

An Exploration of Small Molecules That Bind Human Single-Stranded DNA Binding Protein 1
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
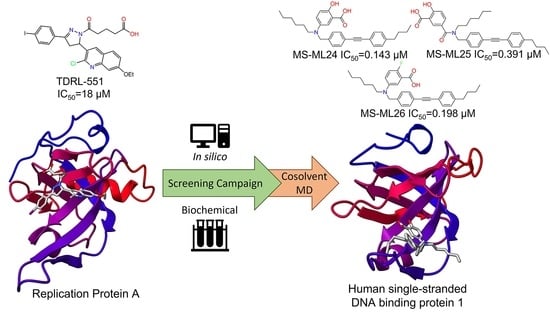
1. Introduction

2. Methodology
2.1. Structure-Based Design of Compounds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | R3 |
|---|---|---|
 |  |  |
 |  |  |
 |  |  |
 |  |  |
2.2. Compound Synthesis
2.2.1. Synthesis of TDRL-551
2.2.2. Synthesis of Intermediate

2.2.3. Synthesis of DAZLN-51

2.2.4. Synthesis of DAZLN-55

2.2.5. Synthesis of DAZLN-56

2.3. Expression and Purification of Recombinant Wild-Type hSSB1
2.4. AlphaLISA Assay Design
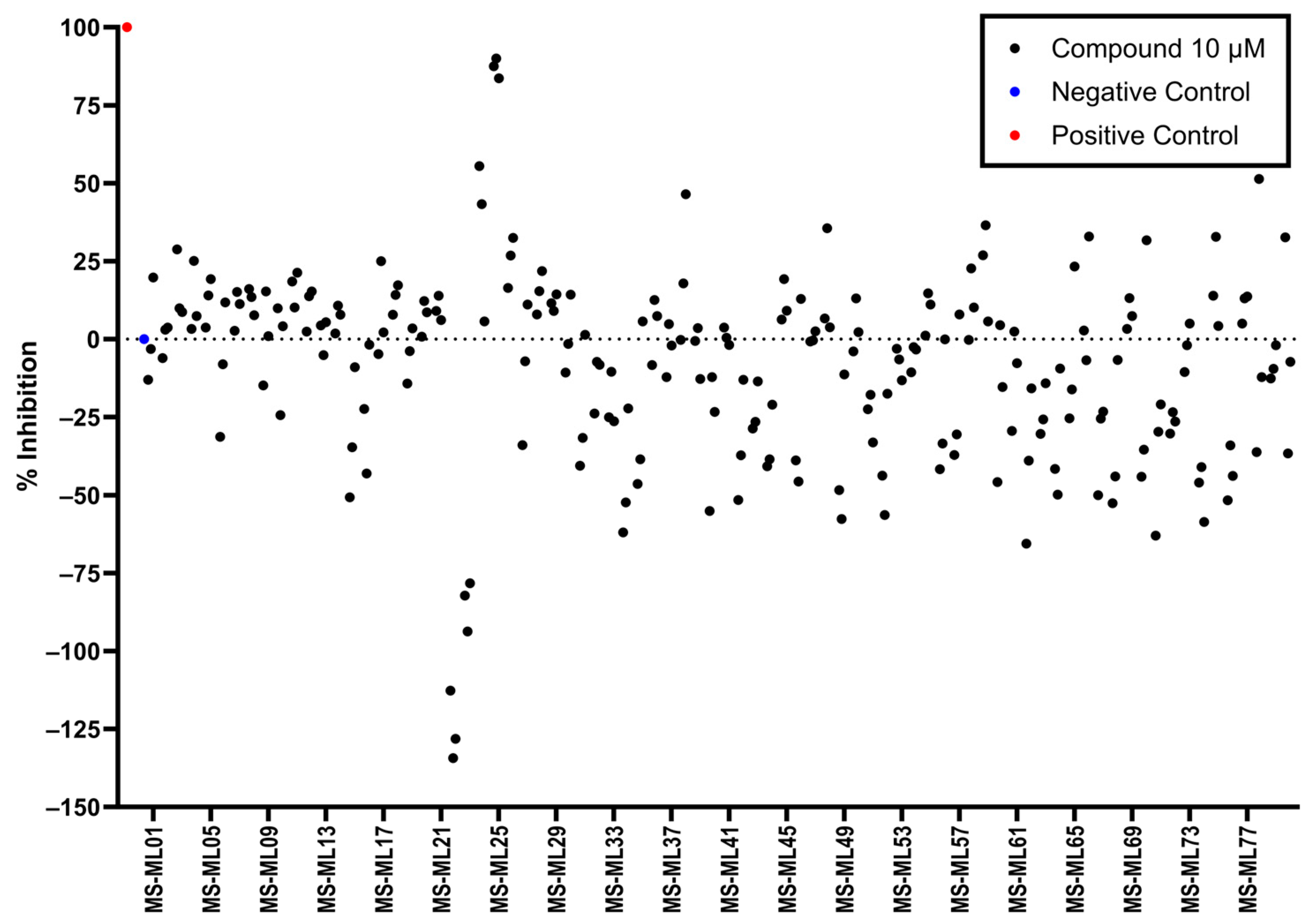
2.5. Mini Library Compound Library Screen
2.6. AlphaLISA Dose–Response Curves
2.7. TruHits Counter Assay
2.8. Electrophoretic Mobility Shift Assays
2.9. Cell Culture and Cytotoxicity Assay
2.10. Cosolvent Molecular Dynamics Simulations
3. Results
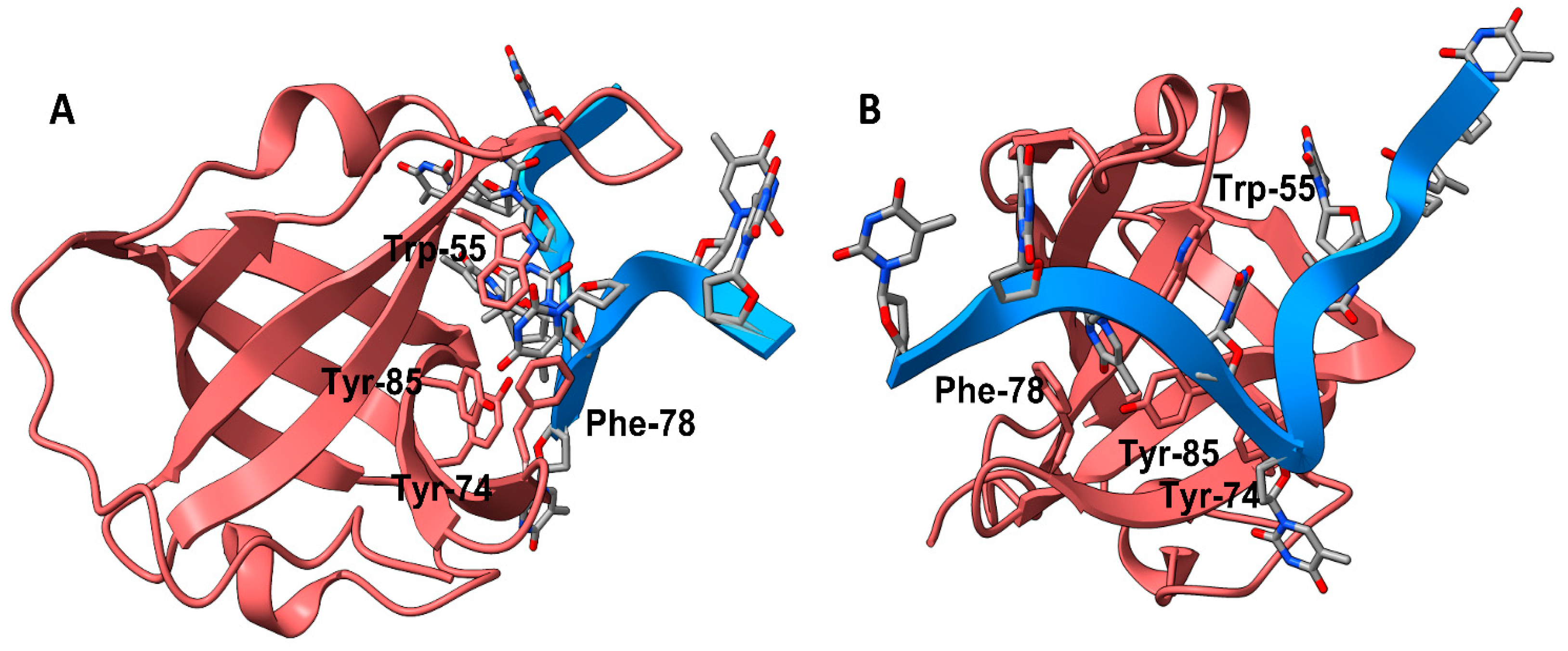
3.1. Recognition of Small Molecule-hSSB1 Binding Site
3.2. Combinatorial Virtual Library Screen
3.3. Synthesis of Compounds
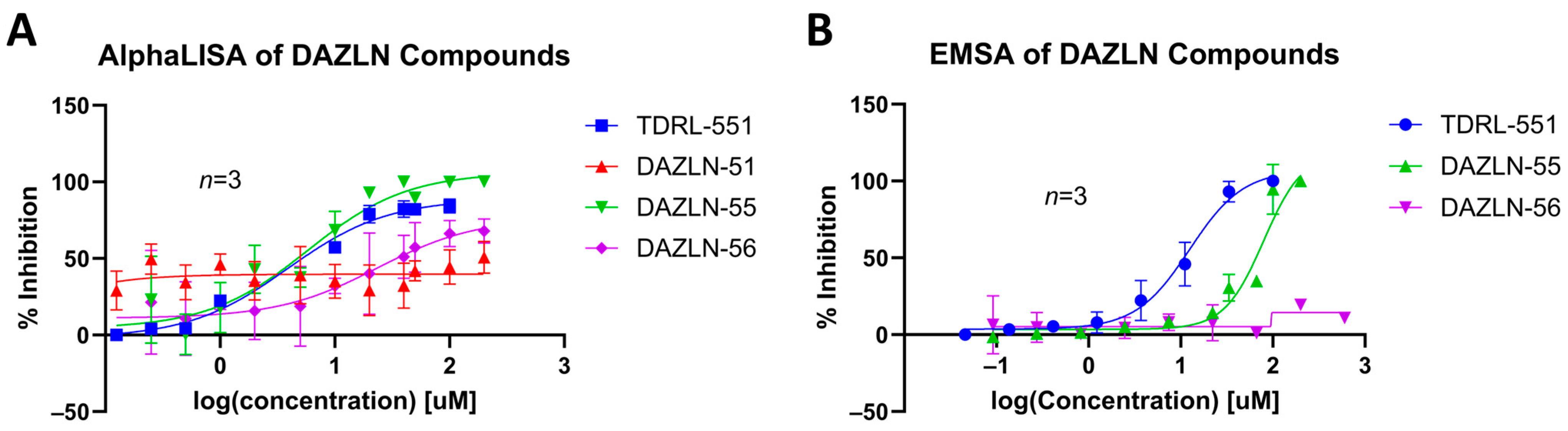
3.4. Dose–Response of DAZLN Compounds and TDRL-551
3.5. AlphaLISA Library Screen
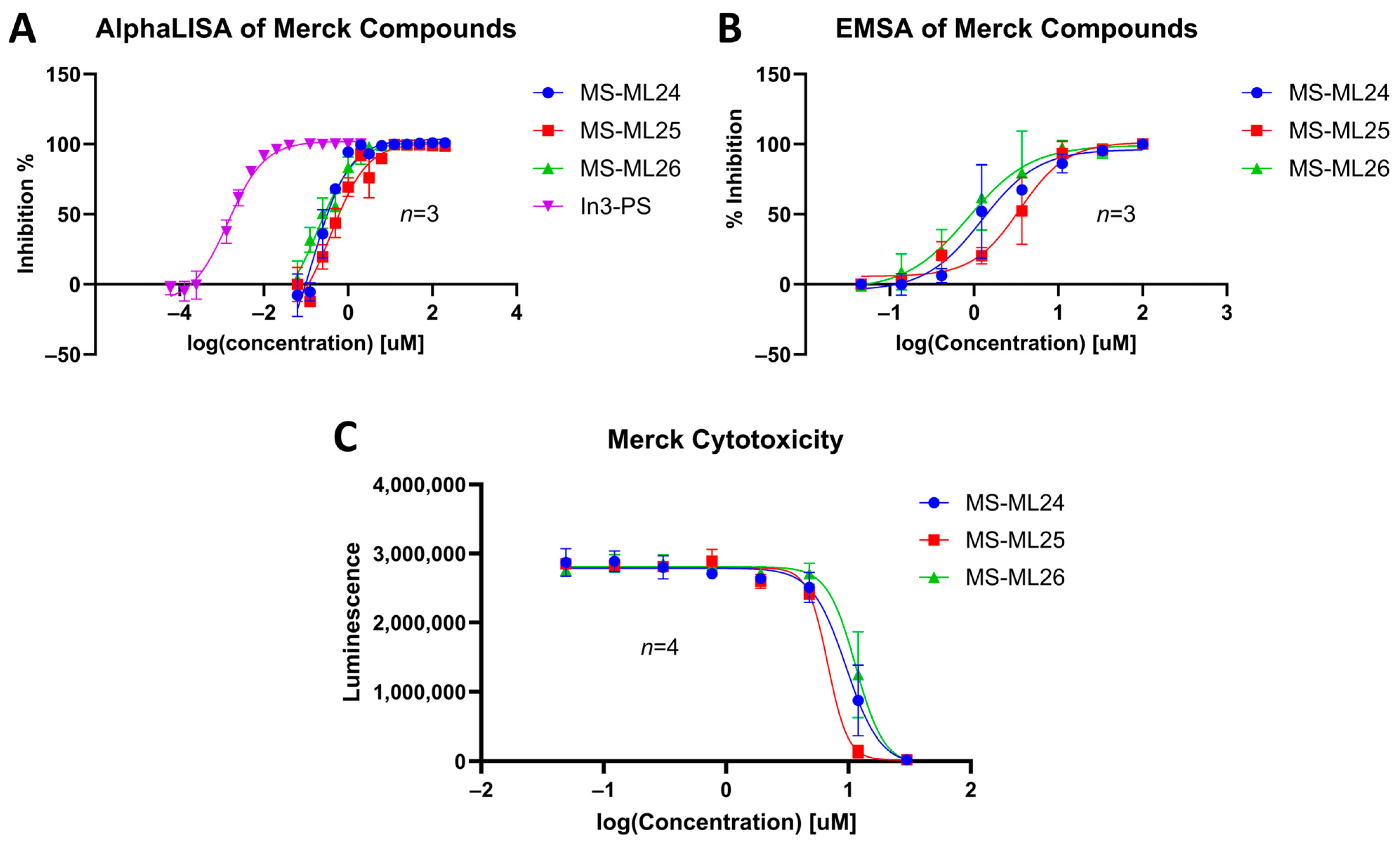
3.6. Dose–Response of Merck Compounds
3.7. In Vitro Cytotoxicity
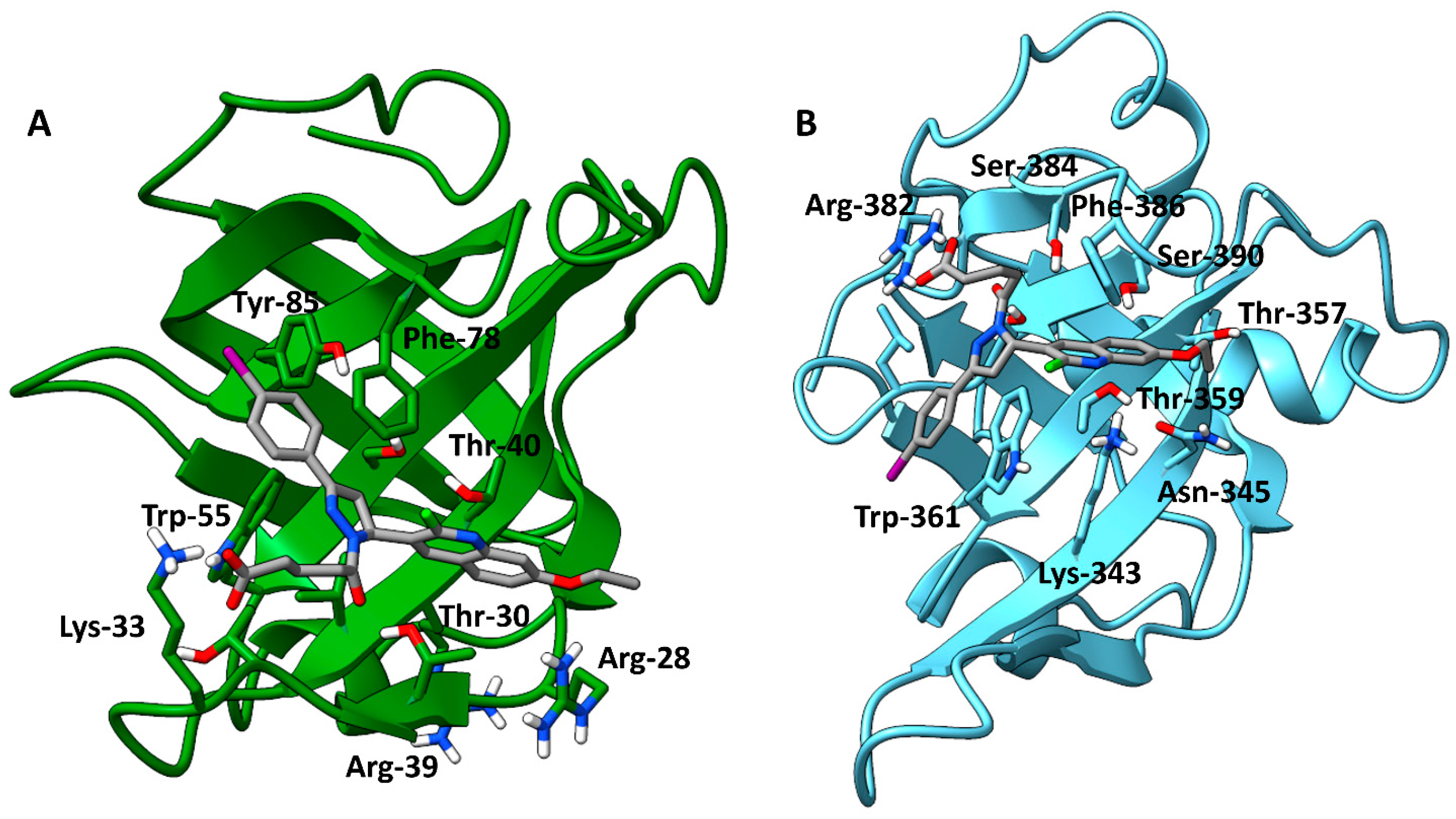
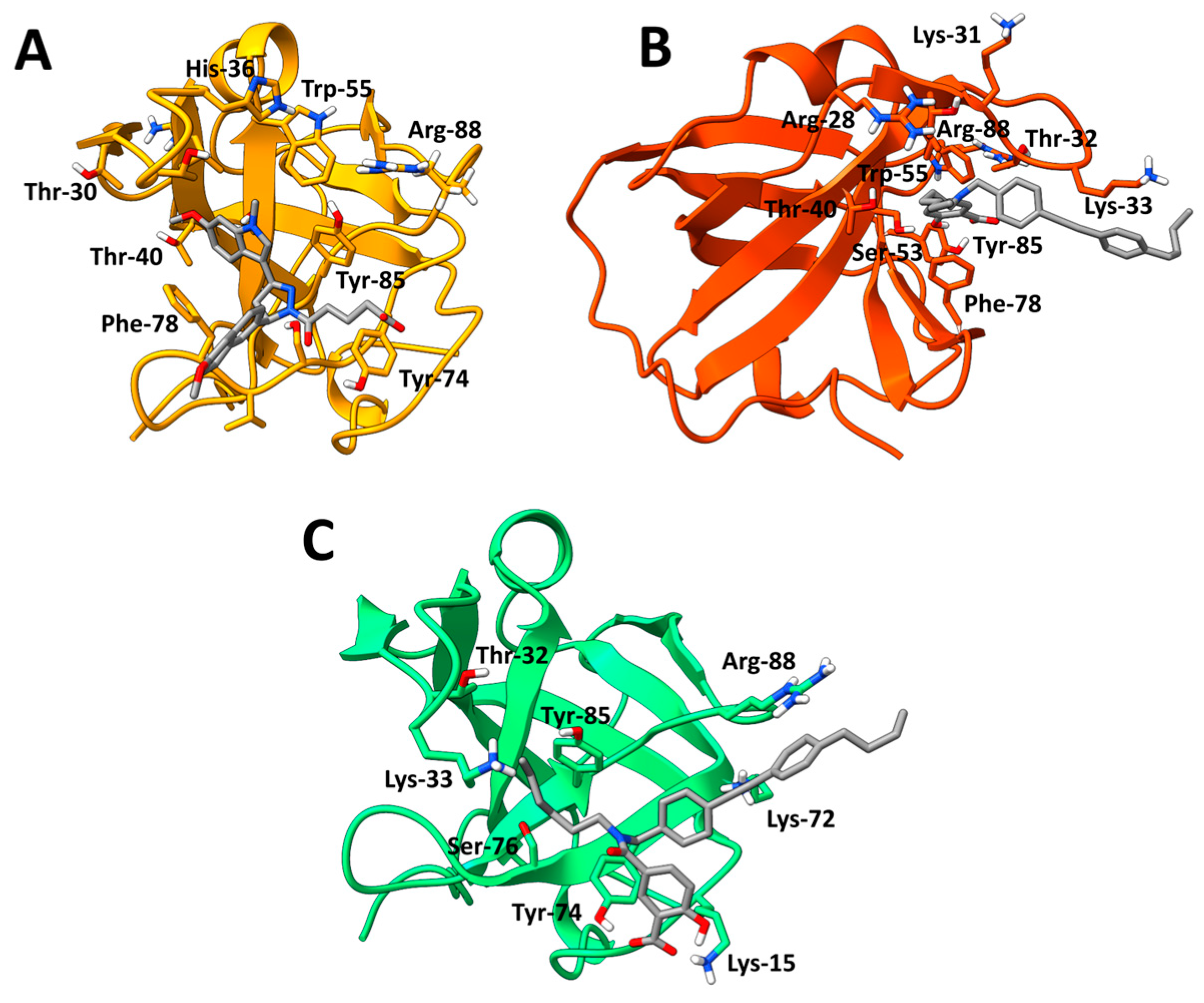
3.8. Cosolvent MD Simulations
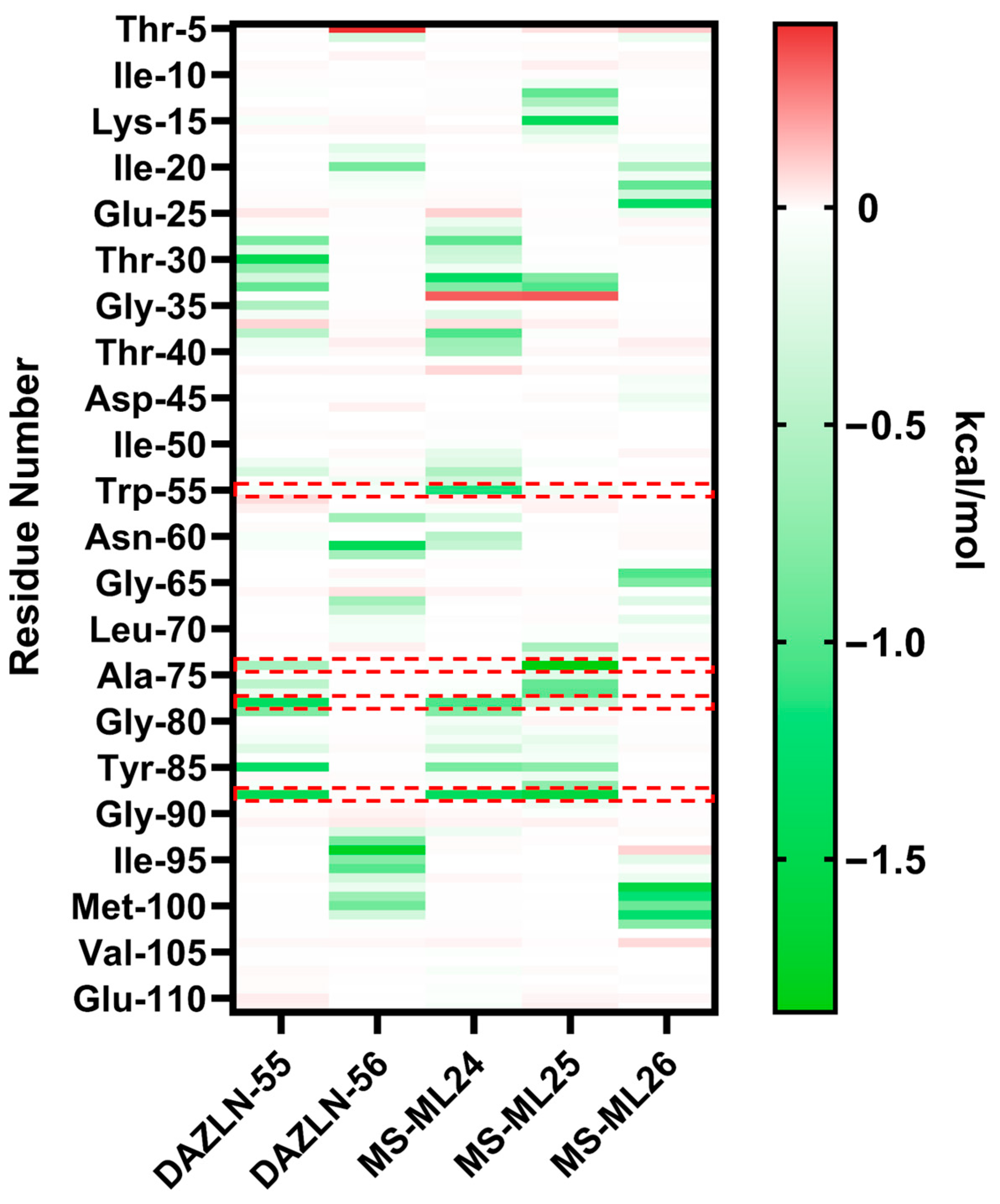
