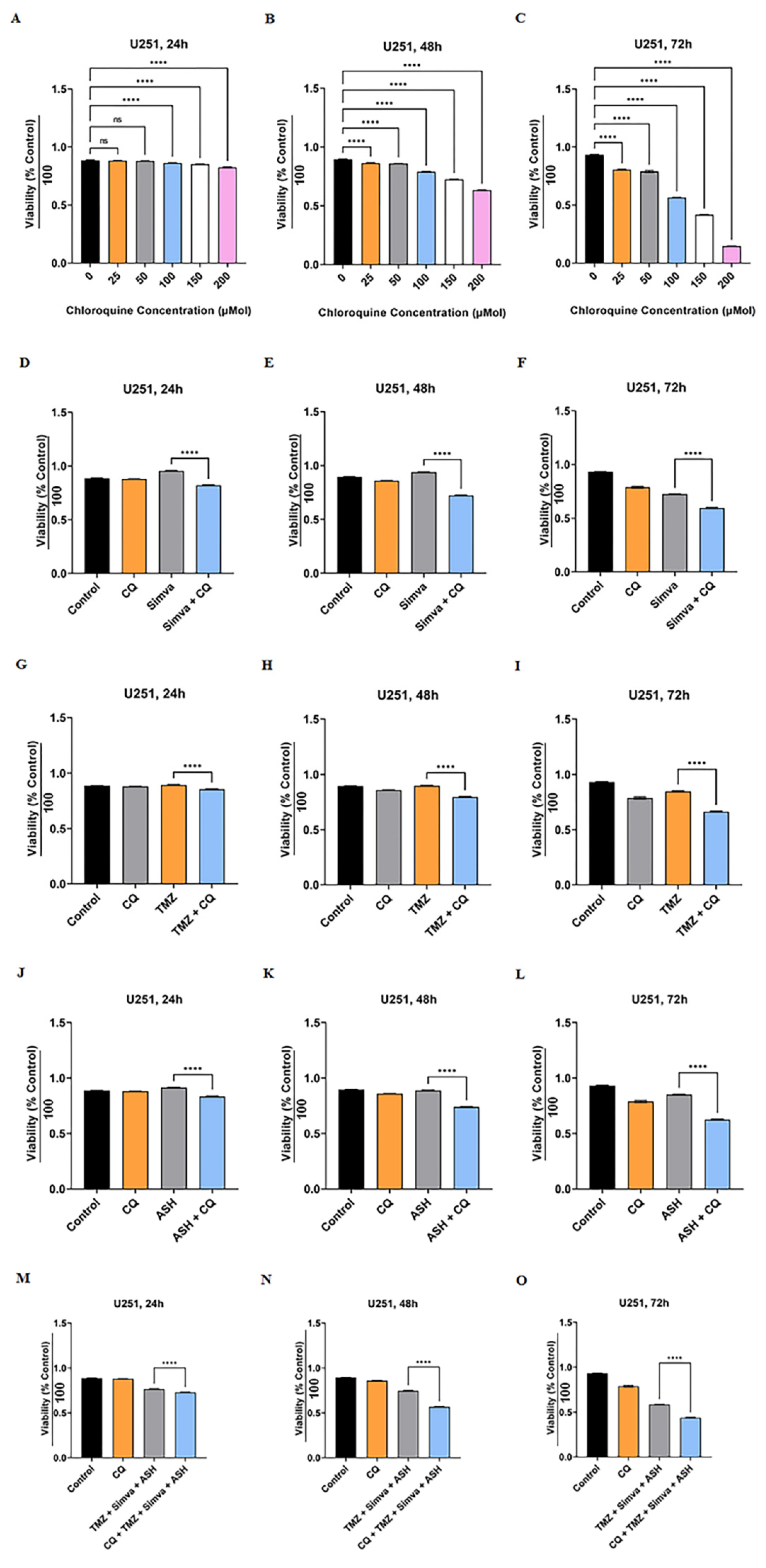
3.5. Autophagy Flux Inhibition Increases Simva, TMZ, ASH and Combination Triple Treatment-Induced Cell Death in GBM Cells
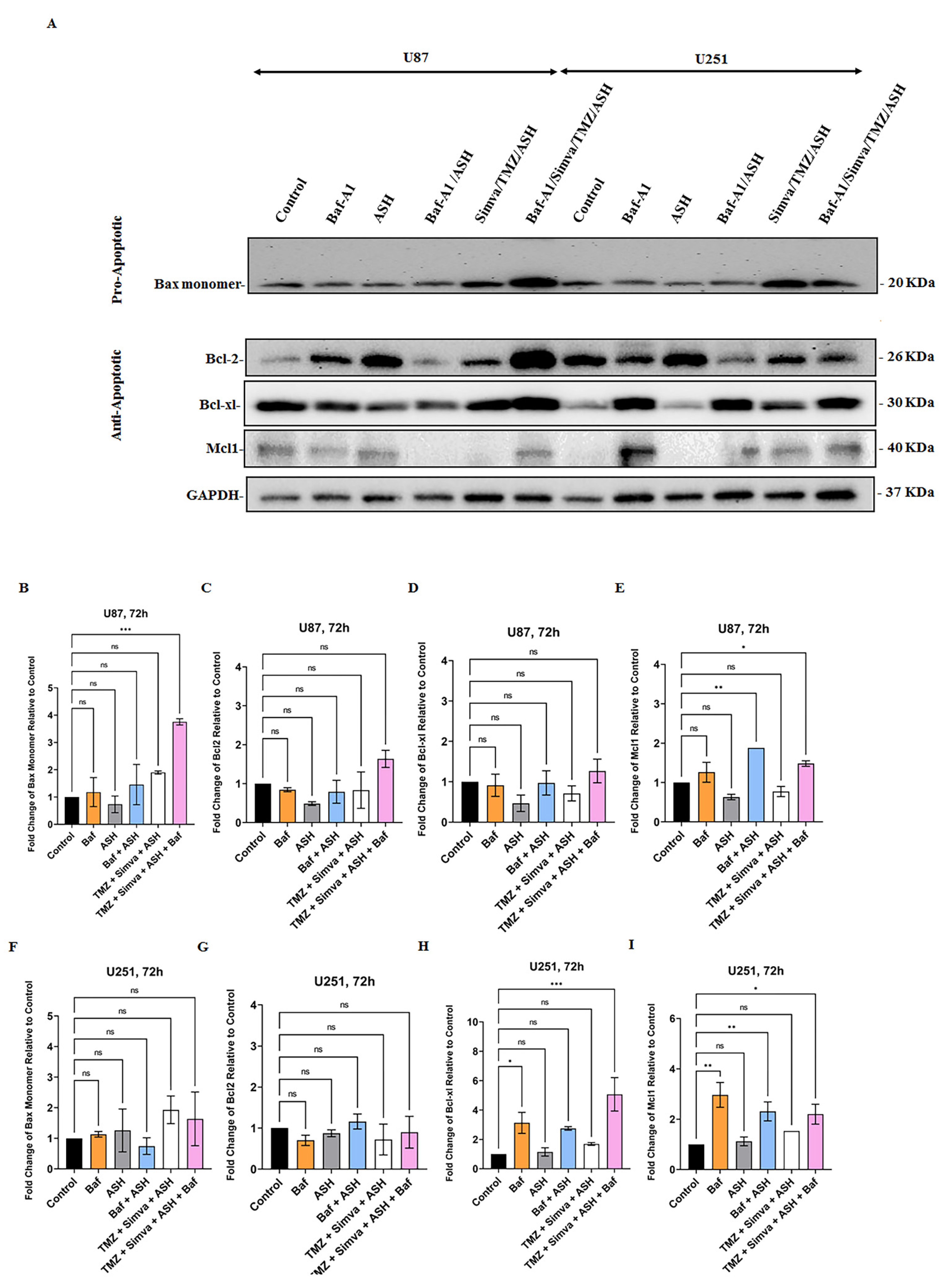
3.6. The Role of Bcl2 Pro- and Anti-Apoptotic Family Proteins in TMZ/Simva/ASH-Induced Cell Death in GBM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pirmoradi, L.; Seyfizadeh, N.; Ghavami, S.; Zeki, A.A.; Shojaei, S. Targeting cholesterol metabolism in glioblastoma: A new therapeutic approach in cancer therapy. J. Investig. Med. 2019, 67, 715–719. [Google Scholar] [CrossRef]
- Johannessen, T.C.; Hasan-Olive, M.M.; Zhu, H.; Denisova, O.; Grudic, A.; Latif, M.A.; Saed, H.; Varughese, J.K.; Røsland, G.V.; Yang, N. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int. J. Cancer 2019, 144, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Liao, W.; Fan, S.; Zheng, Y.; Liao, S.; Xiong, Y.; Li, Y.; Liu, J. Recent advances on glioblastoma multiforme and nano-drug carriers: A review. Curr. Med. Chem. 2019, 26, 5862–5874. [Google Scholar] [CrossRef]
- Cruz, J.V.R.; Batista, C.; Afonso, B.d.H.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Moura Neto, V.; Mendes, F.d.A. Obstacles to glioblastoma treatment two decades after temozolomide. Cancers 2022, 14, 3203. [Google Scholar] [CrossRef]
- McMahon, D.; Gleeson, J.; O’Reilly, S.; Bambury, R. Management of newly diagnosed glioblastoma multiforme: Current state of the art and emerging therapeutic approaches. Med. Oncol. 2022, 39, 129. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, H.R.; Choi, Y.W. Acetylshikonin inhibits growth of oral squamous cell carcinoma by inducing apoptosis. Arch. Oral Biol. 2016, 70, 149–157. [Google Scholar]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef]
- Shojaei, S.; Alizadeh, J.; Thliveris, J.; Koleini, N.; Kardami, E.; Hatch, G.M.; Xu, F.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J. Investig. Med. 2018, 66, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.r.; Albokashy, M.; Butterfield, Y. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Lin, C.-J.; Lee, C.-C.; Shih, Y.-L.; Lin, T.-Y.; Wang, S.-H.; Lin, Y.-F.; Shih, C.-M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Zamani, M.; Ahmadi, M.; Erfani, M.; Dastghaib, S.; Darbandi, M.; Darbandi, S.; Vakili, O.; Siri, M.; Grabarek, B.O. Epigenetic regulation of autophagy in gastrointestinal cancers. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2022, 1868, 166512. [Google Scholar] [CrossRef]
