

Identifying Fibroblast Growth Factor Receptor 3 as a Mediator of Periosteal Osteochondral Differentiation through the Construction of microRNA-Based Interaction Networks

Abstract
:Simple Summary
Abstract
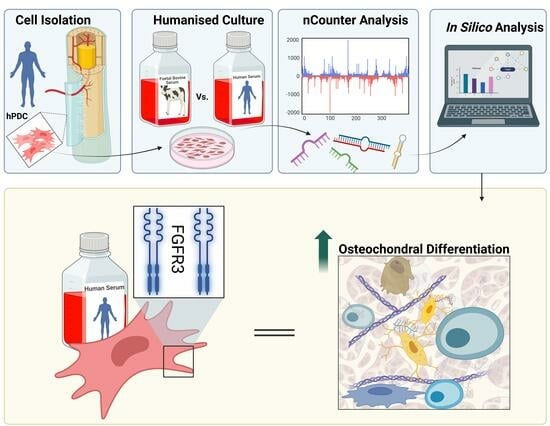
{kind=link}
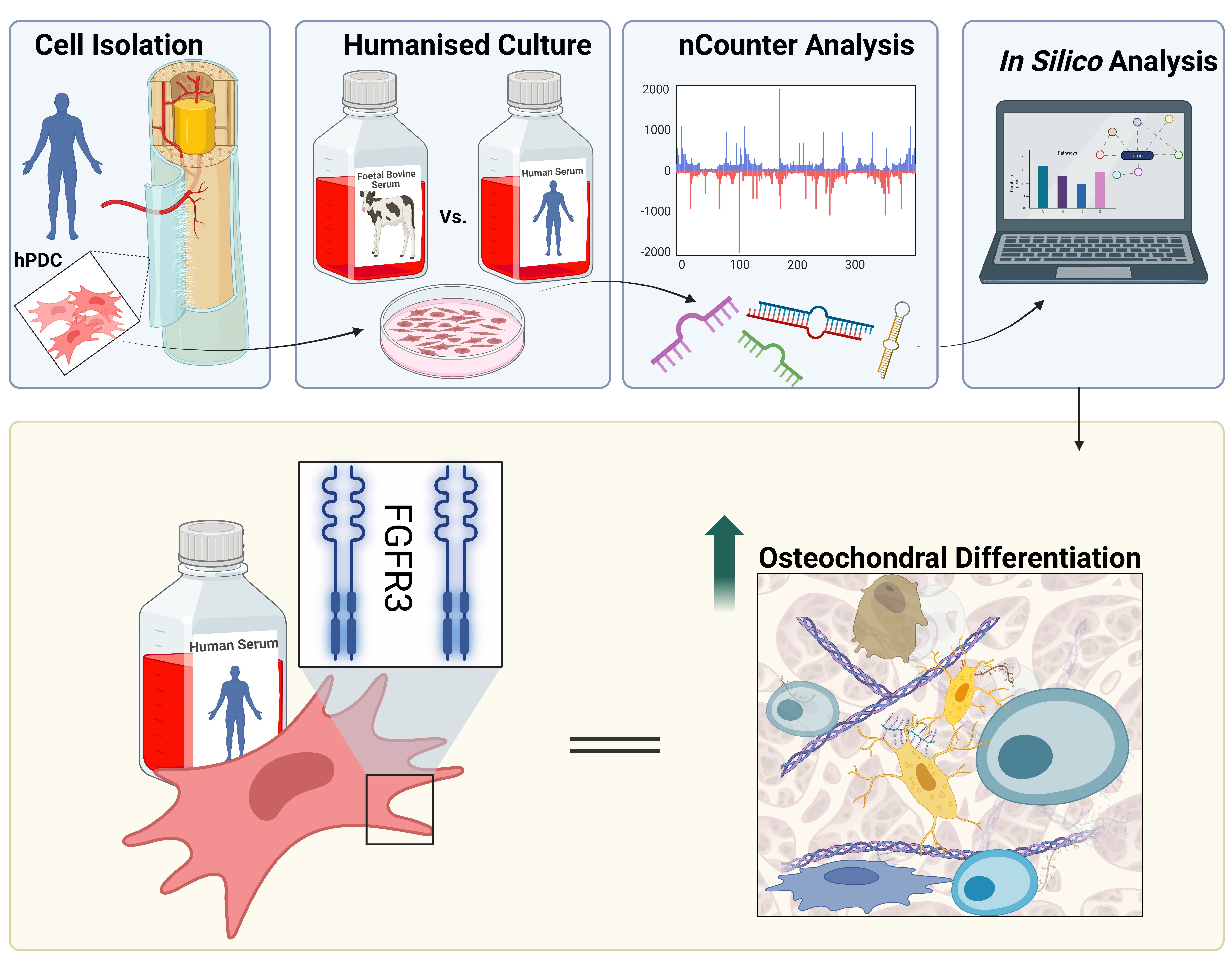
{kind=link}
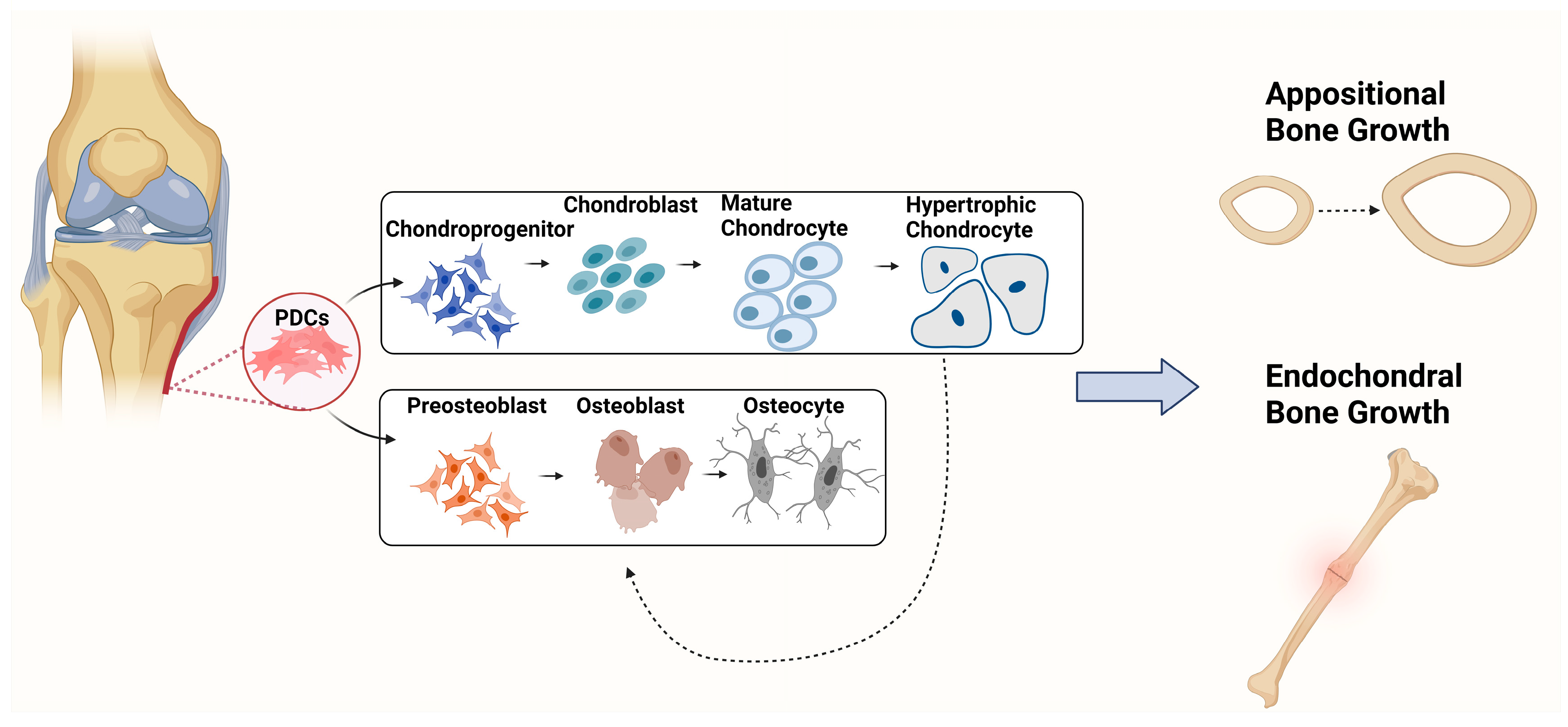
{kind=link}
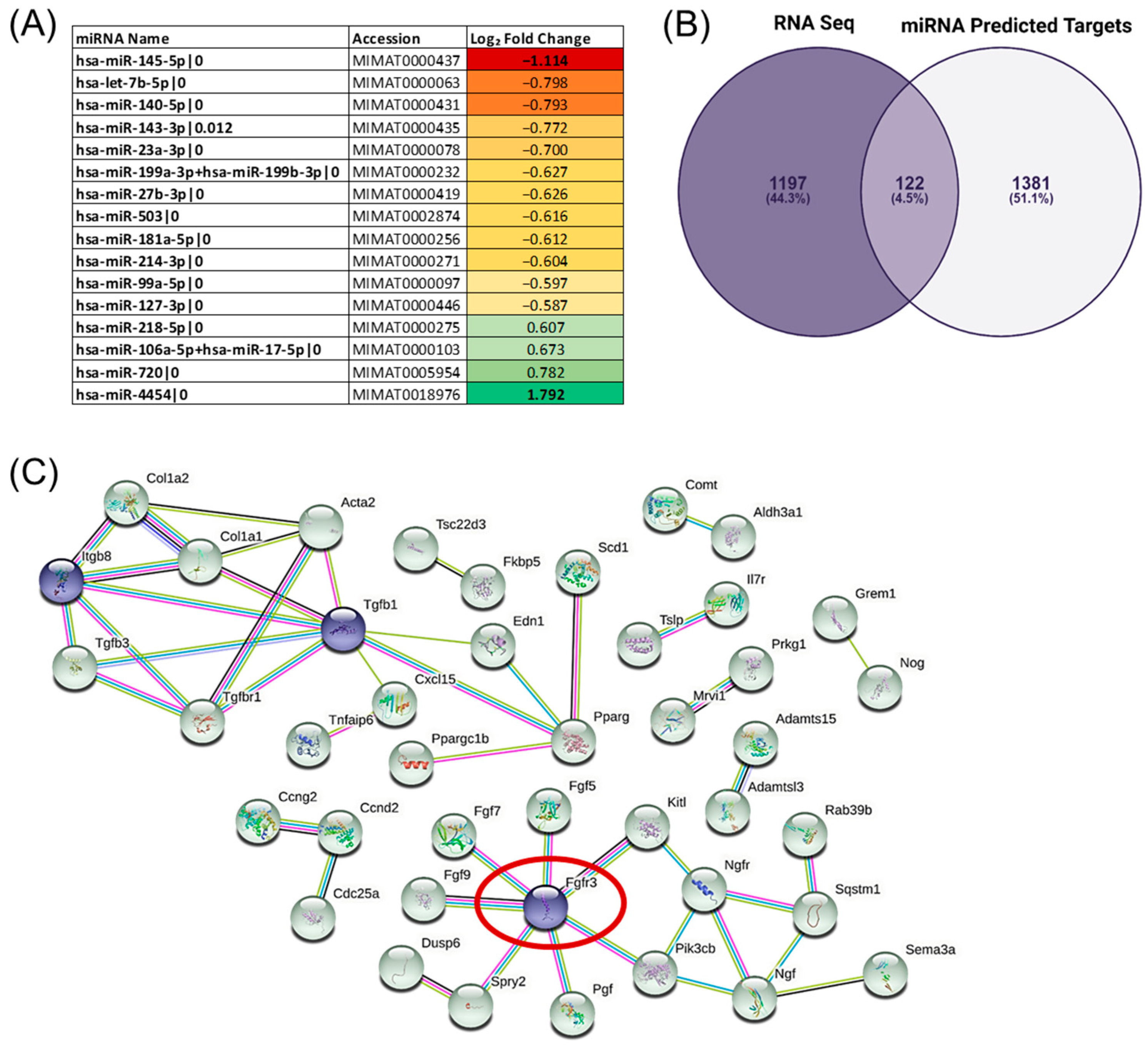
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. hPDC Isolation and Culture
2.2. hPDC RNAseq and miRNA Analysis
2.3. Identifying High-Confidence Differentially Expressed miRNA-Regulated Genes
2.4. PPI Network Construction
2.5. Pathway Enrichment Analysis
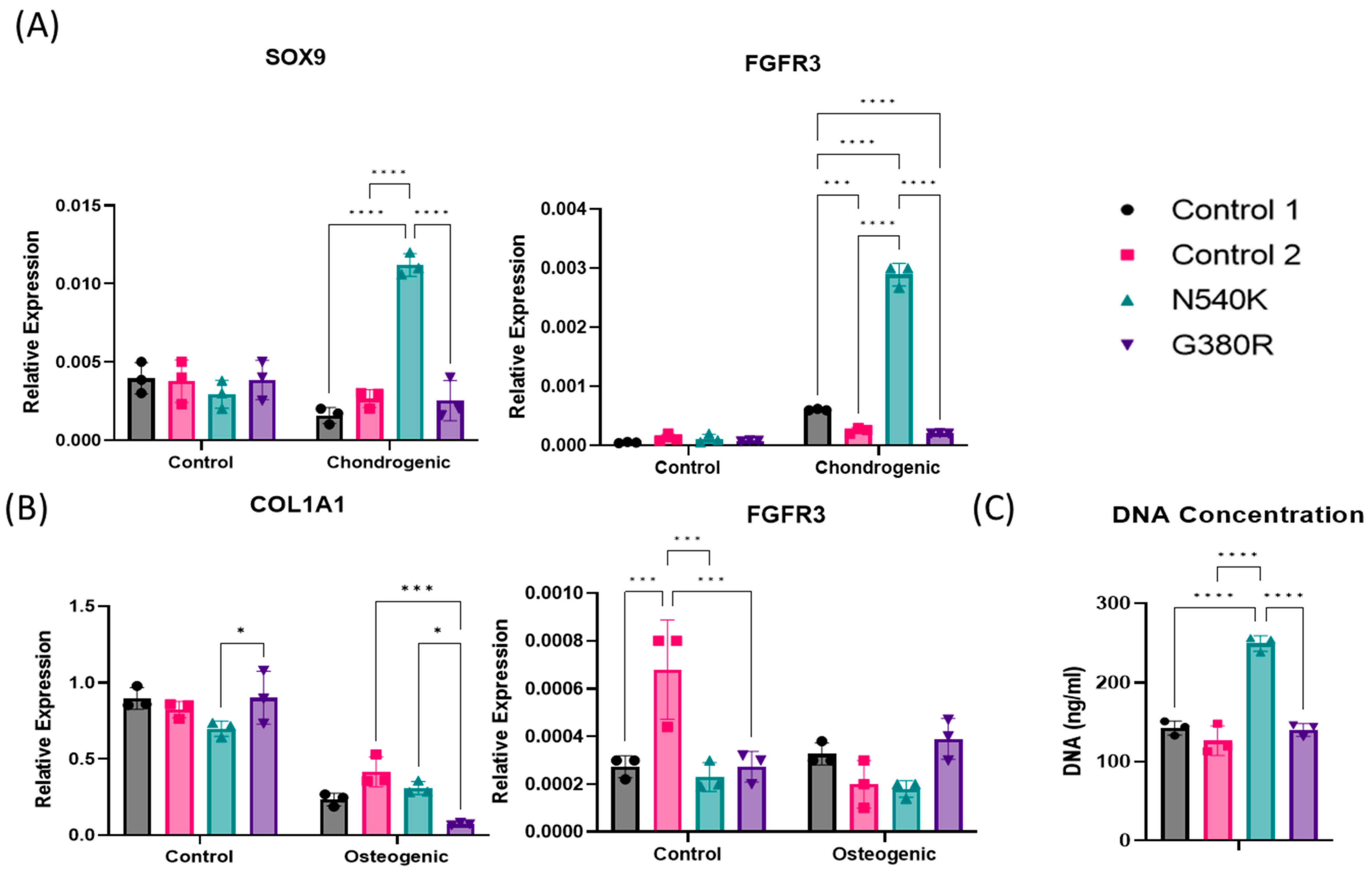
2.6. hPDC-FGFR3(ca) In Vitro Cell Quantification via DNA Analysis
2.7. hPDC-FGFR3(ca) In Vitro Gene Expression Analysis of Chondrogenic Markers
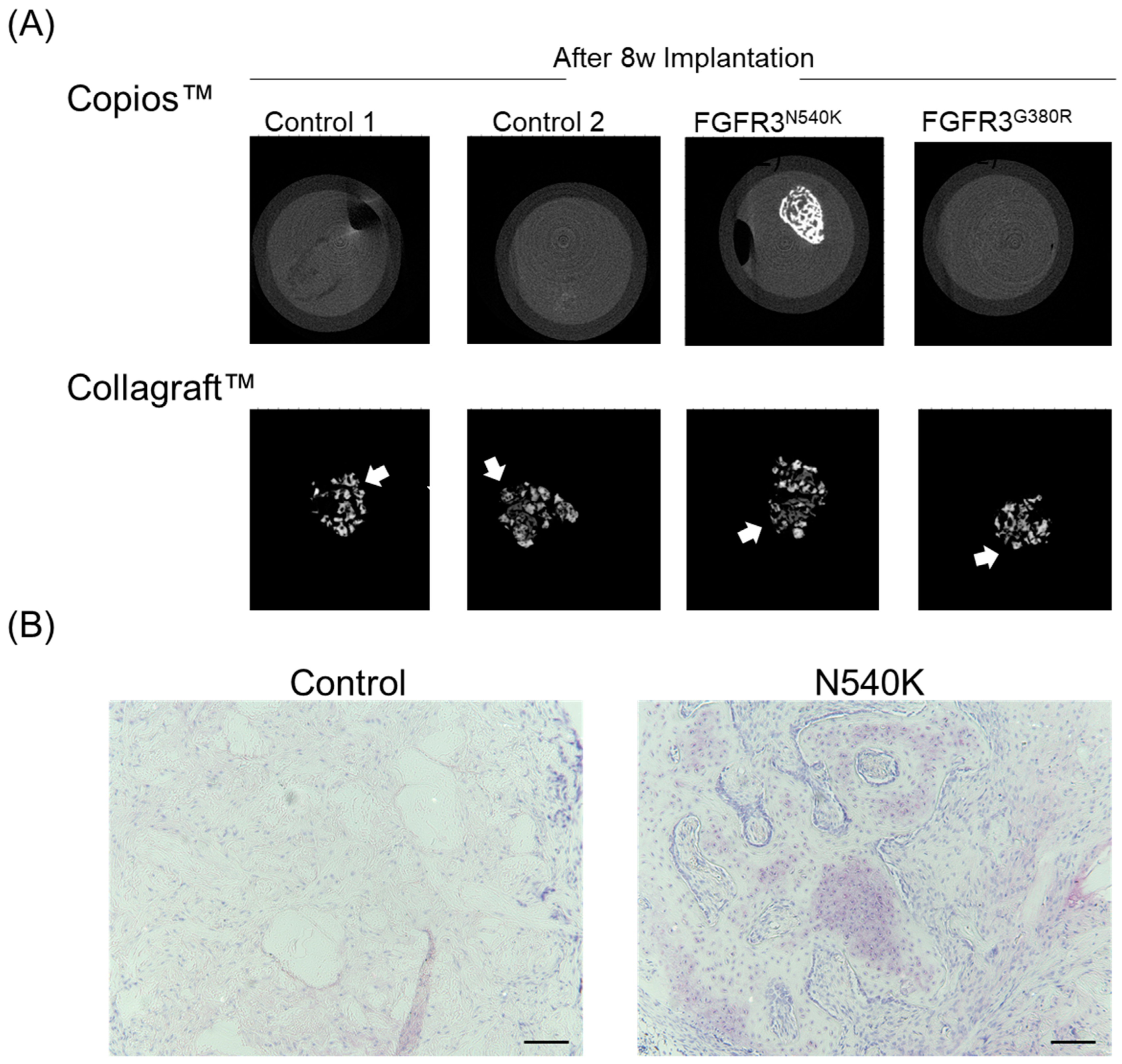