3.9. Relative Binding Free Energy Calculations and Residue Decomposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Torgovnick, A.; Schumacher, B. DNA Repair Mechanisms in Cancer Development and Therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guan, Y.; Chen, X.; Yang, J.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Richard, D.J.; Bolderson, E.; Cubeddu, L.; Wadsworth, R.I.M.; Savage, K.; Sharma, G.G.; Nicolette, M.L.; Tsvetanov, S.; McIlwraith, M.J.; Pandita, R.K.; et al. Single-Stranded DNA-Binding Protein hSSB1 Is Critical for Genomic Stability. Nature 2008, 453, 677–681. [Google Scholar] [CrossRef]
- Paquet, N.; Adams, M.N.; Ashton, N.W.; Touma, C.; Gamsjaeger, R.; Cubeddu, L.; Leong, V.; Beard, S.; Bolderson, E.; Botting, C.H.; et al. hSSB1 (NABP2/OBFC2B) Is Regulated by Oxidative Stress. Sci. Rep. 2016, 6, 27446. [Google Scholar] [CrossRef]
- Richard, D.J.; Savage, K.; Bolderson, E.; Cubeddu, L.; So, S.; Ghita, M.; Chen, D.J.; White, M.F.; Richard, K.; Prise, K.M.; et al. hSSB1 Rapidly Binds at the Sites of DNA Double-Strand Breaks and Is Required for the Efficient Recruitment of the MRN Complex. Nucleic Acids Res. 2011, 39, 1692–1702. [Google Scholar] [CrossRef]
- Bianco, P.R. The Mechanism of Action of the SSB Interactome Reveals It Is the First OB-Fold Family of Genome Guardians in Prokaryotes. Protein Sci. 2021, 30, 1757–1775. [Google Scholar] [CrossRef]
- Ren, W.; Chen, H.; Sun, Q.; Tang, X.; Lim, S.C.; Huang, J.; Song, H. Structural Basis of SOSS1 Complex Assembly and Recognition of ssDNA. Cell Rep. 2014, 6, 982–991. [Google Scholar] [CrossRef]
- Bolderson, E.; Petermann, E.; Croft, L.; Suraweera, A.; Pandita, R.K.; Pandita, T.K.; Helleday, T.; Khanna, K.K.; Richard, D.J. Human Single-Stranded DNA Binding Protein 1 (hSSB1/NABP2) Is Required for the Stability and Repair of Stalled Replication Forks. Nucleic Acids Res. 2014, 42, 6326–6336. [Google Scholar] [CrossRef]
