
Exploring the Role of Immune System and Inflammatory Cytokines in SARS-CoV-2 Induced Lung Disease: A Narrative Review
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
3. Results
3.1. SARS-CoV-2 Infection
3.2. Innate Immune Response to SARS-CoV-2 Infection and COVID-19
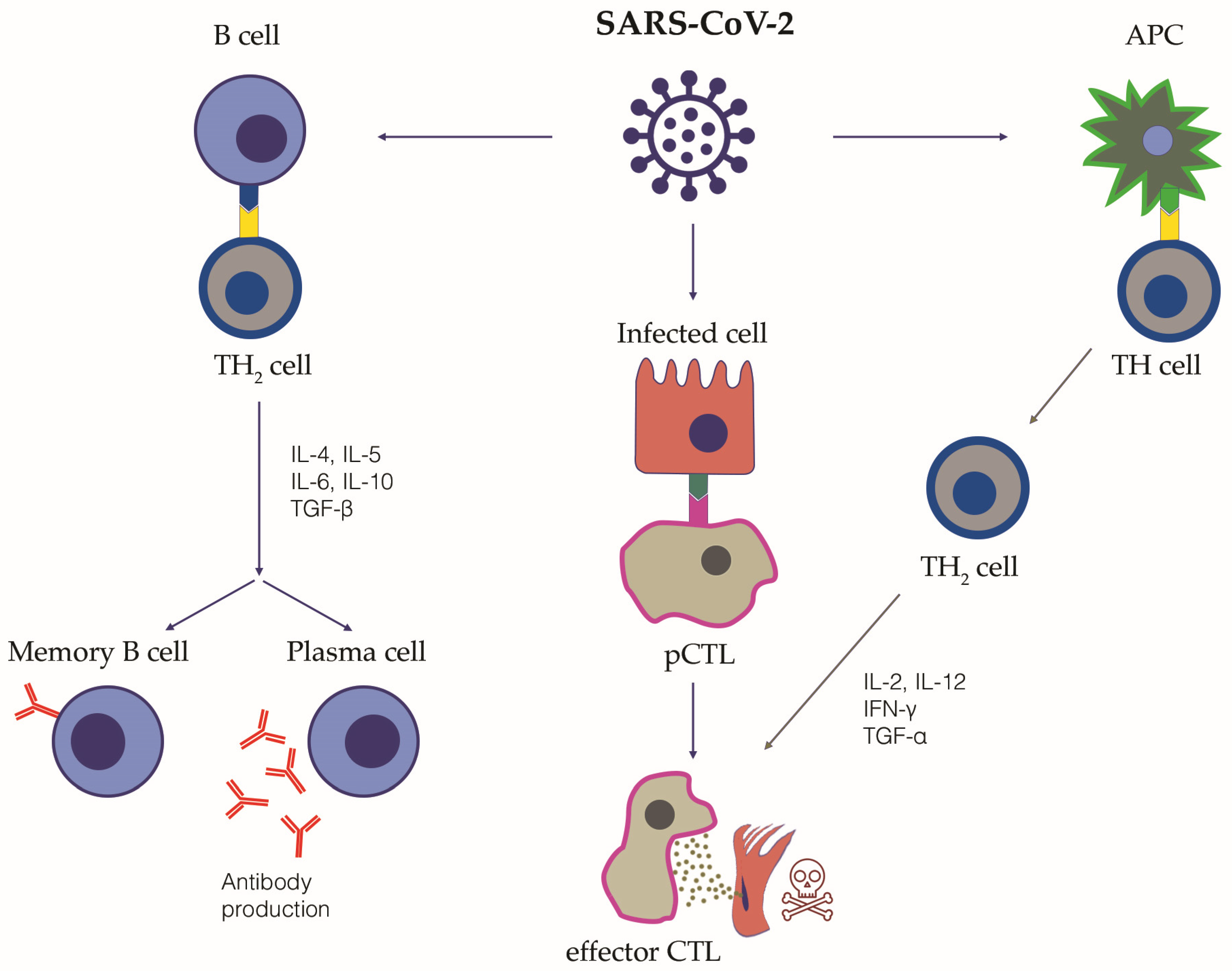
3.3. Adaptive Immune Response to SARS-CoV-2 Infection and COVID-19
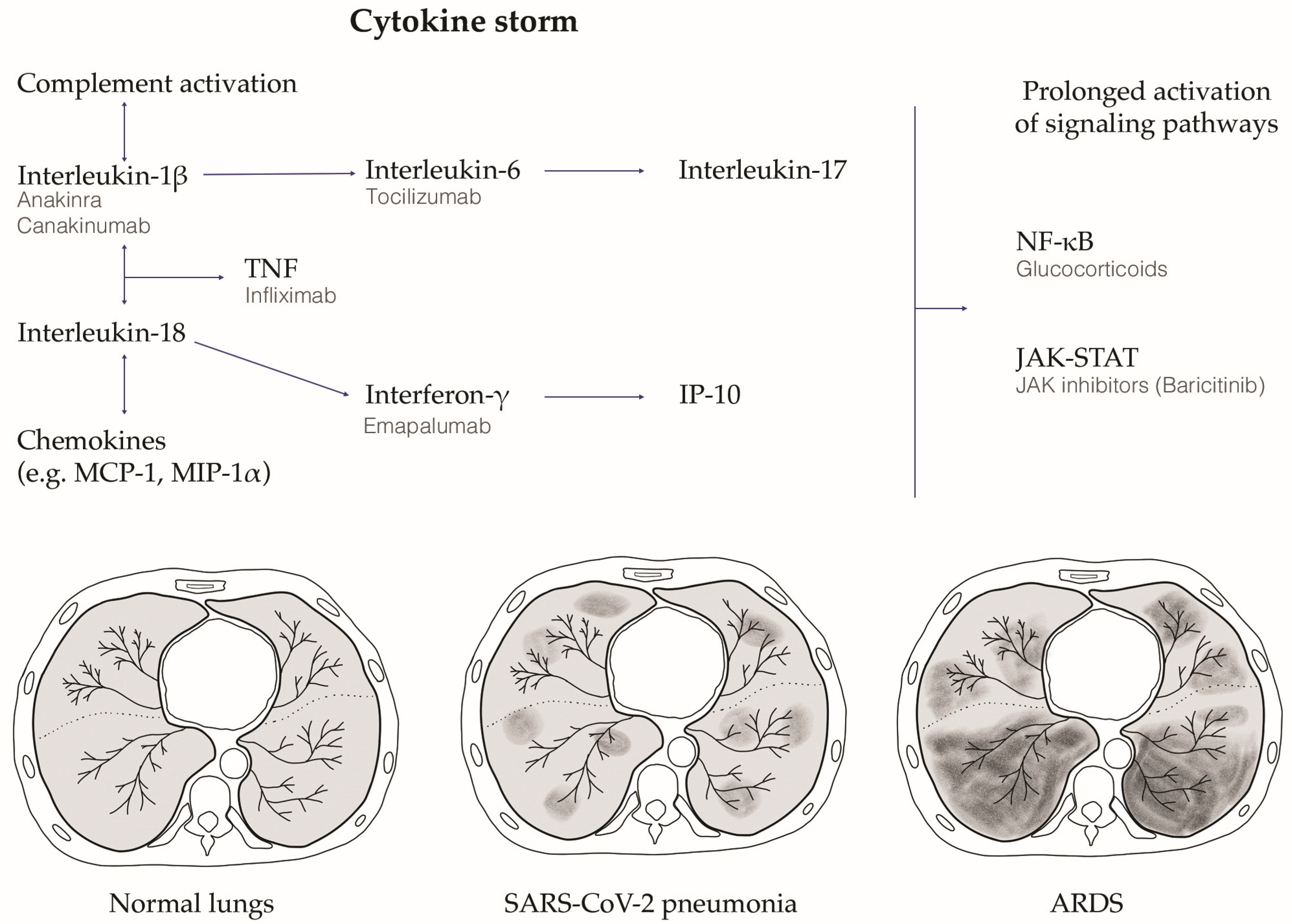
3.4. Cytokine Storm
3.5. Therapy: Targeting the Modulation of Cytokine Storm
3.6. Vaccines
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; von der Thüsen, J.H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: Estimation and application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, E.S.; Kashani, N.R.; Nikzad, H.; Azadbakht, J.; Bafrani, H.H.; Kashani, H.H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.-P. Pulmonary embolism in patients with COVID-19 pneumonia. Eur. Respir. J. 2020, 56, 2001365. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef]
- Heymann, D.L.; Shindo, N.; WHO Scientific and Technical Advisory Group for Infectious Hazards. COVID-19: What is next for public health? Lancet 2020, 395, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Otter, J.A.; Donskey, C.; Yezli, S.; Douthwaite, S.; Goldenberg, S.D.; Weber, D.J. Transmission of SARS and MERS coronaviruses and influenza virus in healthcare settings: The possible role of dry surface contamination. J. Hosp. Infect. 2016, 92, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.; Lau, E.H.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Valsecchi, P.; Colaneri, M.; Zuccaro, V.; Asperges, E.; Costanzo, F.; Mariani, B.; Roda, S.; Minucci, R.; Bertuccio, F.; Fraoilini, E.; et al. Impact of Pneumococcal Urinary Antigen Testing in COVID-19 Patients: Outcomes from the San Matteo COVID-19 Registry (SMACORE). J. Pers. Med. 2021, 11, 762. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boechat, J.L.; Chora, I. The immune response to SARS-CoV-2 and COVID-19 immunopathology—Current perspectives. Pulmonology 2021, 27, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, 423. [Google Scholar]
- Yong, Y.-Y.; Zhang, L.; Hu, Y.-J.; Wu, J.-M.; Yan, L.; Pan, Y.-R.; Tang, Y.; Yu, L.; Law, B.Y.-K.; Yu, C.-L.; et al. Targeting autophagy regulation in NLRP3 inflammasome-mediated lung inflammation in COVID-19. Clin. Immunol. 2022, 244, 109093. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Zhao, N.; Di, B.; Xu, L.L. The NLRP3 inflammasome and COVID 19: Activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. 2021, 61, 2–15. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Gustine, J.N.; Jones, D. Immunopathology of Hyperinflammation in COVID-19. Am. J. Pathol. 