
A Combined in Silico and Structural Study Opens New Perspectives on Aliphatic Sulfonamides, a Still Poorly Investigated Class of CA Inhibitors
 , ,
, ,  , , and
, , and
Abstract
:Simple Summary
Abstract
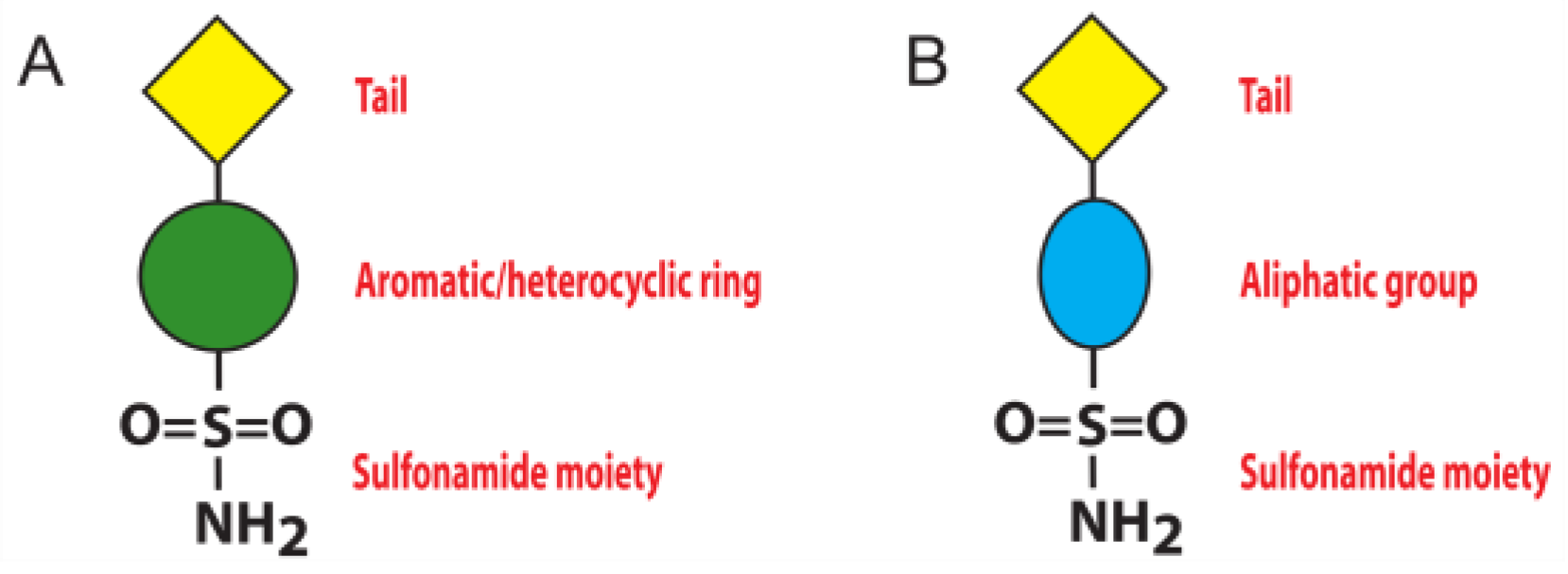
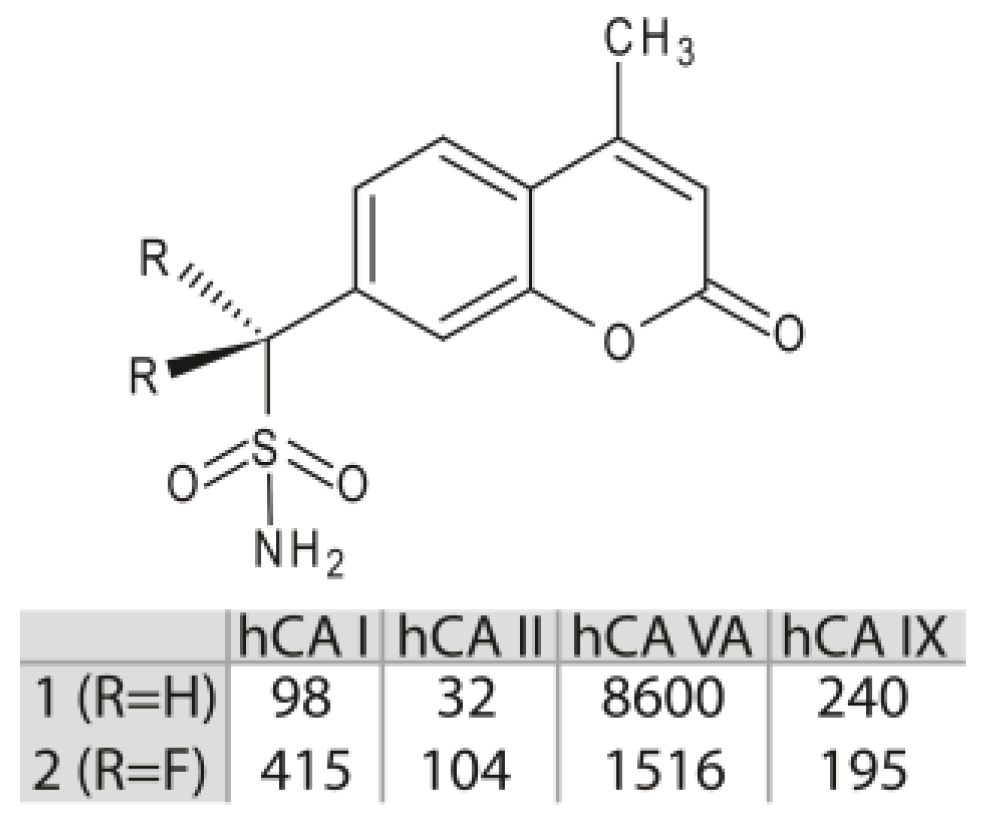
1. Introduction
2. Materials and Methods
2.1. Crystallization, Data Collection, and Structure Refinement
2.2. Computational Study
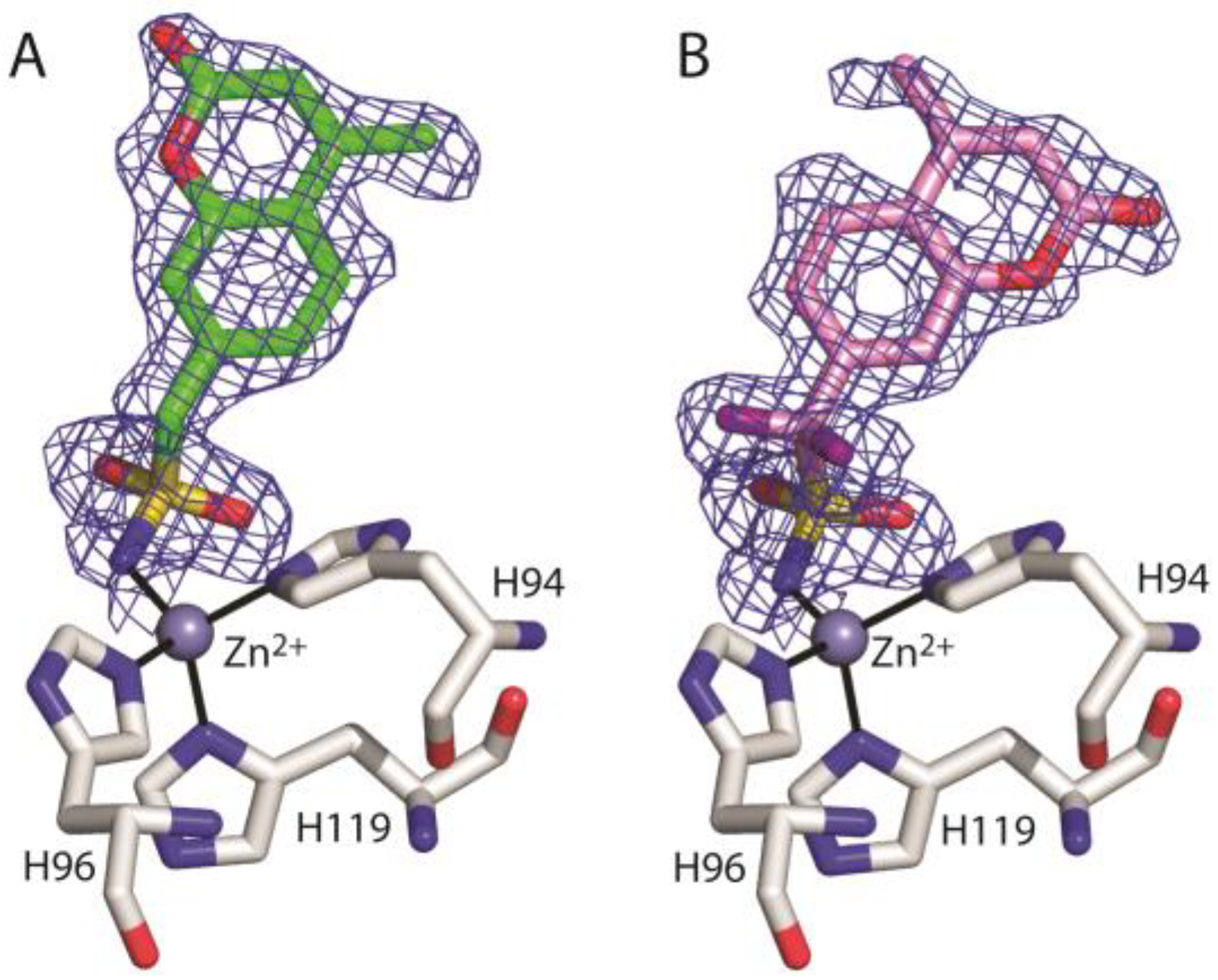
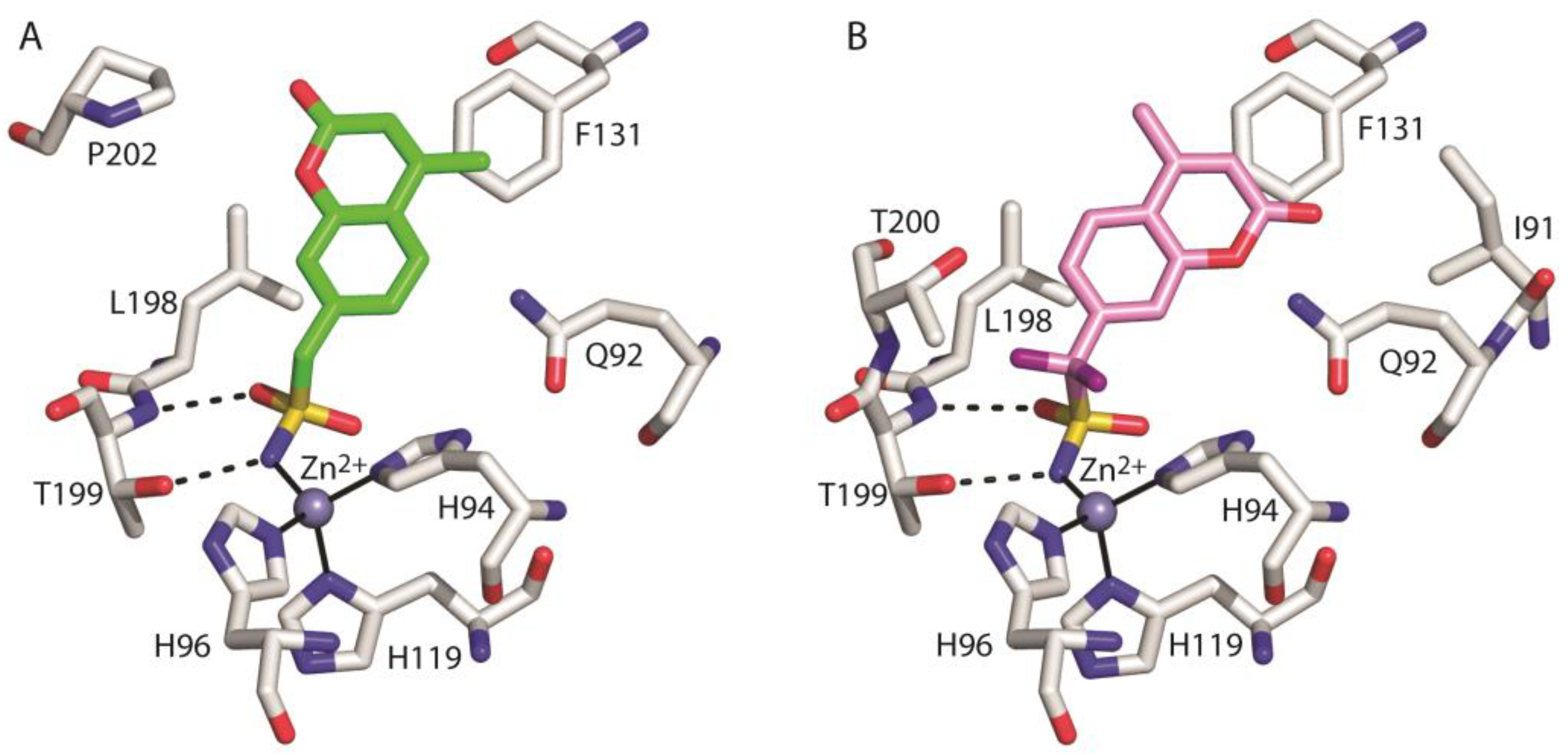
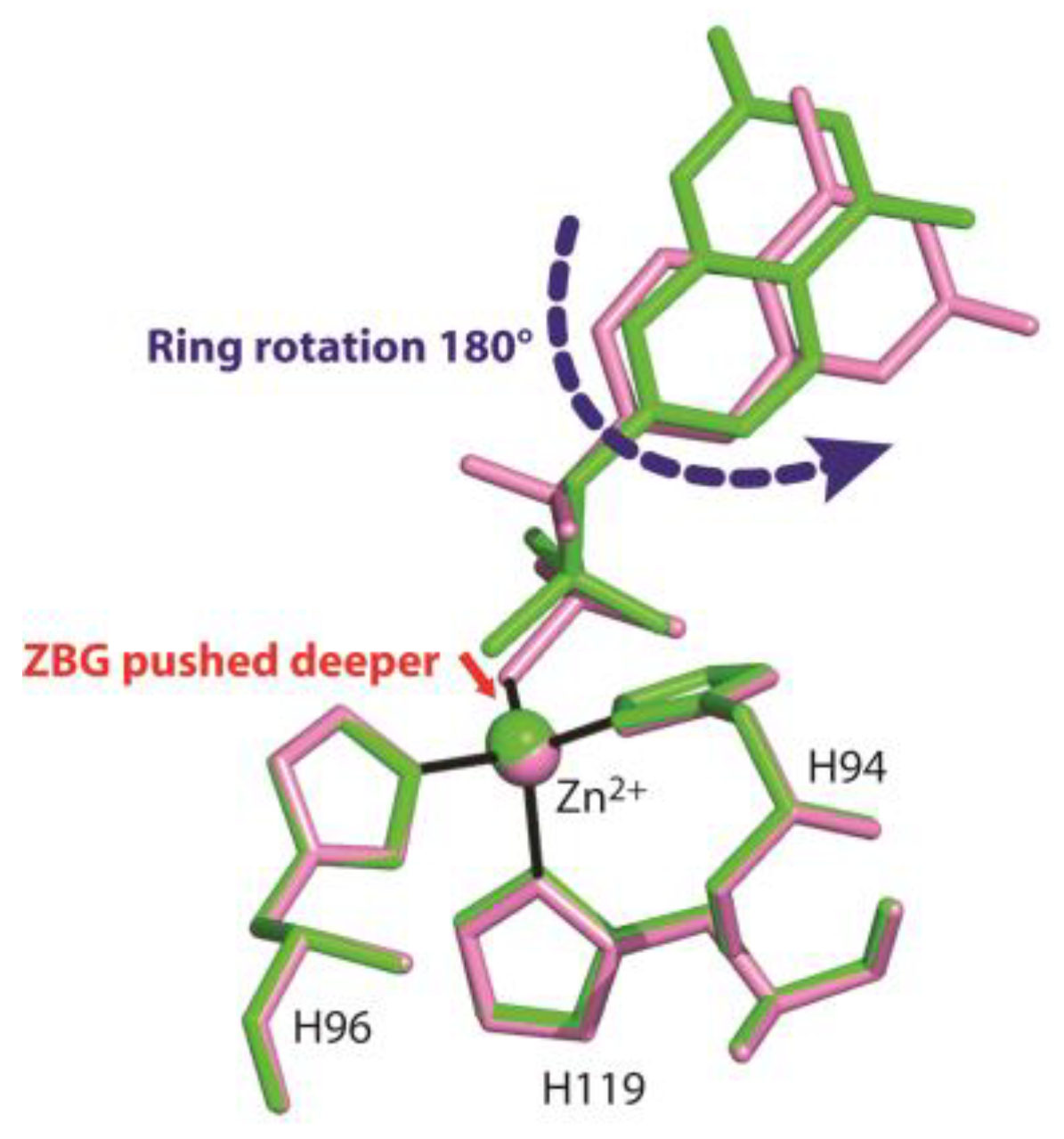
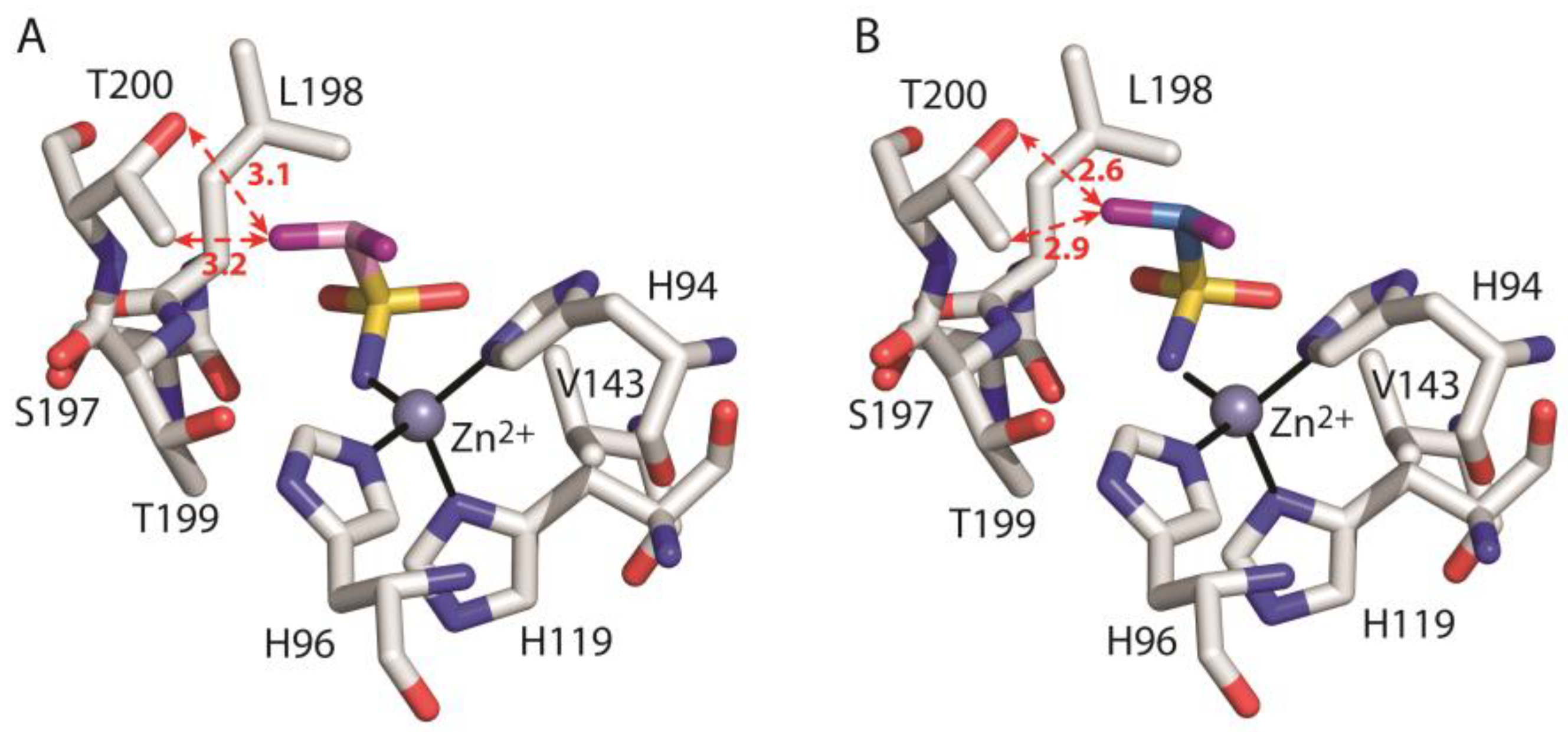
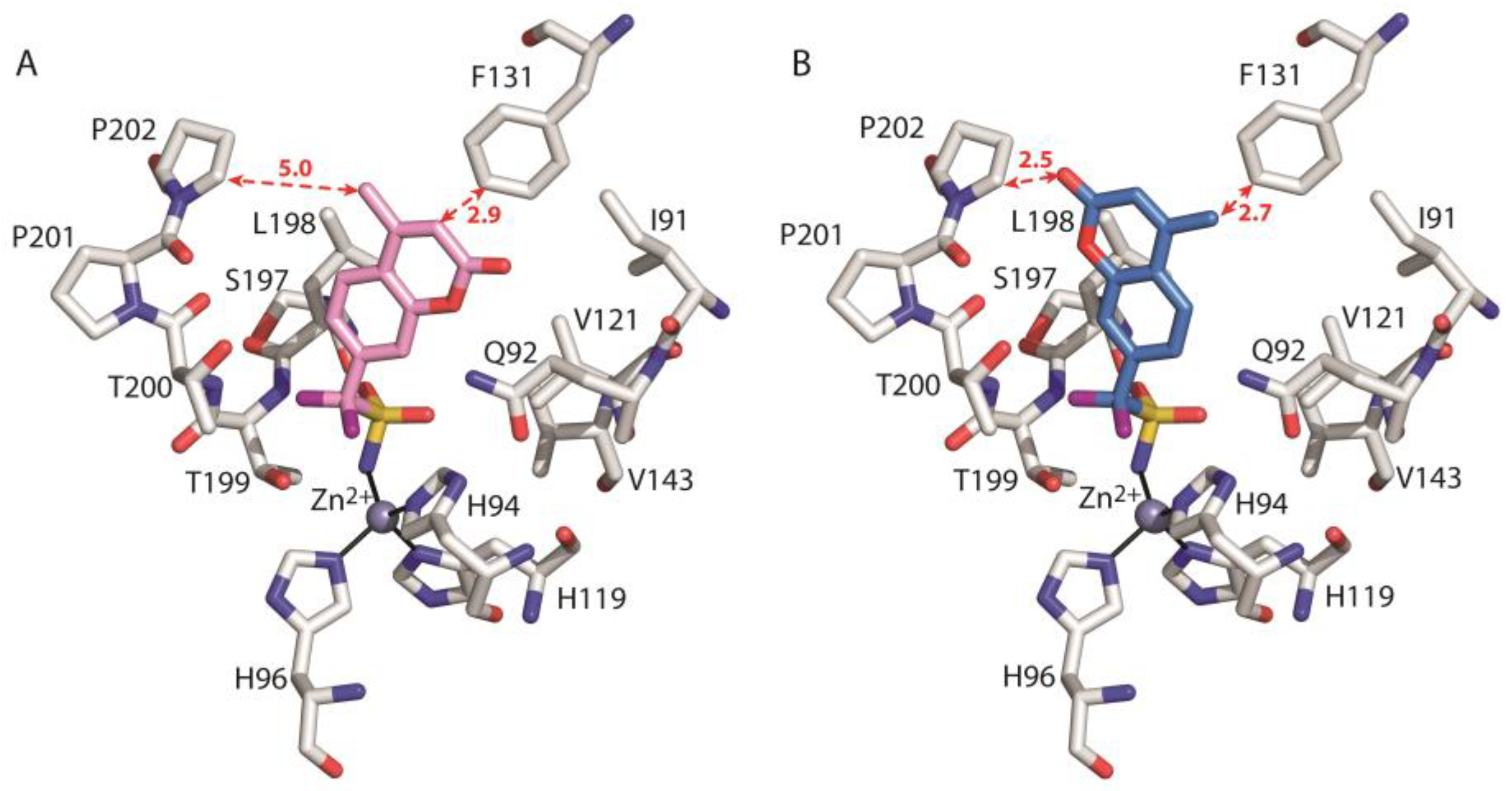
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Langella, E.; Di Fiore, A.; Alterio, V.; Monti, S.M.; De Simone, G.; D’Ambrosio, K. α-CAs from Photosynthetic Organisms. Int. J. Mol. Sci. 2022, 23, 12045. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; Buonanno, M.; Esposito, D.; Nocentini, A.; Berrino, E.; Bua, S.; Polentarutti, M.; Supuran, C.T.; Monti, S.M.; et al. Zeta-Carbonic Anhydrases Show CS2 Hydrolase Activity: A New Metabolic Carbon Acquisition Pathway in Diatoms? Comput. Struct. Biotechnol. J. 2021, 19, 3427–3436. [Google Scholar] [CrossRef]
- Jensen, E.L.; Clement, R.; Kosta, A.; Maberly, S.C.; Gontero, B. A New Widespread Subclass of Carbonic Anhydrase in Marine Phytoplankton. ISME J. 2019, 13, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Kikutani, S.; Nakajima, K.; Nagasato, C.; Tsuji, Y.; Miyatake, A.; Matsuda, Y. Thylakoid Luminal θ-Carbonic Anhydrase Critical for Growth and Photosynthesis in the Marine Diatom Phaeodactylum Tricornutum. Proc. Natl. Acad. Sci. USA 2016, 113, 9828–9833. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.G. The γ-Class of Carbonic Anhydrases. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 374–381. [Google Scholar] [CrossRef]