- Richard, D.J.; Cubeddu, L.; Urquhart, A.J.; Bain, A.; Bolderson, E.; Menon, D.; White, M.F.; Khanna, K.K. hSSB1 Interacts Directly with the MRN Complex Stimulating Its Recruitment to DNA Double-Strand Breaks and Its Endo-Nuclease Activity. Nucleic Acids Res. 2011, 39, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Galeaz, C.; Totis, C.; Bisio, A. Radiation Resistance: A Matter of Transcription Factors. Front. Oncol. 2021, 11, 662840. [Google Scholar] [CrossRef]
- Adams, M.N.; Croft, L.V.; Urquhart, A.; Saleem, M.A.M.; Rockstroh, A.; Duijf, P.H.G.; Thomas, P.B.; Ferguson, G.P.; Najib, I.M.; Shah, E.T.; et al. hSSB1 (NABP2/OBFC2B) Modulates the DNA Damage and Androgen-Induced Transcriptional Response in Prostate Cancer. Prostate 2023, 83, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Croft, L.V.; Bolderson, E.; Adams, M.N.; El-Kamand, S.; Kariawasam, R.; Cubeddu, L.; Gamsjaeger, R.; Richard, D.J. Human Single-Stranded DNA Binding Protein 1 (hSSB1, OBFC2B), a Critical Component of the DNA Damage Response. Semin. Cell Dev. Biol. 2019, 86, 121–128. [Google Scholar] [CrossRef]
- Touma, C.; Kariawasam, R.; Gimenez, A.X.; Bernardo, R.E.; Ashton, N.W.; Adams, M.N.; Paquet, N.; Croll, T.I.; O’Byrne, K.J.; Richard, D.J.; et al. A Structural Analysis of DNA Binding by hSSB1 (NABP2/OBFC2B) in Solution. Nucleic Acids Res. 2016, 44, 7963–7973. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zheng, L.; Hu, K.; Wang, X.; Zhang, R.; Zou, Y.; Zhong, L.; Wang, S.; Wu, Y.; Kang, T. SUMOylation Stabilizes hSSB1 and Enhances the Recruitment of NBS1 to DNA Damage Sites. Signal Transduct. Target. Ther. 2020, 5, 80. [Google Scholar] [CrossRef]
