
microRNA-mRNA Analysis Reveals Tissue-Specific Regulation of microRNA in Mangrove Clam (Geloina erosa)


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Preparation of Animal Material
2.2. miRNA Extraction and Quality Control
2.3. Library Preparation, Library Quality Control, and Sequencing
2.4. Bioinformatics Analysis
2.4.1. Overview of Bioinformatics Analysis
2.4.2. Quality Control of Sequencing Data
2.4.3. Identification of Novel miRNAs and Homologous miRNAs
2.4.4. miRNA Target Gene Prediction
2.4.5. miRNA Expression Quantification
2.4.6. Differential Expression Analysis
2.4.7. Functional Annotation of Predicted Targets
2.4.8. Gene Set Enrichment Analysis
2.4.9. miRNA Co-Expression Analysis
- (1)
- A functional enrichment analysis was employed to determine whether the functional characteristics and traits identified within each module aligning with the specific aims of the research. This analysis helped gain insights into the biological processes and pathways that are potentially associated with the study objectives.
- (2)
- A correlation analysis was performed to identify the module that exhibited the highest correlation with the traits of interest. By exploring the relationship between miRNA expression patterns within modules and specific traits, the aim was to uncover modules that potentially play crucial roles in the biological processes underlying these traits.
2.4.10. Real-Time Fluorescent Quantitative PCR Validation of miRNA Expression in Tissues
3. Results
3.1. Sequencing Results and Quality Statistics
3.2. miRNA Identification
3.3. miRNA Base Preference Analysis
3.4. miRNA Target Gene Prediction
3.5. miRNA Expression Quantification
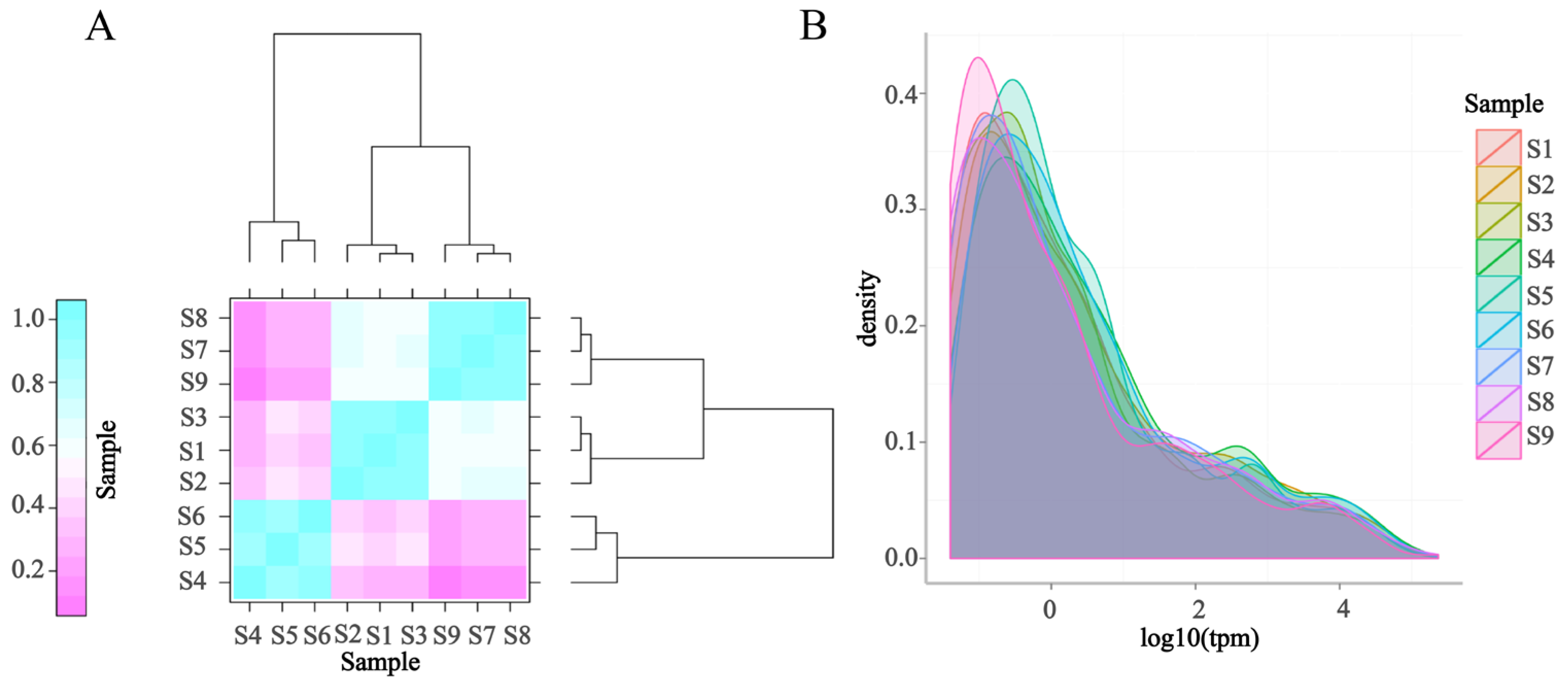
3.5.1. Distribution of Overall Expression in the Sample
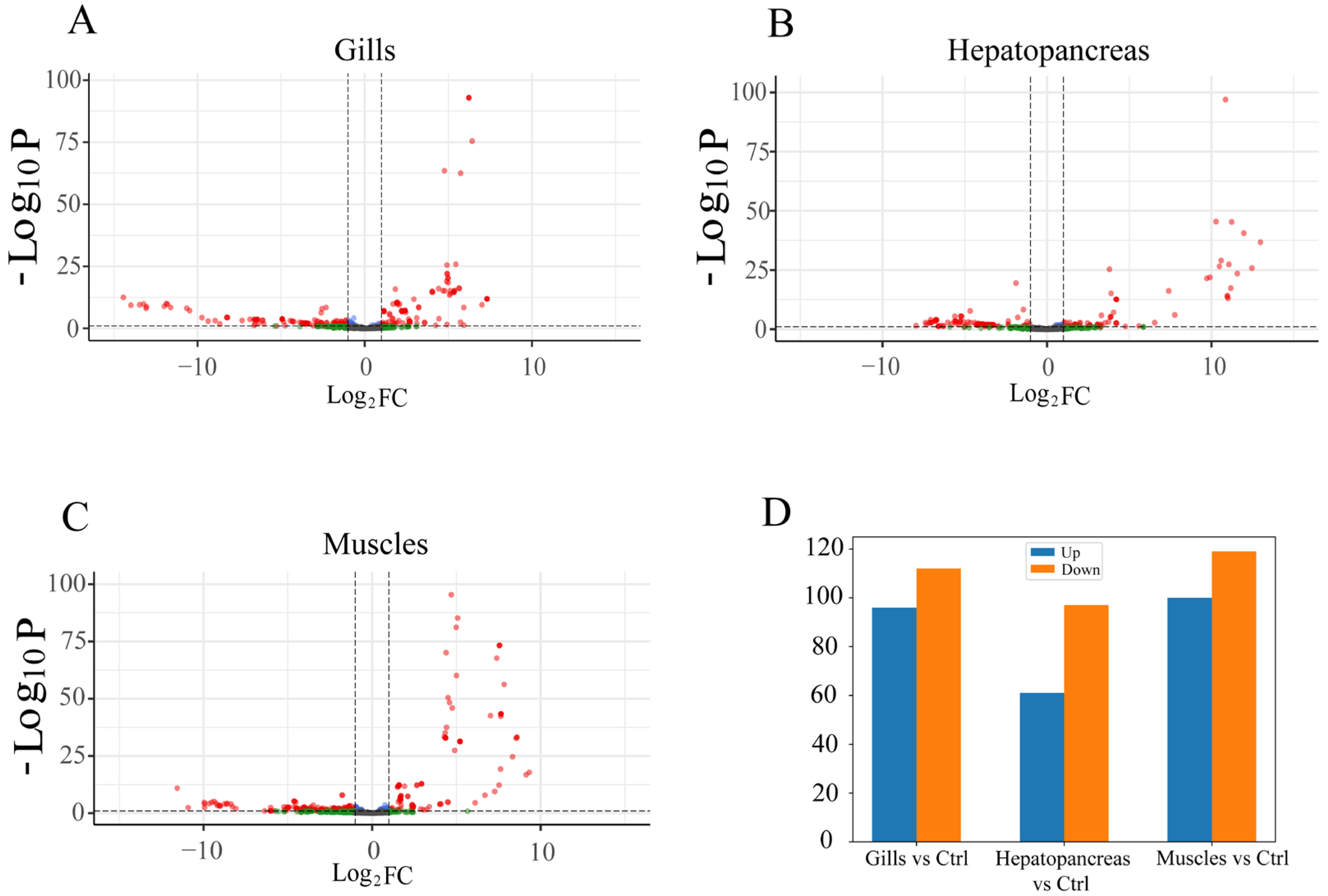
3.5.2. Differential Expression Analysis
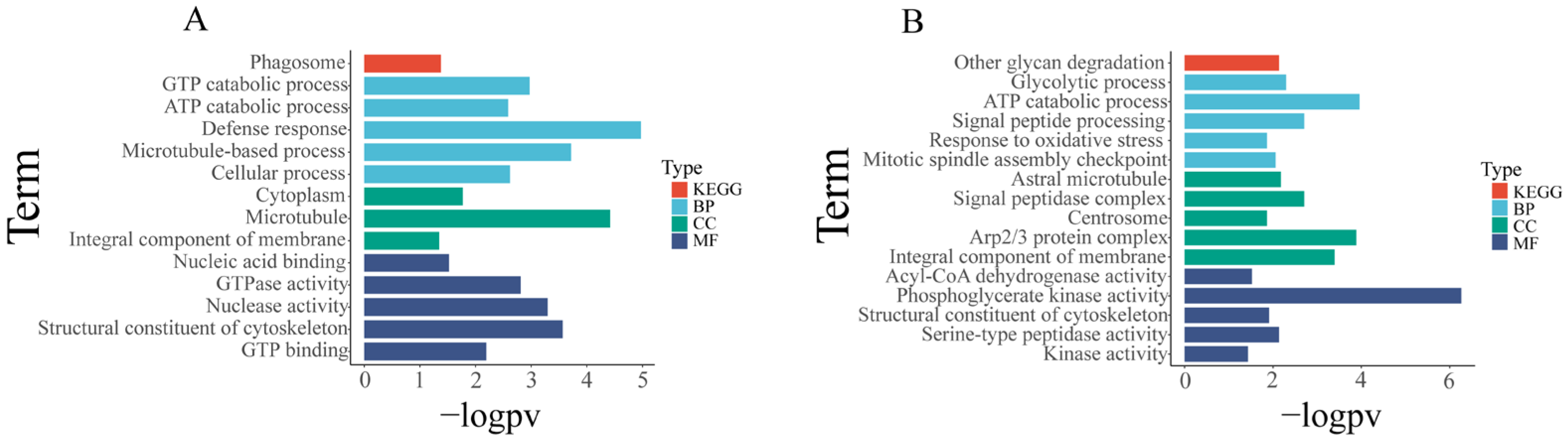
3.5.3. Functional Analysis of Differentially Expressed miRNA Predicted Targets
3.5.4. miRNA Co-Expression Analysis
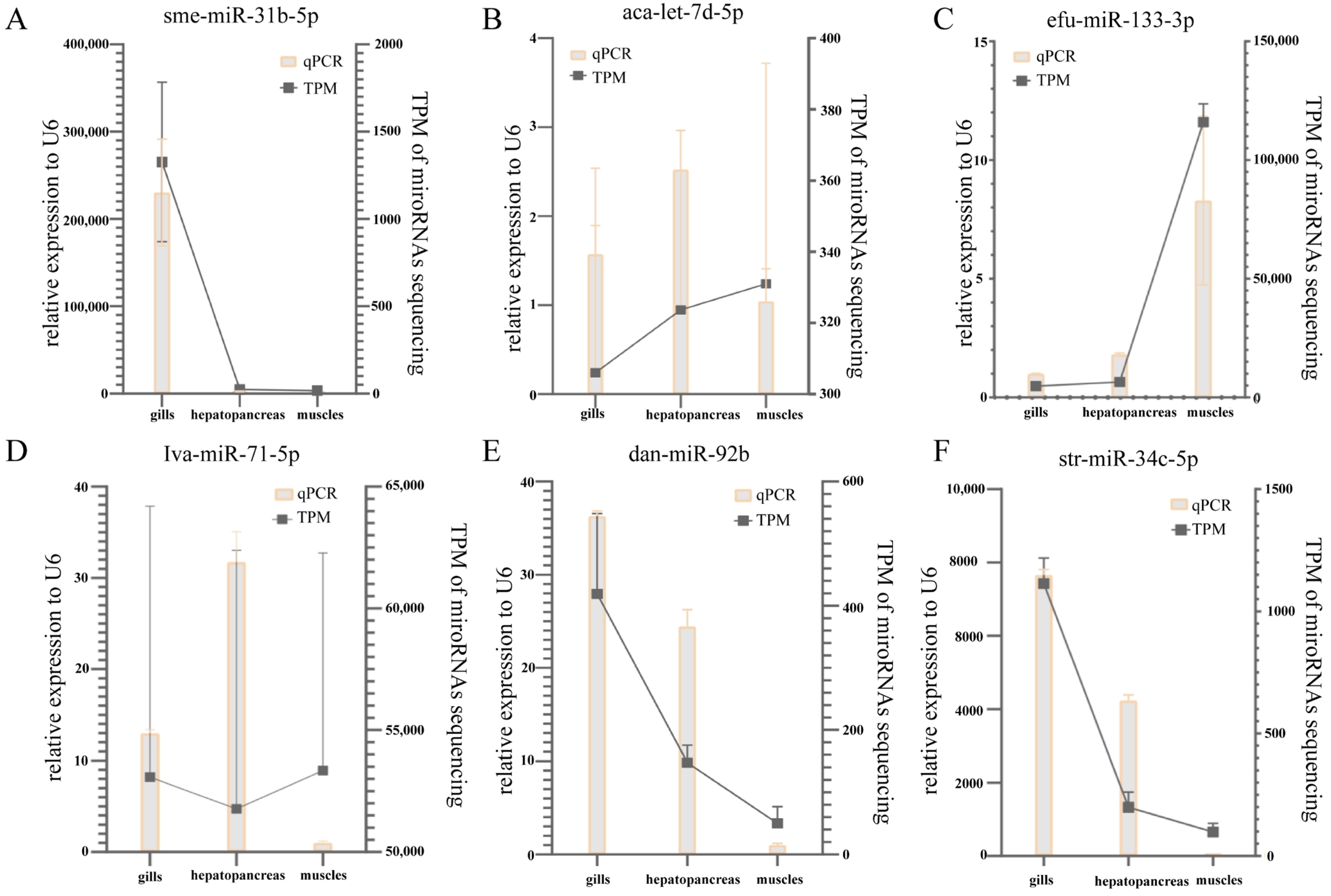
3.5.5. qRT-PCR and RNA-seq Consistency for Selected miRNAs
4. Discussion
4.1. Gill-Specific miRNA and Target Gene Functions
4.2. Hepatopancreatic-Specific miRNA and Target Gene Functions