- Zimmerman, S.; Ferry, J. The β- and γ-Classes of Carbonic Anhydrase. Curr. Pharm. Des. 2008, 14, 716–721. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Di Fiore, A.; Capasso, C.; Supuran, C.T. The Zinc Coordination Pattern in the η-Carbonic Anhydrase from Plasmodium Falciparum Is Different from All Other Carbonic Anhydrase Genetic Families. Bioorg. Med. Chem. Lett. 2015, 25, 1385–1389. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; Viparelli, F.; Vullo, D.; Ascione, G.; Dathan, N.A.; Morel, F.M.M.; Supuran, C.T.; De Simone, G.; Monti, S.M. Structural and Inhibition Insights into Carbonic Anhydrase CDCA1 from the Marine Diatom Thalassiosira Weissflogii. Biochimie 2012, 94, 1232–1241. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Senda, M.; Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a Novel Type of Carbonic Anhydrase That Acts without Metal Cofactors. BMC Biol. 2021, 19, 105. [Google Scholar] [CrossRef]
- Vullo, D.; Del Prete, S.; Di Fonzo, P.; Carginale, V.; Donald, W.A.; Supuran, C.T.; Capasso, C.; Mcphee, D.J.; Muñoz-Torrero, D. Comparison of the Sulfonamide Inhibition Profiles of the β- and γ-Carbonic Anhydrases from the Pathogenic Bacterium Burkholderia pseudomallei. Molecules 2017, 22, 421. [Google Scholar] [CrossRef]
- Truppo, E.; Supuran, C.T.; Sandomenico, A.; Vullo, D.; Innocenti, A.; Di Fiore, A.; Alterio, V.; De Simone, G.; Monti, S.M. Carbonic Anhydrase VII Is S-Glutathionylated without Loss of Catalytic Activity and Affinity for Sulfonamide Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Langella, E.; Buonanno, M.; Vullo, D.; Dathan, N.; Leone, M.; Supuran, C.T.; De Simone, G.; Monti, S.M. Biochemical, Biophysical and Molecular Dynamics Studies on the Proteoglycan-like Domain of Carbonic Anhydrase IX. Cell. Mol. Life Sci. 2018, 75, 3283–3296. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T.; De Simone, G. (Eds.) Carbonic Anhydrases as Biocatalysts, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780444632586. [Google Scholar]
- De Simone, G.; Alterio, V.; Supuran, C.T. Exploiting the Hydrophobic and Hydrophilic Binding Sites for Designing Carbonic Anhydrase Inhibitors. Expert Opin. Drug Discov. 2013, 8, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Maren, T.H.; Conroy, C.W. A New Class of Carbonic Anhydrase Inhibitor. J. Biol. Chem. 1993, 268, 26233–26239. [Google Scholar] [CrossRef]
- Remko, M.; Von Der Lieth, C.W. Theoretical Study of Gas-Phase Acidity, PKa, Lipophilicity, and Solubility of Some Biologically Active Sulfonamides. Bioorg. Med. Chem. 2004, 12, 5395–5403. [Google Scholar] [CrossRef]
- Dvořanová, J.; Kugler, M.; Holub, J.; Šícha, V.; Das, V.; Nekvinda, J.; El Anwar, S.; Havránek, M.; Pospíšilová, K.; Fábry, M.; et al. Sulfonamido Carboranes as Highly Selective Inhibitors of Cancer-Specific Carbonic Anhydrase IX. Eur. J. Med. Chem. 2020, 200, 112460. [Google Scholar] [CrossRef]
- Grüner, B.; Kugler, M.; El Anwar, S.; Holub, J.; Nekvinda, J.; Bavol, D.; Růžičková, Z.; Pospíšilová, K.; Fábry, M.; Král, V.; et al. Cobalt Bis(Dicarbollide) Alkylsulfonamides: Potent and Highly Selective Inhibitors of Tumor Specific Carbonic Anhydrase IX. ChemPlusChem 2021, 86, 352–363. [Google Scholar] [CrossRef]