- Li, Y.; Bolderson, E.; Kumar, R.; Muniandy, P.A.; Xue, Y.; Richard, D.J.; Seidman, M.; Pandita, T.K.; Khanna, K.K.; Wang, W. HSSB1 and hSSB2 Form Similar Multiprotein Complexes That Participate in DNA Damage Response. J. Biol. Chem. 2009, 284, 23525–23531. [Google Scholar] [CrossRef]
- Mishra, A.K.; Dormi, S.S.; Turchi, A.M.; Woods, D.S.; Turchi, J.J. Chemical Inhibitor Targeting the Replication Protein A-DNA Interaction Increases the Efficacy of Pt-Based Chemotherapy in Lung and Ovarian Cancer. Biochem. Pharmacol. 2015, 93, 25–33. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Vernon, T.L.; Jordan, M.R.; Turchi, J.J. Structure-Guided Optimization of Replication Protein A (RPA)–DNA Interaction Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1118–1124. [Google Scholar] [CrossRef]
- Voter, A.F.; Killoran, M.P.; Ananiev, G.E.; Wildman, S.A.; Hoffmann, F.M.; Keck, J.L. A High-Throughput Screening Strategy to Identify Inhibitors of SSB Protein–Protein Interactions in an Academic Screening Facility. SLAS Discov. 2018, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Endres, J.L.; Byrne, B.M.; Liu, S.; Bayles, K.W.; Oakley, G.G. Identification of Inhibitors for Single-Stranded DNA-Binding Proteins in Eubacteria. J. Antimicrob. Chemother. 2016, 71, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Theobald, D.L.; Mitton-Fry, R.M.; Wuttke, D.S. Nucleic Acid Recognition by OB-Fold Proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 115–133. [Google Scholar] [CrossRef]