- Sharma, P.; Alizadeh, J.; Juarez, M.; Samali, A.; Halayko, A.J.; Kenyon, N.J.; Ghavami, S.; Zeki, A.A. Autophagy, apoptosis, the unfolded protein response, and lung function in idiopathic pulmonary fibrosis. Cells 2021, 10, 1642. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Burrough, E. The effects of swine coronaviruses on ER stress, autophagy, apoptosis, and alterations in cell morphology. Pathogens 2022, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Saptarshi, N.; Porter, L.F.; Paraoan, L. PERK/EIF2AK3 integrates endoplasmic reticulum stress-induced apoptosis, oxidative stress and autophagy responses in immortalised retinal pigment epithelial cells. Sci. Rep. 2022, 12, 13324. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-J.; Chien, S.-Y.; Lin, J.-T.; Yang, S.-F.; Chen, M.-K. Polyphyllin G induces apoptosis and autophagy cell death in human oral cancer cells Phytomedicine 23 (2016) 1545–1554. Phytomed. Int. J. Phytother. Phytopharm. 2018, 45, 120. [Google Scholar] [CrossRef]
- Kataura, T.; Sedlackova, L.; Otten, E.G.; Kumari, R.; Shapira, D.; Scialo, F.; Stefanatos, R.; Ishikawa, K.-i.; Kelly, G.; Seranova, E. Autophagy promotes cell survival by maintaining NAD levels. Dev. Cell 2022, 57, 2584–2598.e11. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar]
- Kawalec, P.; Martens, M.D.; Field, J.T.; Mughal, W.; Caymo, A.M.; Chapman, D.; Xiang, B.; Ghavami, S.; Dolinsky, V.W.; Gordon, J.W. Differential impact of doxorubicin dose on cell death and autophagy pathways during acute cardiotoxicity. Toxicol. Appl. Pharmacol. 2022, 453, 116210. [Google Scholar] [CrossRef]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin induces unfolded protein response and enhances temozolomide-induced cell death in glioblastoma cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating programmed cell death and tumor invasion in a three-dimensional (3D) microfluidic model of glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. [Google Scholar] [CrossRef] [PubMed]
- Emami, A.; Shojaei, S.; da Silva Rosa, S.C.; Aghaei, M.; Samiei, E.; Vosoughi, A.R.; Kalantari, F.; Kawalec, P.; Thliveris, J.; Sharma, P. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur. J. Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef] [PubMed]
- Nagakannan, P.; Iqbal, M.A.; Yeung, A.; Thliveris, J.A.; Rastegar, M.; Ghavami, S.; Eftekharpour, E. Perturbation of redox balance after thioredoxin reductase deficiency interrupts autophagy-lysosomal degradation pathway and enhances cell death in nutritionally stressed SH-SY5Y cells. Free Radic. Biol. Med. 2016, 101, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Göbel, A.; Riffel, R.M.; Hofbauer, L.C.; Rachner, T.D. The mevalonate pathway in breast cancer biology. Cancer Lett. 2022, 542, 215761. [Google Scholar] [CrossRef]