- De Simone, G.; Di Fiore, A.; Menchise, V.; Pedone, C.; Antel, J.; Casini, A.; Scozzafava, A.; Wurl, M.; Supuran, C.T. Carbonic Anhydrase Inhibitors. Zonisamide Is an Effective Inhibitor of the Cytosolic Isozyme II and Mitochondrial Isozyme V: Solution and X-Ray Crystallographic Studies. Bioorg. Med. Chem. Lett. 2005, 15, 2315–2320. [Google Scholar] [CrossRef]
- Temperini, C.; Cecchi, A.; Boyle, N.A.; Scozzafava, A.; Cabeza, J.E.; Wentworth, P.; Blackburn, G.M.; Supuran, C.T. Carbonic Anhydrase Inhibitors. Interaction of 2-N,N-Dimethylamino-1,3,4-Thiadiazole-5-Methanesulfonamide with 12 Mammalian Isoforms: Kinetic and X-Ray Crystallographic Studies. Bioorg. Med. Chem. Lett. 2008, 18, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, A.; Taylor, S.D.; Liu, Y.; Hill, B.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Inhibition of the Human Isozymes I, II, VA, and IX with a Library of Substituted Difluoromethanesulfonamides. Bioorg. Med. Chem. Lett. 2005, 15, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.; Liu, Y.; Taylor, S.D. Synthesis of α-Fluorosulfonamides by Electrophilic Fluorination. Org. Lett. 2004, 6, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Angeli, A.; Bozdag, M.; Supuran, C.T.; Winum, J.Y.J.-Y.; Monti, S.M.; Alterio, V. Inhibition of Carbonic Anhydrases by a Substrate Analog: Benzyl Carbamate Directly Coordinates the Catalytic Zinc Ion Mimicking Bicarbonate Binding. Chem. Commun. 2018, 54, 10312–10315. [Google Scholar] [CrossRef]
- Alterio, V.; De Simone, G.; Monti, S.M.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Inhibition of Human, Bacterial, and Archaeal Isozymes with Benzene-1,3-Disulfonamides-Solution and Crystallographic Studies. Bioorg. Med. Chem. Lett. 2007, 17, 4201–4207. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Alterio, V.; Tanc, M.; Ivanova, J.; Zalubovskis, R.; Vozny, I.; Monti, S.M.; Di Fiore, A.; De Simone, G.; Supuran, C.T. X-Ray Crystallographic and Kinetic Investigations of 6-Sulfamoyl-Saccharin as a Carbonic Anhydrase Inhibitor. Org. Biomol. Chem. 2015, 13, 4064–4069. [Google Scholar] [CrossRef]
- Jones, T.A.; Zou, J.-Y.; Cowan, S.W.; Kjeldgaard, M. Improved Methods for Building Protein Models in Electron Density Maps and the Location of Errors in These Models. Acta Crystallogr. Sect. A Found. Crystallogr. 1991, 47, 110–119. [Google Scholar] [CrossRef]
- Brunger, A.T. Version 1.2 of the Crystallography and NMR System. Nat. Protoc. 2007, 2, 2728–2733. [Google Scholar] [CrossRef]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; Delano, W.L.; Gros, P.; Grossekunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B: Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y.R.E.D. Server: A Web Service for Deriving RESP and ESP Charges and Building Force Field Libraries for New Molecules and Molecular Fragments. Nucleic Acids Res. 2011, 39, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.W.; King, R.W.; Burgen, A.S.V. Influence of PH on the Kinetics of Complex Formation between Aromatic Sulfonamides and Human Carbonic Anhydrase. Biochemistry 1970, 9, 3894–3920. [Google Scholar] [CrossRef] [PubMed]
- Tsui, V.; Case, D.A. Theory and Applications of the Generalized Born Solvation Model in Macromolecular Simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.I.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 114. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef]