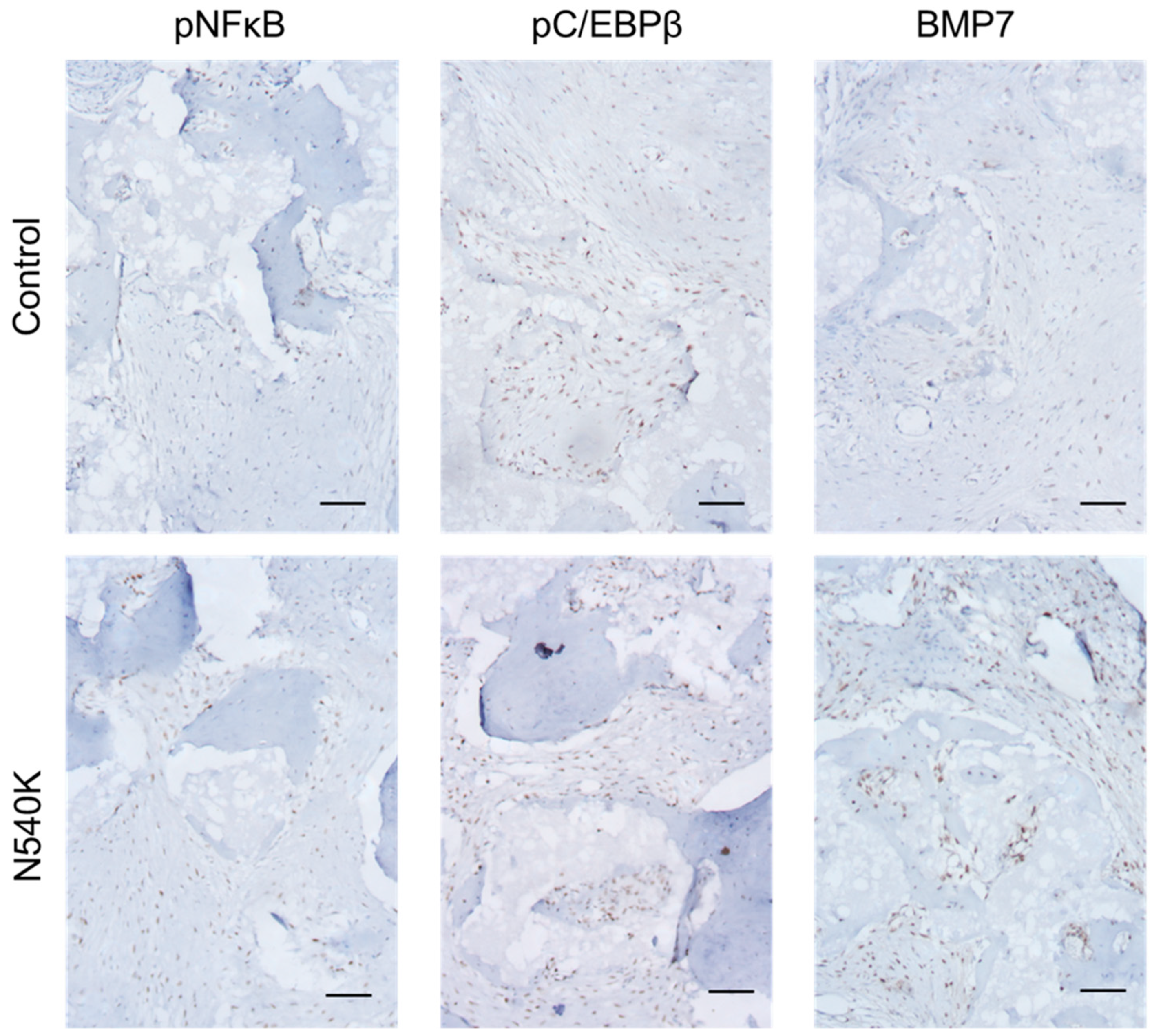
2.8. In Vivo Analysis of hPDC-FGFR3(ca) Osteochondral Differentiation
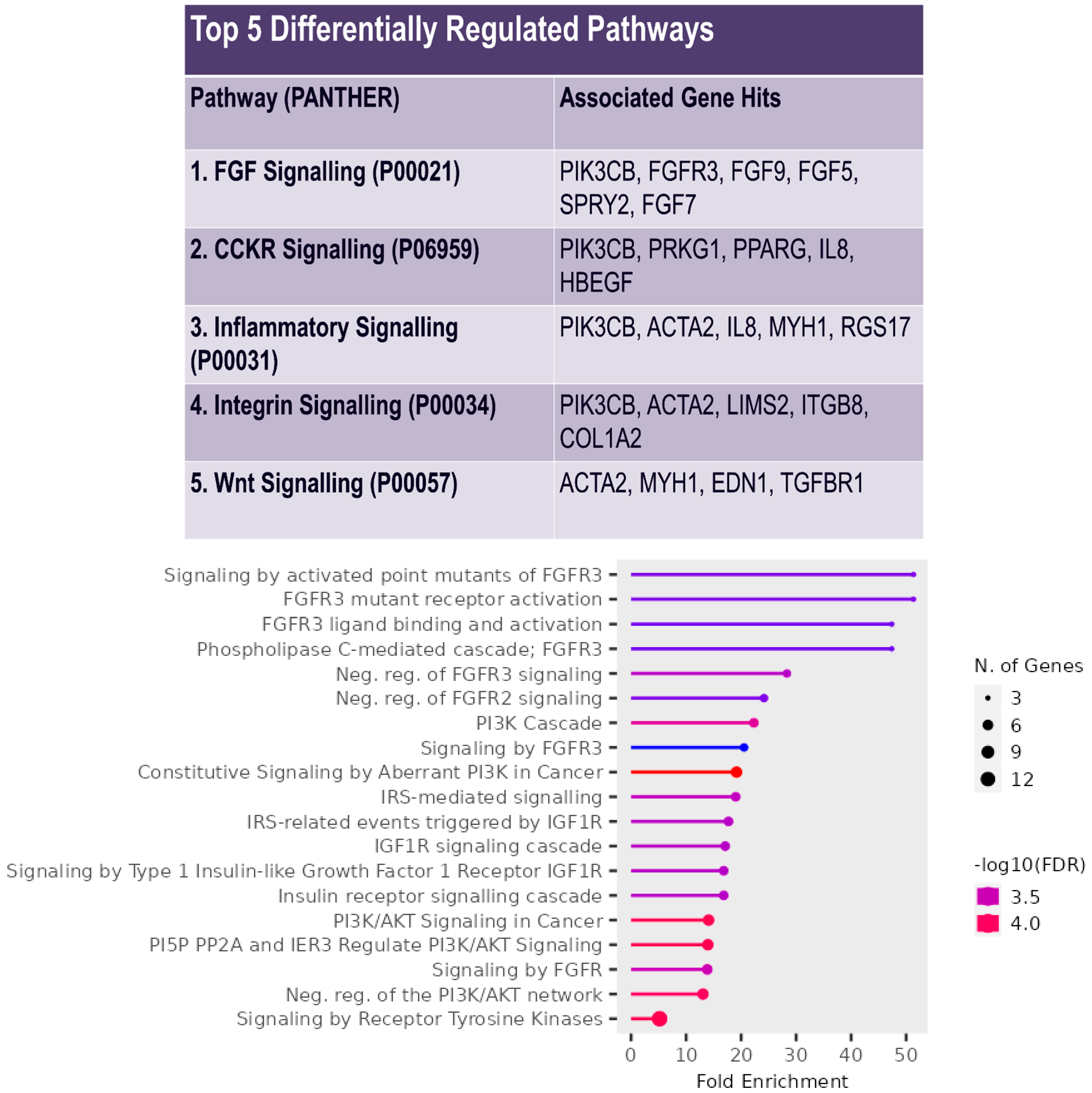
3. Results
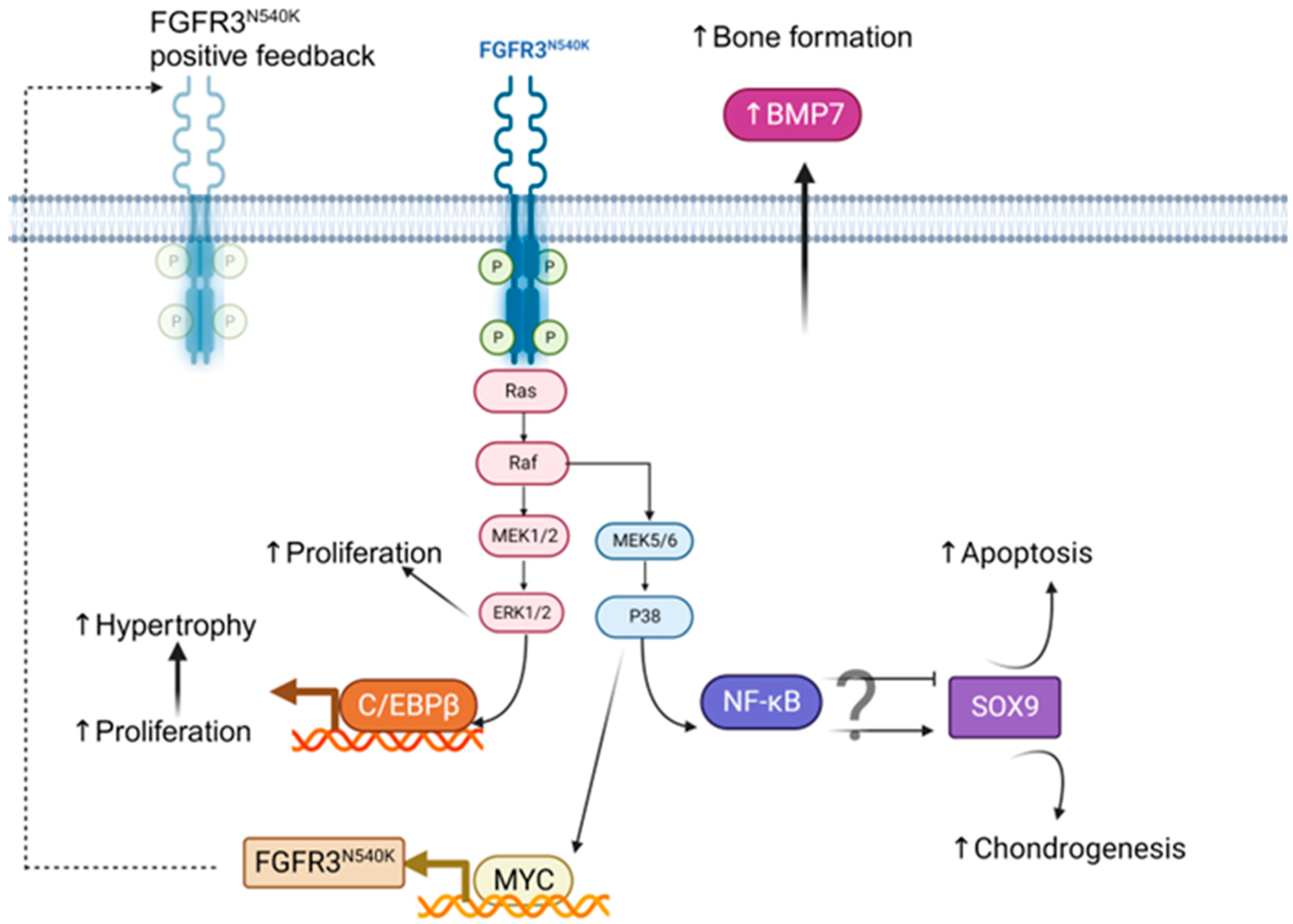
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Squier, C.A.; Ghoneim, S.; Kremenak, C.R. Ultrastructure of the periosteum from membrane bone. J. Anat. 1990, 171, 233–239. [Google Scholar] [PubMed]
- Roberts, S.J.; Van Gastel, N.; Carmeliet, G.; Luyten, F.P. Uncovering the periosteum for skeletal regeneration: The stem cell that lies beneath. Bone 2015, 70, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Dwek, J.R. The periosteum: What is it, where is it, and what mimics it in its absence? Skelet. Radiol. 2010, 39, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Colnot, C. Skeletal Cell Fate Decisions Within Periosteum and Bone Marrow During Bone Regeneration. J. Bone Miner. Res. 2009, 24, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.; Li, Z.; Tian, Y.; Li, Z.; Lu, A.; Hsu, C.