- Marcianò, G.; Palleria, C.; Casarella, A.; Rania, V.; Basile, E.; Catarisano, L.; Vocca, C.; Bianco, L.; Pelaia, C.; Cione, E. Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role. Pharmaceuticals 2022, 15, 589. [Google Scholar] [CrossRef]
- Gaist, D.; Hallas, J.; Friis, S.; Hansen, S.; Sørensen, H.T. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol. 2014, 38, 722–727. [Google Scholar] [CrossRef]
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Rezaei Moghadam, A.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W. Mevalonate cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef] [PubMed]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin induces apoptosis in medulloblastoma brain tumor cells via mevalonate cascade prenylation substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [Green Version]
- Zorman, J.; Sušjan, P.; Hafner-Bratkovič, I. Shikonin suppresses NLRP3 and AIM2 inflammasomes by direct inhibition of caspase-1. PLoS ONE 2016, 11, e0159826. [Google Scholar]
- Matias, D.; Balça-Silva, J.; Dubois, L.G.; Pontes, B.; Ferrer, V.P.; Rosário, L.; do Carmo, A.; Echevarria-Lima, J.; Sarmento-Ribeiro, A.B.; Lopes, M.C. Dual treatment with shikonin and temozolomide reduces glioblastoma tumor growth, migration and glial-to-mesenchymal transition. Cell. Oncol. 2017, 40, 247–261. [Google Scholar] [CrossRef]
- Chen, Q.; Han, H.; Lin, F.; Yang, L.; Feng, L.; Lai, X.; Wen, Z.; Yang, M.; Wang, C.; Ma, Y. Novel shikonin derivatives suppress cell proliferation, migration and induce apoptosis in human triple-negative breast cancer cells via regulating PDK1/PDHC axis. Life Sci. 2022, 310, 121077. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hu, X.; Fan, S.; Zhou, J.; Ren, S.; Sun, R.; Chen, Y.; Shen, X.; Chen, Y. Inhibition of Mitochondrial Biosynthesis Using a “Right-Side-Out” Membrane-Camouflaged Micelle to Facilitate the Therapeutic Effects of Shikonin on Triple-Negative Breast Cancer. Adv. Healthc. Mater. 2022, 11, 2200742. [Google Scholar] [CrossRef]
- Martelli, A.; Omrani, M.; Zarghooni, M.; Citi, V.; Brogi, S.; Calderone, V.; Sureda, A.; Lorzadeh, S.; da Silva Rosa, S.C.; Grabarek, B.O. New Visions on Natural Products and Cancer Therapy: Autophagy and Related Regulatory Pathways. Cancers 2022, 14, 5839. [Google Scholar] [CrossRef]