- De Simone, G.; Langella, E.; Esposito, D.; Supuran, C.T.; Monti, S.M.; Winum, J.-Y.; Alterio, V. Insights into the Binding Mode of Sulphamates and Sulphamides to hCA II: Crystallographic Studies and Binding Free Energy Calculations. J. Enzyme Inhib. Med. Chem. 2017, 32, 1002–1011. [Google Scholar] [CrossRef]
- Langella, E.; Alterio, V.; D’Ambrosio, K.; Cadoni, R.; Winum, J.-Y.; Supuran, C.T.; Monti, S.M.; De Simone, G.; Di Fiore, A. Exploring Benzoxaborole Derivatives as Carbonic Anhydrase Inhibitors: A Structural and Computational Analysis Reveals Their Conformational Variability as a Tool to Increase Enzyme Selectivity. J. Enzyme Inhib. Med. Chem. 2019, 34, 1498–1505. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in Reproducing the Binding Free Energies to HIV-1 RT of TIBO Derivatives and Predicting the Binding Mode to HIV-1 RT of Efavirenz by Docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Autiero, I.; Saviano, M.; Langella, E. Conformational Studies of Chiral D-Lys-PNA and Achiral PNA System in Binding with DNA or RNA through a Molecular Dynamics Approach. Eur. J. Med. Chem. 2015, 91, 109–117. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins Struct. Funct. Genet. 2004, 55, 383–394. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate Solvent-Accessible Surface Areas from Tetrahedrally Directed Neighbor Densities. Biopolymers 1999, 50, 373–380. [Google Scholar] [CrossRef]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the Mechanism of Carbonic Anhydrase Inhibition with Coumarins and Thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods: I. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Menchise, V.; De Simone, G.; Alterio, V.; Di Fiore, A.; Pedone, C.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Stacking with Phe131 Determines Active Site Binding Region of Inhibitors as Exemplified by the X-Ray Crystal Structure of a Membrane-Impermeant Antitumor Sulfonamide Complexed with Isozyme II. J. Med. Chem. 2005, 48, 5721–5727. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hCA II/1 | hCA II/2 | |