2021, 191, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Melazzini, F.; Colaneri, M.; Fumoso, F.; Freddi, G.; Lenti, M.V.; Pieri, T.C.; Piloni, D.; Noris, P.; Pieresca, C.; Preti, P.S.; et al. Venous thromboembolism and COVID19: A single center experience from an academic tertiary referral hospital of Northern Italy. Intern. Emerg. Med. 2021, 16, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Zietz, M.; Zucker, J.; Tatonetti, N.P. Associations between blood type and COVID-19 infection, intubation, and death. Nat. Commun. 2020, 11, 5761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, G.; Zhi, L.; Yang, H.; Zhai, Y.; Dong, X.; Zhang, X.; Gao, X.; Zhu, Y.; He, F. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 2005, 192, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, W.K.E.; Chan, K.H.; Law, H.K.-W.; Tso, G.H.W.; Kong, E.K.P.; Wong, H.S.W.; To, Y.F.; Yung, R.W.H.; Chow, E.Y.; Au, K.L.; et al. Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 2005, 191, 1697–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.-J.; Desai, J.; Mulay, S.R. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef]
- De Biasi, S.; Tartaro, D.L.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Mattioli, M.; Paolini, A.; Gozzi, L.; et al. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur. J. Immunol. 2020, 50, 1283–1294. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; Li, T.; Li, J.; et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Ibarrondo, F.J.; Fulcher, J.A.; Goodman-Meza, D.; Elliott, J.; Hofmann, C.; Hausner, M.A.; Ferbas, K.G.; Tobin, N.H.; Aldrovandi, G.M.; Yang, O.O. Rapid decay of anti-SARS-CoV-2 antibodies in persons with mild COVID-19. N. Engl. J. Med. 2020, 383, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A possible role for B cells in COVID-19? lesson from patients with Agammaglobulinemia. J. Allergy Clin. Immunol. 2020, 146, 211–213. [Google Scholar] [CrossRef]
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Foca, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr. Allergy Immunol. 2020, 31, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Padoan, A.; Cosma, C.; Sciacovelli, L.; Faggian, D.; Plebani, M. Analytical performances of a chemiluminescence immunoassay for SARS-CoV-2 IgM/IgG and antibody kinetics. Clin. Chem. Lab. Med. 2020, 58, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Monsalvo, A.C.; Batalle, J.P.; Lopez, M.F.; Krause, J.C.; Klemenc, J.; Hernandez, J.Z.; Maskin, B.; Bugna, J.; Rubinstein, C.; Aguilar, L.; et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 2011, 17, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158. [Google