-Y.; Negri, S.; Tang, C.; Tower, R.J.; et al. PDGFRα reporter activity identifies periosteal progenitor cells critical for bone formation and fracture repair. Bone Res. 2022, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; VanHouten, J.N.; Nasiri, A.R.; Tommasini, S.M.; Broadus, A.E. Periosteal PTHrP regulates cortical bone modeling during linear growth in mice. J. Anat. 2014, 225, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Isogai, N.; Tokui, T. 6.23 Finger. In Comprehensive Biomaterials II; Ducheyne, P., Ed.; Elsevier: Oxford, UK, 2017; pp. 416–423. [Google Scholar]
- Jeffery, E.C.; Mann, T.L.A.; Pool, J.A.; Zhao, Z.; Morrison, S.J. Bone marrow and periosteal skeletal stem/progenitor cells make distinct contributions to bone maintenance and repair. Cell Stem Cell 2022, 29, 1547–1561.e6. [Google Scholar] [CrossRef] [PubMed]
- Schindeler, A.; McDonald, M.M.; Bokko, P.; Little, D.G. Bone remodeling during fracture repair: The cellular picture. Semin. Cell Dev. Biol. 2008, 19, 459–466. [Google Scholar] [CrossRef]
- O’Driscoll, S.W.; Saris, D.B.; Ito, Y.; Fitzimmons, J.S. The chondrogenic potential of periosteum decreases with age. J. Orthop. Res. 2001, 19, 95–103. [Google Scholar] [CrossRef]
- Brown, S.; Malik, S.; Aljammal, M.; O’Flynn, A.; Hobbs, C.; Shah, M.; Roberts, S.J.; Logan, M.P.O. The Prrx1eGFP Mouse Labels the Periosteum During Development and a Subpopulation of Osteogenic Periosteal Cells in the Adult. JBMR Plus 2023, 7, e10707. [Google Scholar] [CrossRef]
- Mills, L.A.; Aitken, S.A.; Simpson, A. The risk of non-union per fracture: Current myths and revised figures from a population of over 4 million adults. Acta Orthop. 2017, 88, 434–439. [Google Scholar] [CrossRef] [PubMed]