- Wu, H.; Xie, J.; Pan, Q.; Wang, B.; Hu, D.; Hu, X. Anticancer agent shikonin is an incompetent inducer of cancer drug resistance. PLoS ONE 2013, 8, e52706. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder cancer by inducing necroptosis. Int. J. Biol. Sci. 2018, 14, 1883. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Luo, G.; Zhou, L.; Zeng, Y.; Yang, W. In vitro and in vivo antitumor effects of acetylshikonin isolated from Arnebia euchroma (Royle) Johnst (Ruanzicao) cell suspension cultures. Chin. Med. 2009, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Lyu, C.; Stadlbauer, B.; Schrader, I.; Buchner, A.; Stepp, H.; Sroka, R.; Pohla, H. The role of Shikonin in improving 5-aminolevulinic acid-based photodynamic therapy and chemotherapy on glioblastoma stem cells. Photodiagnosis Photodyn. Ther. 2022, 39, 102987. [Google Scholar] [CrossRef] [PubMed]
- Siri, M.; Behrouj, H.; Dastghaib, S.; Zamani, M.; Likus, W.; Rezaie, S.; Hudecki, J.; Khazayel, S.; Łos, M.J.; Mokarram, P. Casein Kinase-1-alpha inhibitor (D4476) sensitizes microsatellite instable colorectal cancer cells to 5-fluorouracil via authophagy flux inhibition. Arch. Immunol. Ther. Exp. 2021, 69, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Behrouj, H.; Seghatoleslam, A.; Mokarram, P.; Ghavami, S. Effect of casein kinase 1α inhibition on autophagy flux and the AKT/phospho-β-catenin (S552) axis in HCT116, a RAS-mutated colorectal cancer cell line. Can. J. Physiol. Pharmacol. 2021, 99, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Hinton, M.; Eltayeb, E.; Ghavami, S.; Dakshinamurti, S. Effect of pulsatile stretch on unfolded protein response in a new model of the pulmonary hypertensive vascular wall. Biochem. Biophys. Rep. 2021, 27, 101080. [Google Scholar] [CrossRef]
- Alizadeh, J.; Kochan, M.M.; Stewart, V.D.; Drewnik, D.A.; Hannila, S.S.; Ghavami, S. Inhibition of autophagy flux promotes secretion of chondroitin sulfate proteoglycans in primary rat astrocytes. Mol. Neurobiol. 2021, 58, 6077–6091. [Google Scholar] [CrossRef]
- Ghavami, S.; Asoodeh, A.; Klonisch, T.; Halayko, A.J.; Kadkhoda, K.; Kroczak, T.J.; Gibson, S.B.; Booy, E.P.; Naderi-Manesh, H.; Los, M. Brevinin-2R1 semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J. Cell. Mol. Med. 2008, 12, 1005–1022. [Google Scholar] [CrossRef]