4.3. Muscle-Specific miRNA and Target Gene Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rhee, S.; Chae, H.; Kim, S. PlantMirnaT: MiRNA and MRNA Integrated Analysis Fully Utilizing Characteristics of Plant Sequencing Data. Methods 2015, 83, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Qiu, S.B.; Nisbet, A.J.; Fu, J.H.; Shao, C.C.; Zhu, X.Q. Global Characterization of MicroRNAs in Trichomonas Gallinae. Parasit. Vectors 2014, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.W.; MacRae, I.J. The Molecular Machines That Mediate MicroRNA Maturation. J. Cell Mol. Med. 2009, 13, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.J.; Namigai, E.K.O.; Marlétaz, F.; Hui, J.H.L.; Shimeld, S.M. Draft Genome Assemblies and Predicted MicroRNA Complements of the Intertidal Lophotrochozoans Patella Vulgata (Mollusca, Patellogastropoda) and Spirobranchus (Pomatoceros) Lamarcki (Annelida, Serpulida). Mar. Genom. 2015, 24, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Abbadi, M.; Green, T.; Bai, C.M.; Turolla, E.; Arcangeli, G.; Wegner, K.M.; Venier, P. Parallel Analysis of MiRNAs and MRNAs Suggests Distinct Regulatory Networks in Crassostrea Gigas Infected by Ostreid Herpesvirus 1. BMC Genom. 2020, 21, 620. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Sun, L.; Shi, H.; Cheng, Y.; Jiang, D.; Fu, B.; Conte, M.A.; Gammerdinger, W.J.; Kocher, T.D.; Wang, D. Integrated Analysis of MiRNA and MRNA Expression Profiles in Tilapia Gonads at an Early Stage of Sex Differentiation. BMC Genom. 2016, 17, 328. [Google Scholar] [CrossRef]
- Wu, L.M.; Bai, Z.Y.; Liu, X.J.; Jin, C.; Yin, H.; Li, J.L. Comparative Analysis of Shell Color Variety and Genetic Structure among Five High-Quality Freshwater Pearl Mussel Populations. Genet. Mol. Res. 2016, 15, gmr15048390. [Google Scholar] [CrossRef]
- Real, F.M.; Sekido, R.; Lupiánẽz, D.G.; Lovell-Badge, R.; Jiménez, R.; Burgos, M. A MicroRNA (Mmu-MiR-124) Prevents Sox9 Expression in Developing Mouse Ovarian Cells. Biol. Reprod. 2013, 89, 78. [Google Scholar] [CrossRef]
- Chantler, P.D. Scallop Adductor Muscles: Structure and Function. In Developments in Aquaculture and Fisheries Science; Elsevier: Amsterdam, The Netherlands, 2016; Volume 40. [Google Scholar]
- Sugatani, T.; Hruska, K.A. MicroRNA-223 Is a Key Factor in Osteoclast Differentiation. J. Cell Biochem. 2007, 101, 996–999. [Google Scholar] [CrossRef]