Scholar] [CrossRef] [Green Version]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants of the SARS-CoV-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Immunological memory to SARS-CoV-2 infection and COVID-19 vaccines. Immunol. Rev. 2022, 310, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Edwards, R.J.; Manne, K.; Martinez, D.R.; Schäfer, A.; Alam, S.M.; Wiehe, K.; Lu, X.; Parks, R.; Sutherland, L.L.; et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell 2021, 184, 4203–4219. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Peteranderl, C.; Herold, S. The Impact of the Interferon/TNF-related apoptosis-inducing ligand signaling axis on disease progression in respiratory viral infection and beyond. Front. Immunol. 2017, 8, 313. [Google Scholar] [CrossRef] [Green Version]
- Helal, M.A.; Shouman, S.; Abdelwaly, A.; Elmehrath, A.O.; Essawy, M.; Sayed, S.M.; Saleh, A.H.; El-Badri, N. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J. Biomol. Struct. Dyn. 2020, 40, 1109–1119. [Google Scholar] [CrossRef]
- Wang, X.; Gui, J. Cell-mediated immunity to SARS-CoV-2. Pediatr. Investig. 2020, 4, 281–291. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Paton, R.S.; Edmans, M.; Youngs, J.; Wellens, J.; Phalora, P.; Fyfe, A.; Belij-Rammerstorfer, S.; Bolton, J.S.; Ball, J.; et al. Fatal COVID-19 outcomes are associted with an antibody response targeting epitops shared with endemic coronaviruses. JCI Insight 2022, 7, e156372. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic changes and SARS-CoV-2 immunostaining in the lung of a patient with COVID-19. Ann. Intern. Med. 2020, 172, 629–632. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- St John, A.L.; Rathore, A.P. Early insights into immune responses during COVID-19. J. Immunol. 2020, 205, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Qiu, Y.; Feng, F.; Feng, J.; Jia, Y.; Zhu, H.; Hu, K.; Liu, J.; Liu, Z.; et al. Clinical characteristics of 82 death cases with COVID-19. PLoS ONE 2020, 15, e0235458. [Google Scholar]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. HCA Lung Biological Network: SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Chuquimia, O.D.; Petursdottir, D.H.; Periolo, N.; Fernández, C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect. Immun. 2013, 81, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate Coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Rochwerg, B.; Siemieniuk, R.A.; Agoritsas, T.; Lamontagne, F.; Askie, L.; Lytvyn, L.; Agarwal, A.; Leo, Y.-S.; Macdonald, H.; Zeng, L.; et al. A living WHO guideline on drugs for COVID-19. Br. Med. J. 2020, 370, m3379. [Google Scholar]