- Ghavami, S.; Cunnington, R.H.; Yeganeh, B.; Davies, J.J.; Rattan, S.G.; Bathe, K.; Kavosh, M.; Los, M.J.; Freed, D.H.; Klonisch, T. Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 2274–2286. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, J.; Wang, J.; Ju, X.; Zhang, H. Molecular mechanism of shikonin inhibiting tumor growth and potential application in cancer treatment. Toxicol. Res. 2021, 10, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S. Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac. J. Cancer Prev. 2015, 16, 7261–7266. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yu, M.; Hao, C.; Yang, W. Shikonin induces tumor apoptosis in glioma cells via endoplasmic reticulum stress, and Bax/Bak mediated mitochondrial outer membrane permeability. J. Ethnopharmacol. 2020, 263, 113059. [Google Scholar] [CrossRef]
- Paskeh, M.D.A.; Entezari, M.; Clark, C.; Zabolian, A.; Ranjbar, E.; Farahani, M.V.; Saleki, H.; Sharifzadeh, S.O.; Far, F.B.; Ashrafizadeh, M. Targeted regulation of autophagy using nanoparticles: New insight into cancer therapy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2022, 1868, 166326. [Google Scholar] [CrossRef] [PubMed]
- Yeganeh, B.; Moghadam, A.R.; Alizadeh, J.; Wiechec, E.; Alavian, S.M.; Hashemi, M.; Geramizadeh, B.; Samali, A.; Lankarani, K.B.; Post, M. Hepatitis B and C virus-induced hepatitis: Apoptosis, autophagy, and unfolded protein response. World J. Gastroenterol. 2015, 21, 13225. [Google Scholar] [CrossRef]
- Moosavi, M.A.; Sharifi, M.; Ghafary, S.M.; Mohammadalipour, Z.; Khataee, A.; Rahmati, M.; Hajjaran, S.; Łos, M.J.; Klonisch, T.; Ghavami, S. Photodynamic N-TiO2 nanoparticle treatment induces controlled ROS-mediated autophagy and terminal differentiation of leukemia cells. Sci. Rep. 2016, 6, 34413. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Eshragi, M.; Ande, S.R.; Chazin, W.J.; Klonisch, T.; Halayko, A.J.; Mcneill, K.D.; Hashemi, M.; Kerkhoff, C.; Los, M. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 2010, 20, 314–331. [Google Scholar] [CrossRef]
- da Silva Rosa, S.C.; Martens, M.D.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; Hai, Y.; Chapman, D.; Diehl-Jones, W.; Aliani, M.; West, A.R. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021, 17, 2257–2272. [Google Scholar] [CrossRef]
- Chaabane, W.; Cieślar-Pobuda, A.; El-Gazzah, M.; Jain, M.V.; Rzeszowska-Wolny, J.; Rafat, M.; Stetefeld, J.; Ghavami, S.; Łos, M.J. Human-gyrovirus-Apoptin triggers mitochondrial death pathway—Nur77 is required for apoptosis triggering. Neoplasia 2014, 16, 679–693. [Google Scholar] [CrossRef]
- Ghavami, S.; Mutawe, M.M.; Schaafsma, D.; Yeganeh, B.; Unruh, H.; Klonisch, T.; Halayko, A.J. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L420–L428. [Google Scholar] [CrossRef]