- Kang, I.H.; Jeong, B.C.; Hur, S.W.; Choi, H.; Choi, S.H.; Ryu, J.H.; Hwang, Y.C.; Koh, J.T. MicroRNA-302a Stimulates Osteoblastic Differentiation by Repressing COUP-TFII Expression. J. Cell Physiol. 2015, 230, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Chen, C.; Chen, P.; Xie, H.; Luo, X. MicroRNAs and Their Roles in Osteoclast Differentiation. Front. Med. China 2011, 5, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Abo-Al-Ela, H.G.; Faggio, C. MicroRNA-Mediated Stress Response in Bivalve Species. Ecotoxicol. Environ. Saf. 2021, 208, 111442. [Google Scholar] [CrossRef] [PubMed]
- Morton, B. The Biology and Functional Morphology of the Southeast Asian Mangrove Bivalve, Polymesoda (Geloina) Erosa (Solander, 1786) (Bivalvia: Corbiculidae). Can. J. Zool. 1976, 54, 482–500. [Google Scholar] [CrossRef]
- Cai, Y.; Huang, X.; Wu, D. Studies on the Ecology of Polymesoda Erosa (Solander). Trop. Oceanol. 1995, 14, 94–98. [Google Scholar]
- Ingole, B.S.; Naik, S.; Furtado, R.; Ansari, Z.A.; Chatterji, A. Population Characteristics of the Mangrove Clam Polymesoda (Geloina) Erosa (Solander, 1786) in the Chorao Mangrove, Goa. Indian Council of Agricultural Research; Old Goa; India. 2002. Available online: https://drs.nio.res.in/drs/handle/2264/1410 (accessed on 20 February 2023).