- Salton, F.; Confalonieri, P.; Centanni, S.; Mondoni, M.; Petrosillo, N.; Bonfanti, P.; Lapadula, G.; Lacedonia, D.; Voza, A.; Carpenè, N.; et al. Prolonged higher dose methylprednisolone vs. conventional dexamethasone in COVID-19 pneumonia: A randomised controlled trial (MEDEAS). Eur. Respir. J. 2022, 61, 2201514. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, X.; Liu, X.; Jiang, X. The immunology and immunotherapy for COVID-19. Expert Rev. Mol. Med. 2021, 23, e24. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.M.; Bézie, Y.; Laplanche, S.; et al. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef]
- Kooistra, E.J.; Waalders, N.J.B.; Grondman, I.; Janssen, N.A.F.; de Nooijer, A.H.; Netea, M.G.; van de Veerdonk, F.L.; Ewalds, E.; van der Hoeven, J.G.; Kox, M.; et al. Anakinra treatment in critically ill COVID-19 patients: A prospective cohort study. Crit. Care 2020, 24, 688. [Google Scholar] [CrossRef]
- Katia, F.; Myriam, D.P.; Ucciferri, C.; Auricchio, A.; Di Nicola, M.; Marchioni, M.; Eleonora, C.; Emanuela, S.; Cipollone, F.; Vecchiet, J. Efficacy of canakinumab in mild or severe COVID-19 pneumonia. Immun. Inflamm. Dis. 2021, 9, 399–405. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Assessing the importance of interleukin-6 in COVID-19. Lancet Respir. Med. 2021, 9, e13. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, 30–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Ugel, S.; Tinazzi, E.; Vella, A.; De Sanctis, F.; Canè, S.; Batani, V.; Trovato, R.; Fiore, A.; Petrova, V.; et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Investig. 2020, 130, 6409–6416. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Nannini, C.; Matarrese, D.; Natale, M.E.D.; Lotti, P.; Aquilini, D.; Landini, G.; Cimolato, B.; Pietro, M.A.D.; et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J. Infect. 2020, 81, 647–679. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Monk, P.D.; Marsden, R.J.; Tear, V.J.; Brookes, J.; Batten, T.N.; Mankowski, M.; Gabbay, F.J.; Davies, D.E.; Holgate, S.T.; Ho, L.P.; et al. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021, 9, 196–206. [Google Scholar] [CrossRef]
- Locatelli, F.; Jordan, M.B.; Allen, C.; Cesaro, S.; Rizzari, C.; Rao, A.; Degar, B.; Garrington, T.P.; Sevilla, J.; Putti, M.-C.; et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J. Med. 2020, 382, 1811. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef]
- Wood, E.M.; Estcourt, L.J.; McQuilten, Z.K. How should we use convalescent plasma therapies for the management of COVID-19? Blood 2021, 137, 1573–1581. [Google Scholar] [CrossRef]