|---|---|---|
| Crystal parameters | ||
| Space group | P21 | P21 |
| a (Å) | 42.3 | 42.3 |
| b (Å) | 41.5 | 41.5 |
| c (Å) | 72.2 | 72.1 |
| β (°) | 104.4 | 104.4 |
| Data collection statistics | ||
| Resolution (Å) | 25.4–1.67 (1.70–1.67) | 24.6–1.56 (1.59–1.56) |
| Temperature (K) | 100 | 100 |
| Total reflections | 104,361 | 141,168 |
| Unique reflections | 26,790 | 33,655 |
| Completeness (%) | 94.0 (81.3) | 96.4 (71.9) |
| <I>/<σ(I)> | 15.2 (2.1) | 15.9 (2.5) |
| Redundancy (%) | 3.9 (2.9) | 4.2 (2.3) |
| Rmerge a | 0.073 (0.591) | 0.070 (0.359) |
| Rmeas b | 0.082 (0.703) | 0.077 (0.448) |
| Rpim c | 0.038 (0.374) | 0.032 (0.261) |
| CC1/2 d | 0.998 (0.622) | 0.998 (0.801) |
| Refinement statistics | ||
| Resolution (Å) | 25.4–1.67 | 24.6–1.56 |
| Rwork e (%) | 17.5 | 17.7 |
| Rfree e (%) | 21.8 | 20.1 |
| r.m.s.d. from ideal geometry: | ||
| Bond lengths (Å) | 0.009 | 0.009 |
| Bond angles (°) | 1.6 | 1.6 |
| Number of protein atoms | 2076 | 2072 |
| Number of inhibitor atoms | 17 | 19 |
| Number of water molecules | 219 | 238 |
| Average B factor (Å2) | ||
| All atoms | 15.5 | 12.7 |
| Protein atoms | 14.7 | 11.7 |
| Inhibitor atoms | 18.1 | 16.9 |
| Water molecules | 23.2 | 20.9 |
| hCA II/2ZBG | hCA II/2*ZBG | ||
|---|---|---|---|
| ΔGbind-Val143 | −1.234 | −0.988 | |
| ΔGbind-Ser197 | −0.968 | −1.130 | |
| ΔGbind-Leu198 | −5.170 | −5.644 | |
| ΔGbind-Thr199 | −1.890 | −0.715 | |
| ΔGbind-Thr200 | −0.512 | 2.238 | |
| ΔEvdW | −0.003 | 2.310 | |
| ΔEelec | 2.510 | 3.008 | |