- Clemente, S.; Ingole, B. Recruitment of Mud Clam Polymesoda Erosa (Solander, 1876) in a Mangrove Habitat of Chorao Island, Goa. Braz. J. Oceanogr. 2011, 59, 153–162. [Google Scholar] [CrossRef]
- Ma, B.; Ran, Z.; Xu, X.; Xu, J.; Liao, K.; Cao, J.; Yan, X. Comparative Transcriptome Analyses Provide Insights into the Adaptation Mechanisms to Acute Salt Stresses in Juvenile Sinonovacula Constricta. Genes. Genom. 2019, 41, 599–612. [Google Scholar] [CrossRef]
- Zhang, T.; Yan, Z.; Zheng, X.; Wang, S.; Fan, J.; Liu, Z. Effects of Acute Ammonia Toxicity on Oxidative Stress, DNA Damage and Apoptosis in Digestive Gland and Gill of Asian Clam (Corbicula Fluminea). Fish. Shellfish. Immunol. 2020, 99, 514–525. [Google Scholar] [CrossRef]
- Cappello, T.; Giannetto, A.; Parrino, V.; Maisano, M.; Oliva, S.; De Marco, G.; Guerriero, G.; Mauceri, A.; Fasulo, S. Baseline Levels of Metabolites in Different Tissues of Mussel Mytilus Galloprovincialis (Bivalvia: Mytilidae). Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 26, 32–39. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. II. Error Probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef]
- Friedländer, M.R.; MacKowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. MiRDeep2 Accurately Identifies Known and Hundreds of Novel MicroRNA Genes in Seven Animal Clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.; Chen, M. MTide: An Integrated Tool for the Identification of MiRNA-Target Interaction in Plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The MicroRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and Effective Prediction of MicroRNA/Target Duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-Throughput Sequencing of Arabidopsis MicroRNAs: Evidence for Frequent Birth and Death of MIRNA Genes. PLoS ONE 2007, 2, e0000219. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating Some Redundancy Significantly Speeds up Clustering of Large Protein Databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG Database: A Tool for Genome-Scale Analysis of Protein Functions and Evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Database and Its Supplement TrEMBL in 2000. Nucleic Acids Res 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.; Hu, X.; Xun, X.; Zhang, J.; Guo, X.; Jiao, W.; Zhang, L.; Liu, W.; Wang, J.; et al. Scallop Genome Reveals Molecular Adaptations to Semi-Sessile Life and Neurotoxins. Nat. Commun. 2017, 8, 1721. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef]
- Tsai, W.C.; Da Hsu, S.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 Plays a Critical Role in Liver Homeostasis and Hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; MacDonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A Pancreatic Islet-Specific MicroRNA Regulates Insulin Secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Christgen, S.L.; Becker, D.F. Role of Proline in Pathogen and Host Interactions. Antioxid. Redox Signal. 2019, 30, 683–709. [Google Scholar] [CrossRef]