- Tabarsi, P.; Barati, S.; Jamaati, H.; Haseli, S.; Marjani, M.; Moniri, A.; Abtahian, Z.; Dastan, A.; Yousefian, S.; Eskandari, R.; et al. Evaluating the effects of Intravenous Immunoglobulin (IVIg) on the management of severe COVID-19 cases: A randomized controlled trial. Int. Immunopharmacol. 2021, 90, 107205. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Wang, J.; Zheng, X. The humoral and cellular immune evasion of SARS-CoV-2 Omicron and sub-lineages. Virol. Sin. 2022, 37, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Kebria, M.M.; Milan, P.B.; Peyravian, N.; Kiani, J.; Khatibi, S.; Mozafari, M. Stem cell therapy for COVID-19 pneumonia. Mol. Biomed. 2022, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Hydroxychloroquine in patients with COVID-19: A Systematic Review and meta-analysis. Diabetes Obes. Metab. 2020, 14, 589–596. [Google Scholar] [CrossRef]
- Batty, C.J.; Heise, M.T.; Bachelder, E.M.; Ainslie, K.M. Vaccine formulations in clinical development for the prevention of severe acute respiratory syndrome coronavirus 2 infection. Adv. Drug Deliv. Rev. 2020, 169, 168–189. [Google Scholar] [CrossRef]
- Fathizadeh, H.; Afshar, S.; Masoudi, M.R.; Gholizadeh, P.; Asgharzadeh, M.; Ganbarov, K.; Köse, Ş.; Yousefi, M.; Kafil, H.S. SARS-CoV-2 (Covid-19) vaccines structure, mechanisms and effectiveness: A review. Int. J. Biol. Macromol. 2021, 188, 740–750. [Google Scholar] [CrossRef]
- Darnell, M.E.; Subbarao, K.; Feinstone, S.M.; Taylor, D.R. Inactivation of the coronavirus that induces severe acute respiratory syndrome, SARS-CoV. J. Virol. Methods 2004, 121, 85–91. [Google Scholar] [CrossRef]
- Kumar, A.; Meldgaard, T.S.; Bertholet, S. Novel platforms for the development of a universal influenza vaccine. Front. Immunol. 2018, 9, 600. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Y.; Gao, G.; Tan, W.; Wu, G.; Xu, M.; Lou, Z. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: A randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect. Dis. 2021, 21, 39–51. [Google Scholar] [CrossRef]
- Benihoud, K.; Yeh, P.; Perricaudet, M. Adenovirus vectors for gene delivery. Curr. Opin. Biotechnol. 1999, 10, 440–447. [Google Scholar] [CrossRef]
- Folegatti, P.; Ewer, K.; Aley, P.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef] [PubMed]
- FZhu, C.; Guan, X.-H.; Li, Y.-H.; Huang, J.-Y.; Jiang, T.; Hou, L.-H.; Li, J.-X.; Yang, B.F.; Wang, L.; Wang, W.-J. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar]
- Dicks, M.; Spencer, A.; Edwards, N.; Wadell, G.; Bojang, K.; Gilbert, S.; Hill, A.V.; Cottingham, M. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS ONE 2012, 7, e40385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisbert, T.; Bailey, M.; Hensley, L.; Asiedu, C.; Geisbert, J.; Stanley, D.; Honko, A.; Johnson, J.; Mulangu, S.; Pau, M. Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J. Virol. 2011, 85, 4222–4233. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.; Walker, S.; Schultheis, K.; Purwar, M. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef]