- van der Kraan, P.M.; van den Berg, W.B. Osteophytes: Relevance and biology. Osteoarthr. Cartil. 2007, 15, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.-J.; Leng, X.-M. Dynamic miRNA–mRNA paradigms: New faces of miRNAs. Biochem. Biophys. Rep. 2015, 4, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.J.; Owen, H.C.; Tam, W.L.; Solie, L.; Van Cromphaut, S.J.; Van den Berghe, G.; Luyten, F.P. Humanized culture of periosteal progenitors in allogeneic serum enhances osteogenic differentiation and in vivo bone formation. Stem Cells Transl. Med. 2014, 3, 218–228. [Google Scholar] [CrossRef] [PubMed]
- De Bari, C.; Dell’Accio, F.; Vanlauwe, J.; Eyckmans, J.; Khan, I.M.; Archer, C.W.; Jones, E.A.; McGonagle, D.; Mitsiadis, T.A.; Pitzalis, C.; et al. Mesenchymal multipotency of adult human periosteal cells demonstrated by single-cell lineage analysis. Arthritis Rheum. 2006, 54, 1209–1221. [Google Scholar] [CrossRef]
- Al Hosni, R.; Shah, M.; Cheema, U.; Roberts, H.C.; Luyten, F.P.; Roberts, S.J. Mapping human serum-induced gene networks as a basis for the creation of biomimetic periosteum for bone repair. Cytotherapy 2020, 22, 424–435. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chen, Y.; Sonnaert, M.; Roberts, S.J.; Luyten, F.P.; Schrooten, J. Validation of a PicoGreen-Based DNA Quantification Integrated in an RNA Extraction Method for Two-Dimensional and Three-Dimensional Cell Cultures. Tissue Eng. Part C Methods 2012, 18, 444–452. [Google Scholar] [CrossRef]
- Roberts, S.J.; Geris, L.; Kerckhofs, G.; Desmet, E.; Schrooten, J.; Luyten, F.P. The combined bone forming capacity of human periosteal derived cells and calcium phosphates. Biomaterials 2011, 32, 4393–4405. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: www.bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 10 March 2022).