- Williams, M.; Baxter, R. The Structure and Function of Thioester-Containing Proteins in Arthropods. Biophys. Rev. 2014, 6, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Duval, D.; Pichon, R.; Lassalle, D.; Laffitte, M.; Gourbal, B.; Galinier, R. A New Assessment of Thioester-Containing Proteins Diversity of the Freshwater Snail Biomphalaria Glabrata. Genes 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, A.A.; Andreenko, T.I.; Sysoeva, I.V.; Sysoev, A.A. Tissue Specificity of Metabolism in the Bivalve Mollusc Anadara Inaequivalvis Br. under Conditions of Experimental Anoxia. J. Evol. Biochem. Physiol. 2009, 45, 349–355. [Google Scholar] [CrossRef]
- Hosoi, M.; Yoshinaga, Y.; Toyohara, M.; Shiota, F.; Toyohara, H. Freshwater Bivalve Corbicula Sandai Uses Free Amino Acids as Osmolytes under Hyperosmotic Condition. Fish. Sci. 2008, 74, 1339–1341. [Google Scholar] [CrossRef]
- Ellington, W.R. The Recovery from Anaerobic Metabolism in Invertebrates. J. Exp. Zool. 1983, 228, 431–444. [Google Scholar] [CrossRef]
- Yu, J.; Wang, H.; Liu, B. Changes in Gluconeogenesis Pathways and Key Genes Associated with Mass Mortality in the Clam Meretrix Petechialis upon Vibrio Infection. Aquaculture 2022, 548, 737691. [Google Scholar] [CrossRef]
- Ma, L.; Su, L.; Liu, H.; Zhao, F.; Zhou, D.; Duan, D. Norovirus Contamination and the Glycosphingolipid Biosynthesis Pathway in Pacific Oyster: A Transcriptomics Study. Fish. Shellfish. Immunol. 2017, 66, 26–34. [Google Scholar] [CrossRef]
- Zhao, X.; Duan, X.; Wang, Z.; Zhang, W.; Li, Y.; Jin, C.; Xiong, J.; Li, C. Comparative Transcriptome Analysis of Sinonovacula Constricta in Gills and Hepatopancreas in Response to Vibrio Parahaemolyticus Infection. Fish. Shellfish. Immunol. 2017, 67, 523–535. [Google Scholar] [CrossRef]
- Gomes, A.M.; Kozlowski, E.O.; Pomin, V.H.; De Barros, C.M.; Zaganeli, J.L.; Pavão, M.S.G. Unique Extracellular Matrix Heparan Sulfate from the Bivalve Nodipecten Nodosus (Linnaeus, 1758) Safely Inhibits Arterial Thrombosis after Photochemically Induced Endothelial Lesion. J. Biol. Chem. 2010, 285, 7312–7323. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, H.; Kong, L.; Liu, S.; Li, Q. High Throughput Sequencing of Small RNAs Transcriptomes in Two Crassostrea Oysters Identifies MicroRNAs Involved in Osmotic Stress Response. Sci. Rep. 2016, 6, 22687. [Google Scholar] [CrossRef]
- Ventoso, P.; Pazos, A.J.; Blanco, J.; Pérez-Parallé, M.L.; Triviño, J.C.; Sánchez, J.L. Transcriptional Response in the Digestive Gland of the King Scallop (Pecten Maximus) after the Injection of Domoic Acid. Toxins 2021, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Kirsikka Sillanpää, J.; Sundh, H.; Sundell, K.S. Calcium Transfer across the Outer Mantle Epithelium in the Pacific Oyster, Crassostrea Gigas. Proc. R. Soc. B Biol. Sci. 2018, 285, 20181676. [Google Scholar] [CrossRef]
- Martins, J.C.; Campos, A.; Osório, H.; Da Fonseca, R.; Vasconcelos, V. Proteomic Profiling of Cytosolic Glutathione Transferases from Three Bivalve Species: Corbicula Fluminea, Mytilus Galloprovincialis and Anodonta Cygnea. Int. J. Mol. Sci. 2014, 15, 1887. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Liu, Y.; Han, T.; Yang, M.; Liu, W.; Wang, Y.; He, C.; Lu, Z. Full-Length Transcriptome Sequencing Reveals Tissue-Specific Gene Expression Profile of Mangrove Clam Geloina Erosa. Front. Physiol. 2022, 13, 851957. [Google Scholar] [CrossRef]
- Xu, R.; Pan, L.; Yang, Y.; Zhou, Y. Characterizing Transcriptome in Female Scallop Chlamys Farreri Provides New Insights into the Molecular Mechanisms of Reproductive Regulation during Ovarian Development and Spawn. Gene 2020, 758, 144967. [Google Scholar] [CrossRef]