- Anciano Granadillo, V.J.; Earley, J.N.; Shuck, S.C.; Georgiadis, M.M.; Fitch, R.W.; Turchi, J.J. Targeting the OB-Folds of Replication Protein A with Small Molecules. J. Nucleic Acids 2010, 2010, 304035. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Bernstein, D.A.; Walsh, B.W.; Shapiro, W.; Simmons, L.A.; Keck, J.L. Protein Interactions in Genome Maintenance as Novel Antibacterial Targets. PLoS ONE 2013, 8, e58765. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef]
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A.S.J.S.; Mackey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60, 3120–3130. [Google Scholar] [CrossRef]
- Bielefeld-Sevigny, M. AlphaLISA Immunoassay Platform--the “No-Wash” High-Throughput Alternative to ELISA. ASSAY Drug Dev. Technol. 2009, 7, 90–92. [Google Scholar] [CrossRef]
- Determining Kd with an Alpha Assay. Available online: https://www.perkinelmer.com/lab-products-and-services/application-support-knowledgebase/alphalisa-alphascreen-no-wash-assays/determining-kd.html (accessed on 21 September 2023).
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, M.K.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Turchi, J.J. Materials and Method for Inhibiting Replication Protein A and Uses Thereof. U.S. Patent US10774063, 15 September 2020. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Gobert, R.P.; van den Eijnden, M.; Szyndralewiez, C.; Jorand-Lebrun, C.; Swinnen, D.; Chen, L.; Gillieron, C.; Pixley, F.; Juillard, P.; Gerber, P.; et al. GLEPP1/Protein-Tyrosine Phosphatase φ Inhibitors Block Chemotaxis in Vitro and in Vivo and Improve Murine Ulcerative Colitis. J. Biol. Chem. 2009, 284, 11385–11395. [Google Scholar] [CrossRef]