- García-Mato, Á.; Cervantes, B.; Rodríguez-de la Rosa, L.; Varela-Nieto, I. IGF-1 Controls Metabolic Homeostasis and Survival in HEI-OC1 Auditory Cells through AKT and mTOR Signaling. Antioxidants 2023, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.; Atkins, J.; Janku, F.; Moulder, S.; Stephens, P.; Yelensky, R.; Valero, V.; Miller, V.; Meric-Bernstam, F. Presence of both alterations in FGFR/FGF and PI3K/AKT/mTOR confer improved outcomes for patients with metastatic breast cancer treated with PI3K/AKT/mTOR inhibitors. Oncoscience 2016, 3, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Eyckmans, J.; Roberts, S.J.; Bolander, J.; Schrooten, J.; Chen, C.S.; Luyten, F.P. Mapping calcium phosphate activated gene networks as a strategy for targeted osteoinduction of human progenitors. Biomaterials 2013, 34, 4612–4621. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, V.; Giannoni, P.; Gentili, C.; Cancedda, R.; Descalzi, F. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J. Cell Biochem. 2008, 104, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gade, P.; Xiao, W.; Kalvakolanu, D.V. The interferon signaling network and transcription factor C/EBP-beta. Cell Mol. Immunol. 2007, 4, 407–418. [Google Scholar]
- Matsushita, T.; Wilcox, W.R.; Chan, Y.Y.; Kawanami, A.; Bükülmez, H.; Balmes, G.; Krejci, P.; Mekikian, P.B.; Otani, K.; Yamaura, I.; et al. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum. Mol. Genet. 2009, 18, 227–240. [Google Scholar] [CrossRef]
- Julien, A.; Perrin, S.; Duchamp de Lageneste, O.; Carvalho, C.; Bensidhoum, M.; Legeai-Mallet, L.; Colnot, C. FGFR3 in Periosteal Cells Drives Cartilage-to-Bone Transformation in Bone Repair. Stem Cell Rep. 2020, 15, 955–967. [Google Scholar] [CrossRef]
- Sharrocks, A.D. Cell Cycle: Sustained ERK Signalling Represses the Inhibitors. Curr. Biol. 2006, 16, R540–R542. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, S.; Ozaki, Y.-i.; Fujita, K.; Kuroda, S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 2005, 7, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Loisay, L.; Komla-Ebri, D.; Morice, A.; Heuzé, Y.; Viaut, C.; de La Seiglière, A.; Kaci, N.; Chan, D.; Lamouroux, A.; Baujat, G.; et al. Hypochondroplasia gain-of-function mutation in FGFR3 causes defective bone mineralization in mice. JCI Insight 2023, 8, 168796. [Google Scholar] [CrossRef] [PubMed]
- Schliermann, A.; Nickel, J. Unraveling the Connection between Fibroblast Growth Factor and Bone Morphogenetic Protein Signaling. Int. J. Mol. Sci. 2018, 19, 3220. [Google Scholar] [CrossRef] [PubMed]
- Kozmikova, I.; Candiani, S.; Fabian, P.; Gurska, D.; Kozmik, Z. Essential role of Bmp signaling and its positive feedback loop in the early cell fate evolution of chordates. Dev. Biol. 2013, 382, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Seidel, C.W.; Gibson, M.C. The feedback regulator Nord controls Dpp/BMP signaling via extracellular interaction with Dally in the Drosophila wing. Dev. Biol. 2022, 488, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Mahe, M.; Dufour, F.; Neyret-Kahn, H.; Moreno-Vega, A.; Beraud, C.; Shi, M.; Hamaidi, I.; Sanchez-Quiles, V.; Krucker, C.; Dorland-Galliot, M.; et al. An FGFR3/MYC positive feedback loop provides new opportunities for targeted therapies in bladder cancers. EMBO Mol. Med. 2018, 10, 8163. [Google Scholar] [CrossRef] [PubMed]
- Klomp, J.E.; Klomp, J.A.; Der, C.J. The ERK mitogen-activated protein kinase signaling network: The final frontier in RAS signal transduction. Biochem. Soc. Trans. 2021, 49, 253–267. [Google Scholar] [CrossRef]
- Cui, T.X.; Lin, G.; LaPensee, C.R.; Calinescu, A.-A.; Rathore, M.; Streeter, C.; Piwien-Pilipuk, G.; Lanning, N.; Jin, H.; Carter-Su, C.; et al. C/EBPβ Mediates Growth Hormone-Regulated Expression of Multiple Target Genes. Mol. Endocrinol. 2011, 25, 681–693. [Google Scholar] [CrossRef]
- Sebastian, T.; Johnson, P.F. Stop and Go: Anti-Proliferative and Mitogenic Functions of the Transcription Factor C/EBP&beta. Cell Cycle 2006, 5, 953–957. [Google Scholar] [CrossRef]
- Ushijima, T.; Okazaki, K.; Tsushima, H.; Iwamoto, Y. CCAAT/enhancer-binding protein β regulates the repression of type II collagen expression during the differentiation from proliferative to hypertrophic chondrocytes. J. Biol. Chem. 2014, 289, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Stanton, L.A.; Sabari, S.; Sampaio, A.V.; Underhill, T.M.; Beier, F. p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem. J. 2004, 378 Pt 1, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Jimi, E.; Fei, H.; Nakatomi, C. NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis. Int. J. Mol. Sci. 2019, 20, 6275. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Brockmueller, A.; Mueller, A.L.; Shayan, P.; Shakibaei, M. Curcumin Attenuates Environment-Derived Osteoarthritis by Sox9/NF-kB Signaling Axis. Int. J. Mol. Sci. 2021, 22, 7645. [Google Scholar] [CrossRef]