- Chen, C.; Xie, B.; Sun, W.; Gu, Z.; Huang, J.; Qi, P.; Liao, Z.; Zhang, X.; Yan, X. Examination of the Role of Resveratrol in Attenuating Oxidative Damage Induced by Starvation Stress in the Marine Mussel, Mytilus Coruscus, through Regulation of the Sirt1-Mediated Signaling Pathway. Aquaculture 2023, 564, 739047. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Live Weight (g) | Shell Weight (g) | Shell Length (mm) | Shell Height (mm) | Shell Width (mm) | Experimental Usage |
|---|---|---|---|---|---|
| 57.80 | 25.50 | 54.05 | 50.50 | 34.71 | Sequencing |
| 49.20 | 23.74 | 51.94 | 49.46 | 30.30 | Sequencing |
| 47.40 | 20.22 | 52.11 | 48.57 | 31.16 | Sequencing |
| 38.80 | 17.00 | 50.71 | 47.53 | 27.49 | qRT-PCR |
| 49.50 | 21.61 | 54.95 | 51.76 | 30.35 | qRT-PCR |
| 47.00 | 22.41 | 52.43 | 48.45 | 30.21 | qRT-PCR |
| miRNA | Sequence | Forward Primer 5′—3′ |
|---|---|---|
| str-miR-34c-5p | TGGCAGTGTGATTAGCTGGTTG | GCAGTGTGATTAGCTGGTTGAAA |
| sme-miR-31b-5p | AGGCAAGATGCTGGCATAGCTGA | CAAGATGCTGGCATAGCTGAA |
| dan-miR-92b | AATTGCACTAGTCCCGGCCTGC | AATTGCACTAGTCCCGGCC |
| efu-miR-133-3p | TTTGGTCCCCTTCAACCAGCTGTA | GTCCCCTTCAACCAGCTGTAA |
| lva-miR-71-5p | TGAAAGACATGGGTAGTGAGATT | CGGAAAGACATGGGTAGTGAGAT |
| aca-let-7d-5p | AGAGGTAGTAGGTTGCATAGT | AGAGGTAGTAGGTTGCATAGTAA |
| Sample Id | Clean Reads | Q30 (%) | Mapped Reads |
|---|---|---|---|
| S1 | 33,105,265 | 98.04 | 386,457 |
| S2 | 24,612,221 | 98.52 | 138,391 |
| S3 | 33,568,798 | 98.65 | 387,129 |
| S4 | 20,020,543 | 97.71 | 327,220 |
| S5 | 22,796,513 | 97.76 | 330,568 |
| S6 | 24,537,870 | 97.79 | 480,804 |
| S7 | 30,010,373 | 97.84 | 208,711 |
| S8 | 33,494,607 | 97.92 | 239,902 |
| S9 | 32,610,912 | 97.81 | 327,720 |
| Sample Id | Known-miRNAs | Novel-miRNAs | Total |
|---|---|---|---|
| S1 | 796 | 295 | 1091 |
| S2 | 667 | 254 | 921 |
| S3 | 871 | 316 | 1187 |
| S4 | 540 | 286 | 826 |
| S5 | 803 | 283 | 1086 |
| S6 | 593 | 314 | 907 |
| S7 | 602 | 239 | 841 |
| S8 | 610 | 248 | 858 |
| S9 | 648 | 266 | 914 |
| Total | 1047 | 365 | 1412 |
| Types | All miRNA | miRNA with Target | Target Gene |
|---|---|---|---|
| Known miRNA | 1047 | 700 | 5082 |
| Novel miRNA | 365 | 334 | 3223 |
| Total | 1412 | 1034 | 7404 |
| Sample Id | All miRNA | miRNA with Target | Target Gene |
|---|---|---|---|
| S1 | 1091 | 816 | 5694 |
| S2 | 921 | 701 | 4874 |
| S3 | 1187 | 879 | 6623 |
| S4 | 826 | 638 | 4354 |
| S5 | 1086 | 809 | 5818 |
| S6 | 907 | 698 | 4767 |
| S7 | 841 | 636 | 4218 |
| S8 | 858 | 661 | 4372 |
| S9 | 914 | 698 | 4784 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Dong, Z.; Chen, K.; Yang, M.; Shi, N.; Liao, X. microRNA-mRNA Analysis Reveals Tissue-Specific Regulation of microRNA in Mangrove Clam (Geloina erosa). Biology 2023, 12, 1510. https://doi.org/10.3390/biology12121510
Liu Y, Dong Z, Chen K, Yang M, Shi N, Liao X. microRNA-mRNA Analysis Reveals Tissue-Specific Regulation of microRNA in Mangrove Clam (Geloina erosa). Biology. 2023; 12(12):1510. https://doi.org/10.3390/biology12121510
Chicago/Turabian StyleLiu, Yunqing, Ziheng Dong, Kun Chen, Mingliu Yang, Nianfeng Shi, and Xin Liao. 2023. "microRNA-mRNA Analysis Reveals Tissue-Specific Regulation of microRNA in Mangrove Clam (Geloina erosa)" Biology 12, no. 12: 1510. https://doi.org/10.3390/biology12121510