- Bombrun, A.; Huijsduijnen, R.H.V.; Jorand-Lebrun, C.; Vitte, P.-A.; Gerber, P. Glepp-1 Inhibitors in the Treatment of Autoimmune and/or Inflammatory Disorders. U.S. Patent 11/916,097, 28 August 2008. [Google Scholar]
- Szabó, P.B.; Sabanés Zariquiey, F.; Nogueira, J.J. Cosolvent and Dynamic Effects in Binding Pocket Search by Docking Simulations. J. Chem. Inf. Model. 2021, 61, 5508–5523. [Google Scholar] [CrossRef]
- Cele, F.N.; Ramesh, M.; Soliman, M.E. Per-Residue Energy Decomposition Pharmacophore Model to Enhance Virtual Screening in Drug Discovery: A Study for Identification of Reverse Transcriptase Inhibitors as Potential Anti-HIV Agents. DDDT 2016, 10, 1365–1377. [Google Scholar] [CrossRef]
- Touma, C.; Mizzi, M.; El-Kamand, S.; Cubeddu, L.; Gamsjaeger, R.; Richard, D.J.; Adams, M.N.; Ashton, N.W. A Data-Driven Structural Model of hSSB1 (NABP2/OBFC2B) Self-Oligomerization. Nucleic Acids Res. 2017, 45, 8609–8620. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Schapira, M. A Systematic Analysis of Atomic Protein–Ligand Interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of Protein-Ligand Binding Affinity by Hydrogen Bond Pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Skaar, J.R.; Richard, D.J.; Saraf, A.; Toschi, A.; Bolderson, E.; Florens, L.; Washburn, M.P.; Khanna, K.K.; Pagano, M. INTS3 Controls the hSSB1-Mediated DNA Damage Response. J. Cell Biol. 2009, 187, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shinada, N.K.; de Brevern, A.G.; Schmidtke, P. Halogens in Protein–Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef]
- Perkin Elmer. A Practical Guide to Working with AlphaScreenTM; PerkinElmer: Boston, MA, USA, 2004. [Google Scholar]