| ΔGGB | −2.358 | −2.259 | |
| ΔGSA | −0.661 | −0.821 | |
| hCA II/2 | hCA II/2*ring | ||
|---|---|---|---|
| ΔGbind-Ile91 | −1.158 | −0.354 | |
| ΔGbind-Gln92 | −2.726 | −1.271 | |
| ΔGbind-Val121 | −2.332 | −1.622 | |
| ΔGbind-Phe131 | −1.117 | 34.388 | |
| ΔEvdW | −0.131 | 35.858 | |
| ΔEelec | 0.169 | −0.240 | |
| ΔGGB | 0.128 | 0.063 | |
| ΔGSA | −1.283 | −1.293 | |
| ΔGbind-Val143 | −1.266 | −1.254 | |
| ΔGbind-Ser197 | −0.944 | −0.939 | |
| ΔGbind-Leu198 | −7.033 | −6.781 | |
| ΔGbind-Thr199 | −2.203 | −2.146 | |
| ΔGbind-Thr200 | −1.426 | −2.008 | |
| ΔGbind-Pro201 | −0.122 | −0.679 | |
| ΔGbind-Pro202 | −0.766 | 11.394 | |
| ΔEvdW | −0.364 | 12.442 | |
| ΔEelec | −0.340 | −1.797 | |
| ΔGGB | 0.277 | 1.336 | |
| ΔGSA | −0.340 | −0.586 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langella, E.; Esposito, D.; Monti, S.M.; Supuran, C.T.; De Simone, G.; Alterio, V. A Combined in Silico and Structural Study Opens New Perspectives on Aliphatic Sulfonamides, a Still Poorly Investigated Class of CA Inhibitors. Biology 2023, 12, 281. https://doi.org/10.3390/biology12020281
Langella E, Esposito D, Monti SM, Supuran CT, De Simone G, Alterio V. A Combined in Silico and Structural Study Opens New Perspectives on Aliphatic Sulfonamides, a Still Poorly Investigated Class of CA Inhibitors. Biology. 2023; 12(2):281. https://doi.org/10.3390/biology12020281
Chicago/Turabian StyleLangella, Emma, Davide Esposito, Simona Maria Monti, Claudiu T. Supuran, Giuseppina De Simone, and Vincenzo Alterio. 2023. "A Combined in Silico and Structural Study Opens New Perspectives on Aliphatic Sulfonamides, a Still Poorly Investigated Class of CA Inhibitors" Biology 12, no. 2: 281. https://doi.org/10.3390/biology12020281