- Tang, J.; Su, N.; Zhou, S.; Xie, Y.; Huang, J.; Wen, X.; Wang, Z.; Wang, Q.; Xu, W.; Du, X.; et al. Fibroblast Growth Factor Receptor 3 Inhibits Osteoarthritis Progression in the Knee Joints of Adult Mice. Arthritis Rheumatol. 2016, 68, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Klag, K.A.; Horton, W.A. Advances in treatment of achondroplasia and osteoarthritis. Hum. Mol. Genet. 2015, 25, R2–R8. [Google Scholar] [CrossRef] [PubMed]
- Hallett, S.A.; Ono, W.; Ono, N. The hypertrophic chondrocyte: To be or not to be. Histol. Histopathol. 2021, 36, 1021–1036. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef]
- Pottenger, L.A.; Phillips, F.M.; Draganich, L.F. The effect of marginal osteophytes on reduction of varus-valgus instability in osteoarthritic knees. Arthritis Rheum. 1990, 33, 853–858. [Google Scholar] [CrossRef]
- Moore, E.E.; Bendele, A.M.; Thompson, D.L.; Littau, A.; Waggie, K.S.; Reardon, B.; Ellsworth, J.L. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthr. Cartil. 2005, 13, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Chen, X.-Y.; Yan, Z.-P.; Li, J.-T.; Liao, T.; Ni, G.-X. Efficacy and safety of sprifermin injection for knee osteoarthritis treatment: A meta-analysis. Arthritis Res. Ther. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Felix, E.; Marc, C.H.; Hans, G.; Flavie, M.; Victor, O.; Asger Reinstrup, B.; Inger, B.; Jeppe Ragnar, A.; Benjamin, D.; Oliver, G.; et al. Long-term structural and symptomatic effects of intra-articular sprifermin in patients with knee osteoarthritis: 5-year results from the FORWARD study. Ann. Rheum. Dis. 2021, 80, 1062. [Google Scholar] [CrossRef]
- Jin, Y.; Hong, F.; Bao, Q.; Xu, Q.; Duan, R.; Zhu, Z.; Zhang, W.; Ma, C. MicroRNA-145 suppresses osteogenic differentiation of human jaw bone marrow mesenchymal stem cells partially via targeting semaphorin 3A. Connect. Tissue Res. 2020, 61, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Guo, H.; Zhang, Y.; Chen, L.; Ying, D.; Dong, S. MicroRNA-145 regulates chondrogenic differentiation of mesenchymal stem cells by targeting Sox9. PLoS ONE 2011, 6, e21679. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, A.; Dudek, K.A.; Murphy, C.L. Regulation of human chondrocyte function through direct inhibition of cartilage master regulator SOX9 by microRNA-145 (miRNA-145). J. Biol. Chem. 2012, 287, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, A.; Lazzarano, S.; Sharma, E.; Lockstone, H.; Murphy, C.L. High-Throughput Identification of MiR-145 Targets in Human Articular Chondrocytes. Life 2020, 10, 58. [Google Scholar] [CrossRef]
- Fu, L.; Li, N.; Ye, Y.; Ye, X.; Xiao, T.; Wu, X.; Ma, Y.; Yu, J. MicroRNA Hsa-Let-7b Regulates the Osteogenic Differentiation of Human Periodontal Ligament Stem Cells by Targeting CTHRC1. Stem. Cells Int. 2021, 2021, 5791181. [Google Scholar] [CrossRef]
- Papaioannou, G.; Inloes, J.B.; Nakamura, Y.; Paltrinieri, E.; Kobayashi, T. let-7 and miR-140 microRNAs coordinately regulate skeletal development. Proc. Natl. Acad. Sci. USA 2013, 110, E3291–E3300. [Google Scholar] [CrossRef]
- Ji, Q.; Qi, D.; Xu, X.; Xu, Y.; Goodman, S.B.; Kang, L.; Song, Q.; Fan, Z.; Maloney, W.J.; Wang, Y. Cryptotanshinone Protects Cartilage against Developing Osteoarthritis through the miR-106a-5p/GLIS3 Axis. Mol. Ther. Nucleic Acids 2018, 11, 170–179. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Wang, S.; Wei, J.; Fan, J.; Li, J.; Han, Q.; Liao, L.; Shao, C.; Zhao, R.C. miR-17-5p and miR-106a are involved in the balance between osteogenic and adipogenic differentiation of adipose-derived mesenchymal stem cells. Stem Cell Res. 2013, 10, 313–324. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wells, L.M.; Roberts, H.C.; Luyten, F.P.; Roberts, S.J. Identifying Fibroblast Growth Factor Receptor 3 as a Mediator of Periosteal Osteochondral Differentiation through the Construction of microRNA-Based Interaction Networks. Biology 2023, 12, 1381. https://doi.org/10.3390/biology12111381
Wells LM, Roberts HC, Luyten FP, Roberts SJ. Identifying Fibroblast Growth Factor Receptor 3 as a Mediator of Periosteal Osteochondral Differentiation through the Construction of microRNA-Based Interaction Networks. Biology. 2023; 12(11):1381. https://doi.org/10.3390/biology12111381
Chicago/Turabian StyleWells, Leah M., Helen C. Roberts, Frank P. Luyten, and Scott J. Roberts. 2023. "Identifying Fibroblast Growth Factor Receptor 3 as a Mediator of Periosteal Osteochondral Differentiation through the Construction of microRNA-Based Interaction Networks" Biology 12, no. 11: 1381. https://doi.org/10.3390/biology12111381