- Ghanakota, P.; Carlson, H.A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. [Google Scholar] [CrossRef]
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 00180. [Google Scholar] [CrossRef] [PubMed]
- Paquet, N.; Adams, M.N.; Leong, V.; Ashton, N.W.; Touma, C.; Gamsjaeger, R.; Cubeddu, L.; Beard, S.; Burgess, J.T.; Bolderson, E.; et al. hSSB1 (NABP2/OBFC2B) Is Required for the Repair of 8-Oxo-Guanine by the hOGG1-Mediated Base Excision Repair Pathway. Nucleic Acids Res. 2015, 43, 8817–8829. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Boehm, M.; McClendon, C.L.; Torella, R.; Gohlke, H. Cosolvent-Enhanced Sampling and Unbiased Identification of Cryptic Pockets Suitable for Structure-Based Drug Design. J. Chem. Theory Comput. 2019, 15, 3331–3343. [Google Scholar] [CrossRef] [PubMed]
- Lawson, T.; El-Kamand, S.; Kariawasam, R.; Richard, D.J.; Cubeddu, L.; Gamsjaeger, R. A Structural Perspective on the Regulation of Human Single-Stranded DNA Binding Protein 1 (hSSB1, OBFC2B) Function in DNA Repair. Comput. Struct. Biotechnol. J. 2019, 17, 441–446. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A Fast and High-Quality Charge Model for the next Generation General AMBER Force Field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Mermelstein, D.J.; Lin, C.; Nelson, G.; Kretsch, R.; McCammon, J.A.; Walker, R.C. Fast and Flexible Gpu Accelerated Binding Free Energy Calculations within the Amber Molecular Dynamics Package. J. Comput. Chem. 2018, 39, 1354–1358. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- BIOVIA Discovery Studio Visualizer Version 21.0.0 2022; Dassault Systèmes: San Diego, CA, USA, 2019.