- Hassett, K.; Benenato, K.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.; Ketova, T. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, M.; Lyke, K.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.; Fuhr, R.; Smolenov, I.; Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.; Smith, M.; Almarsson, H.S. Pujar, mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Anderson, E.; Rouphael, N.; Roberts, P.; Makhene, M.; Coler, R.; McCullough, M.; Chappell, J.; Denison, M.; Stevens, L. An mRNA vaccine against SARS-CoV-2—Preliminary report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef]
- Walsh, E.; Frenck, R.; Falsey, A.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.; Bailey, R. RNA-based COVID-19 Vaccine BNT162b2 Selected for a Pivotal Efficacy Study. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Anderson, E.; Rouphael, N.; Widge, A.; Jackson, L.; Roberts, P.; Makhene, M.; Chappell, J.; Denison, M.; Stevens, L.; Pruijssers, A. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.; Moreira, G.; Mendonça, M. DNA vaccines against COVID-19: Perspectives and challenges. Life Sci. 2020, 267, 118919. [Google Scholar] [CrossRef] [PubMed]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.; Zhu, M.; Cloney-Clark, S.; Zhou, H. Phase 1–2 trial of a SARS-CoV-2 recombinant spike protein nanoparticle vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Jiaming, L.; Yanfeng, Y.; Yao, D.; Yawei, H.; Linlin, B.; Baoying, H.; Jinghua, Y.; Gao, G.F.; Chuan, Q.; Wenjie, T. The recombinant N-terminal domain of spike proteins is a potential vaccine against Middle East respiratory syndrome coronavirus (MERS-CoV) infection. Vaccine 2017, 35, 10–18. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategies Experimented in COVID-19 | ||||
|---|---|---|---|---|
| Therapeutic Class | Molecule | Target | Evidence of Efficacy from Trials | Mechanism of Action |
| Corticosteroids | Dexamethasone, Methylprednisolone, Hydrocortisone | T cells; Macrophages | High | * Synthesis inhibition of Th1 and macrophage pro-inflammatory cytokines (IL-1β, IL-2, IL-6, TNF-α, and IL-17) * Gene transcription and protein synthesis of NF-kB and lipocortin-1 inhibitors |
| Inhibitors of IL-1 signaling | Anakinra | IL-1 receptor | Moderate | * Reduction of the cascade release of proinflammatory cytokines (TNF, IL-6, IL-8); * Reduction of fever by modulation of thermoregulatory center in the brain |