- Gupta, S.S.; Zeglinski, M.R.; Rattan, S.G.; Landry, N.M.; Ghavami, S.; Wigle, J.T.; Klonisch, T.; Halayko, A.J.; Dixon, I.M. Inhibition of autophagy inhibits the conversion of cardiac fibroblasts to cardiac myofibroblasts. Oncotarget 2016, 7, 78516. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; McAlinden, K.D.; Ghavami, S.; Deshpande, D.A. Chloroquine: Autophagy inhibitor, antimalarial, bitter taste receptor agonist in fight against COVID-19, a reality check? Eur. J. Pharmacol. 2021, 897, 173928. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Mutawe, M.M.; Hauff, K.; Stelmack, G.L.; Schaafsma, D.; Sharma, P.; McNeill, K.D.; Hynes, T.S.; Kung, S.K.; Unruh, H. Statin-triggered cell death in primary human lung mesenchymal cells involves p53-PUMA and release of Smac and Omi but not cytochrome c. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2010, 1803, 452–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.H.; Schaafsma, D.; Los, M. Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: A dual role for p53. PLoS ONE 2011, 6, e16523. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef]
- Fekrirad, Z.; Barzegar Behrooz, A.; Ghaemi, S.; Khosrojerdi, A.; Zarepour, A.; Zarrabi, A.; Arefian, E.; Ghavami, S. Immunology meets bioengineering: Improving the effectiveness of glioblastoma immunotherapy. Cancers 2022, 14, 3698. [Google Scholar] [CrossRef]
- Nakada, M.; Furuta, T.; Hayashi, Y.; Minamoto, T.; Hamada, J.-i. The strategy for enhancing temozolomide against malignant glioma. Front. Oncol. 2012, 2, 98. [Google Scholar] [CrossRef]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef]
- Yeganeh, B.; Wiechec, E.; Ande, S.R.; Sharma, P.; Moghadam, A.R.; Post, M.; Freed, D.H.; Hashemi, M.; Shojaei, S.; Zeki, A.A. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol. Ther. 2014, 143, 87–110. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, P.; Li, N.; Kiang, K.M.Y.; Cheng, S.Y.; Wong, V.K.-W.; Leung, G.K.-K. Lovastatin enhances cytotoxicity of temozolomide via impairing autophagic flux in glioblastoma cells. BioMed Res. Int. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Palko-Łabuz, A.; Środa-Pomianek, K.; Wesołowska, O.; Kostrzewa-Susłow, E.; Uryga, A.; Michalak, K. MDR reversal and pro-apoptotic effects of statins and statins combined with flavonoids in colon cancer cells. Biomed. Pharmacother. 2019, 109, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Luput, L.; Sesarman, A.; Porfire, A.; Achim, M.; Muntean, D.; Casian, T.; Patras, L.; Rauca, V.F.; Drotar, D.M.; Stejerean, I. Liposomal simvastatin sensitizes C26 murine colon carcinoma to the antitumor effects of liposomal 5-fluorouracil in vivo. Cancer Sci. 2020, 111, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-E.; Park, C.; Kwon, S.