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate−DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Fanning, E.; Klimovich, V.; Nager, A.R. A Dynamic Model for Replication Protein A (RPA) Function in DNA Processing Pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Bochkarev, A.; Pfuetzner, R.A.; Edwards, A.M.; Frappier, L. Structure of the Single-Stranded-DNA-Binding Domain of Replication Protein A Bound to DNA. Nature 1997, 385, 176–181. [Google Scholar] [CrossRef] [PubMed]







| Compound Name | Compound Reference | Structure | Kd (μM) AlphaLISA | IC50 (μM) EMSA |
|---|---|---|---|---|
| TDRL-551 |  | 3.888 95% CI 2.503 to 5.859 | 12.48 95% CI 9.325 to 18.38 | |
| DAZLN-51 | 4 |  | <100 μM | N/A |
| DAZLN-55 | 5 |  | 5.793 95% CI 3.188 to 10.31 | 79.56 95% CI 60.44–751.9 |
| DAZLN-56 | 6 |  | 23.95 95% CI 5.738–88.26 | N/A |
| Compound | Structure | Kd (μM) AlphaLISA | IC50 (μM) EMSA | IC50 (μM) Cytotoxicity |
|---|---|---|---|---|
| MS-ML24 |  | 0.143 95% CI 0.0948 to 0.206 | 1.238 95% CI 0.629 to 2.76 | 9.558 95% CI 8.21 to 11.1 |
| MS-ML25 |  | 0.391 95% CI 0.274 to 0.555 | 3.504 95% CI 2.30 to 5.07 | 6.78 95% CI unbound to 7.47 |
| MS-ML26 |  | 0.198 95% CI 0.1273 to 0.2969 | 0.857 95% CI 0.239 to 1.60 | 11.45 95% CI 10.1 to 13.9 |
| DAZLN-55 | DAZLN-56 | MS-ML24 | MS-ML25 | MS-ML26 | |||||
|---|---|---|---|---|---|---|---|---|---|
| Thr-30 | −1.48 ± 1.98 | Lys-94 | −1.76 ± 1.74 | Arg-88 | −1.38 ± 2.33 | Tyr-74 | −1.85 ± 1.32 | Phe-98 | −1.59 ± 0.984 |
| Arg-88 | −1.46 ± 2.51 | Leu-61 | −1.44 ± 1.65 | Thr-31 | −1.35 ± 1.37 | Arg-88 | −1.61 ± 1.84 | Leu-21 | −1.26 ± 0.928 |
| Phe-78 | −1.37 ± 1.05 | Trp-55 | −1.14 ± 1.59 | Lys-15 | −1.43 ± 1.39 | Val-101 | −1.26 ± 0.918 | ||
| Tyr-85 | −1.36 ± 1.54 | Phe-78 | −1.03 ± 0.857 | Lys-33 | −1.01 ± 1.72 | Cys-99 | −1.21 ± 0.904 | ||
| Val-38 | −1.01 ± 0.708 | Pro-64 | −1.03 ± 0.726 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuurs, Z.P.; Martyn, A.P.; Soltau, C.P.; Beard, S.; Shah, E.T.; Adams, M.N.; Croft, L.V.; O’Byrne, K.J.; Richard, D.J.; Gandhi, N.S. An Exploration of Small Molecules That Bind Human Single-Stranded DNA Binding Protein 1. Biology 2023, 12, 1405. https://doi.org/10.3390/biology12111405
Schuurs ZP, Martyn AP, Soltau CP, Beard S, Shah ET, Adams MN, Croft LV, O’Byrne KJ, Richard DJ, Gandhi NS. An Exploration of Small Molecules That Bind Human Single-Stranded DNA Binding Protein 1. Biology. 2023; 12(11):1405. https://doi.org/10.3390/biology12111405
Chicago/Turabian StyleSchuurs, Zachariah P., Alexander P. Martyn, Carl P. Soltau, Sam Beard, Esha T. Shah, Mark N. Adams, Laura V. Croft, Kenneth J. O’Byrne, Derek J. Richard, and Neha S. Gandhi. 2023. "An Exploration of Small Molecules That Bind Human Single-Stranded DNA Binding Protein 1" Biology 12, no. 11: 1405. https://doi.org/10.3390/biology12111405