| Canakinumab | IL-1β | Mild | * Reduction of the cascade release of proinflammatory cytokines (TNF, IL-6, IL-8); * Reduction of fever by modulation of thermoregulatory center in the brain | |
| Inhibitors of IL-6 signaling | Tocilizumab | IL-6 receptor | Moderate/High | * Reduction of macrophage activation; * Reduction of endothelial leakage and liquid extravasation; * Reduction of acute phase protein synthesis and pyrogens activation. |
| Tyrosine kinase inhibitors | Baricitinib | JAK 1 and JAK 2 | Moderate/High | * Inhibition of the enzymatic activity of JAK1 and JAK2; * Reduction of the phosphorylation and activation of STATs; * Interference with viral endocytosis and intracellular transport through ACE2; * Suppression of cytokine storm |
| IFN-based therapy | Recombinant IFN α and β | Type 1 IFN receptor | Mild/Moderate | * Inhibition of viral replication; * Activation of the cell-mediated response against the virus |
| Emapalumab | IFN-γ | Low | * Stimulation of the innate response against viral infection | |
| TNF-α inhibitors | Etanercept | TNFα receptor | Very Low | * Suppression of cytokine storm |
| Infliximab and Adalimumab | TNFα | Very Low | * Suppression of cytokine storm | |
| Convalescent plasma | Neutralizing antibodies | SARS-CoV-2 | Moderate/Low | * Passive humoral response |
| Intravenous Immunoglobulins | Anti-cytokine autoantibodies (i.e. against IL-1, IL-6, and IFN-γ) | IL-1, IL-6, IFN-γ | Moderate/Low | * Anti-inflammatory and immunomodulatory effects |
| Monoclonal antibodies targeting the spike protein | Tixagevimab, casirivimab, sotrovimab | Spike protein of SARS-CoV-2 | Moderate | * Prevention of viral entry in the host cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirelli, C.; De Amici, M.; Albrici, C.; Mira, S.; Nalesso, G.; Re, B.; Corsico, A.G.; Mondoni, M.; Centanni, S. Exploring the Role of Immune System and Inflammatory Cytokines in SARS-CoV-2 Induced Lung Disease: A Narrative Review. Biology 2023, 12, 177. https://doi.org/10.3390/biology12020177
Tirelli C, De Amici M, Albrici C, Mira S, Nalesso G, Re B, Corsico AG, Mondoni M, Centanni S. Exploring the Role of Immune System and Inflammatory Cytokines in SARS-CoV-2 Induced Lung Disease: A Narrative Review. Biology. 2023; 12(2):177. https://doi.org/10.3390/biology12020177
Chicago/Turabian StyleTirelli, Claudio, Mara De Amici, Cristina Albrici, Sabrina Mira, Giulia Nalesso, Beatrice Re, Angelo Guido Corsico, Michele Mondoni, and Stefano Centanni. 2023. "Exploring the Role of Immune System and Inflammatory Cytokines in SARS-CoV-2 Induced Lung Disease: A Narrative Review" Biology 12, no. 2: 177. https://doi.org/10.3390/biology12020177