-J.; Kim, Y.-S.; Park, D.-S.; Lee, M.-K.; Kim, B.-R.; Park, S.-H.; Yoon, K.-H.; Jeong, E.-T. Synergistic induction of apoptosis by sulindac and simvastatin in A549 human lung cancer cells via reactive oxygen species-dependent mitochondrial dysfunction. Int. J. Oncol. 2013, 43, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.-E.; Kim, Y.-S.; Hwang, Y.-R.; Kwon, S.-J.; Park, D.-S.; Cha, B.-K.; Kim, B.-R.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R. Enhanced apoptosis by pemetrexed and simvastatin in malignant mesothelioma and lung cancer cells by reactive oxygen species-dependent mitochondrial dysfunction and Bim induction. Int. J. Oncol. 2014, 45, 1769–1777. [Google Scholar] [CrossRef]
- Cha, H.S.; Lee, H.K.; Park, S.-H.; Nam, M.J. Acetylshikonin induces apoptosis of human osteosarcoma U2OS cells by triggering ROS-dependent multiple signal pathways. Toxicol. In Vitro 2023, 86, 105521. [Google Scholar] [CrossRef]
- Lim, H.M.; Lee, J.; Yu, S.H.; Nam, M.J.; Cha, H.S.; Park, K.; Yang, Y.-H.; Jang, K.Y.; Park, S.-H. Acetylshikonin, a novel CYP2J2 inhibitor, induces apoptosis in RCC cells via FOXO3 activation and ROS elevation. Oxidative Med. Cell. Longev. 2022, 2022, 1–20. [Google Scholar] [CrossRef]
- Lim, H.M.; Lee, J.; Nam, M.J.; Park, S.-H. Acetylshikonin induces apoptosis in human colorectal cancer HCT-15 and LoVo cells via nuclear translocation of FOXO3 and ROS level elevation. Oxidative Med. Cell. Longev. 2021, 2021, 1–19. [Google Scholar] [CrossRef]
- Lohberger, B.; Glänzer, D.; Kaltenegger, H.; Eck, N.; Leithner, A.; Bauer, R.; Kretschmer, N.; Steinecker-Frohnwieser, B. Shikonin derivatives cause apoptosis and cell cycle arrest in human chondrosarcoma cells via death receptors and MAPK regulation. BMC Cancer 2022, 22, 758. [Google Scholar] [CrossRef]
- Hao, G.; Zhai, J.; Jiang, H.; Zhang, Y.; Wu, M.; Qiu, Y.; Fan, C.; Yu, L.; Bai, S.; Sun, L. Acetylshikonin induces apoptosis of human leukemia cell line K562 by inducing S phase cell cycle arrest, modulating ROS accumulation, depleting Bcr-Abl and blocking NF-κB signaling. Biomed. Pharmacother. 2020, 122, 109677. [Google Scholar] [CrossRef]
- Zhao, Q.; Kretschmer, N.; Bauer, R.; Efferth, T. Shikonin and its derivatives inhibit the epidermal growth factor receptor signaling and synergistically kill glioblastoma cells in combination with erlotinib. Int. J. Cancer 2015, 137, 1446–1456. [Google Scholar] [CrossRef]
- Wang, X.; Welsh, N. Bcl-2 maintains the mitochondrial membrane potential, but fails to affect production of reactive oxygen species and endoplasmic reticulum stress, in sodium palmitate-induced β-cell death. Upsala J. Med. Sci. 2014, 119, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Levin, M.D.; Doonan, P.J.; Petrenko, N.B.; Davis, C.W.; Patel, V.V.; Madesh, M. Mitochondrial complex II prevents hypoxic but not calcium-and proapoptotic Bcl-2 protein-induced mitochondrial membrane potential loss. J. Biol. Chem. 2010, 285, 26494–26505. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; Hedley, D.W.; Pham, N.-A.; Chow, S.; Minden, M.D. BAD induces apoptosis in cells over-expressing Bcl-2 or Bcl-xL without loss of mitochondrial membrane potential. Leuk. Lymphoma 2001, 42, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.; Kashani, H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef]
- Wu, M.D.; Zhang, Y.Y.; Yi, S.Y.; Sun, B.B.; Lan, J.; Jiang, H.M.; Hao, G.P. Acetylshikonin induces autophagy-dependent apoptosis through the key LKB1-AMPK and PI3K/Akt-regulated mTOR signalling pathways in HL-60 cells. J. Cell. Mol. Med. 2022, 26, 1606–1620. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Koh, S.S.; Malilas, W.; Cho, I.-R.; Kaewpiboon, C.; Kaowinn, S.; Lee, K.; Jhun, B.H.; Choi, Y.W.; Chung, Y.-H. Acetylshikonin induces apoptosis of hepatitis B virus X protein-expressing human hepatocellular carcinoma cells via endoplasmic reticulum stress. Eur. J. Pharmacol. 2014, 735, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Madrakian, T.; Ghavami, S. Preparation and characterization of simvastatin nanocapsules: Encapsulation of hydrophobic drugs in calcium alginate. Stem Cell Nanotechnol. Methods Protoc. 2020, 2125, 47–56. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajiahmadi, S.; Lorzadeh, S.; Iranpour, R.; Karima, S.; Rajabibazl, M.; Shahsavari, Z.; Ghavami, S. Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy. Biology 2023, 12, 302. https://doi.org/10.3390/biology12020302
Hajiahmadi S, Lorzadeh S, Iranpour R, Karima S, Rajabibazl M, Shahsavari Z, Ghavami S. Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy. Biology. 2023; 12(2):302. https://doi.org/10.3390/biology12020302
Chicago/Turabian StyleHajiahmadi, Sima, Shahrokh Lorzadeh, Rosa Iranpour, Saeed Karima, Masoumeh Rajabibazl, Zahra Shahsavari, and Saeid Ghavami. 2023. "Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy" Biology 12, no. 2: 302. https://doi.org/10.3390/biology12020302