
Mitochondrial Dysfunction-Associated Mechanisms in the Development of Chronic Liver Diseases

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Factors Impairing Mitochondrial Function and Its Consequences
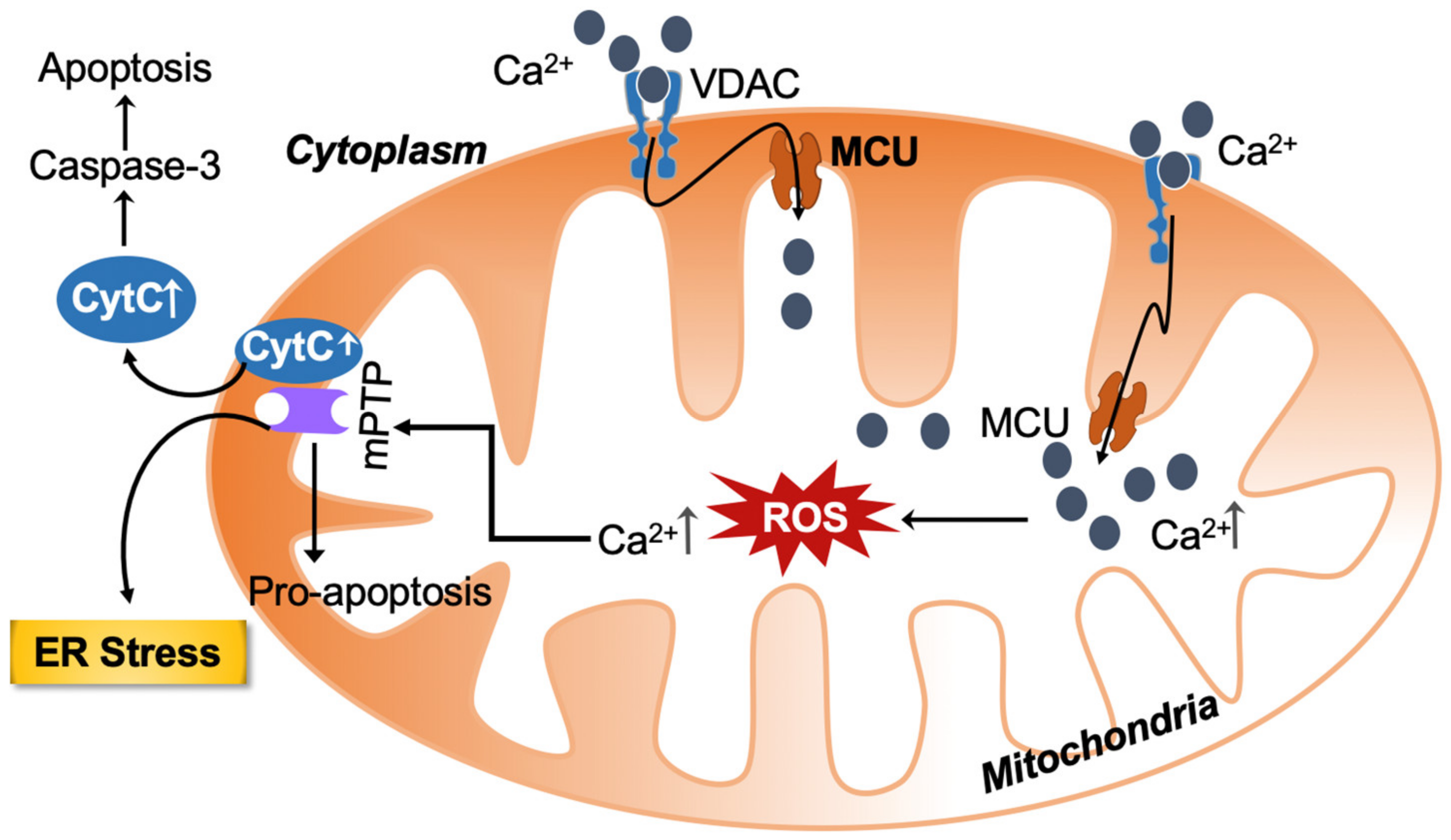
2.1. Calcium Homeostasis under Physiological Conditions and Its Overload-Associated ROS Production and Cell Death
2.2. Dysregulated Iron Homeostasis
2.3. Excessive Carbohydrates and Fatty Acids Intake
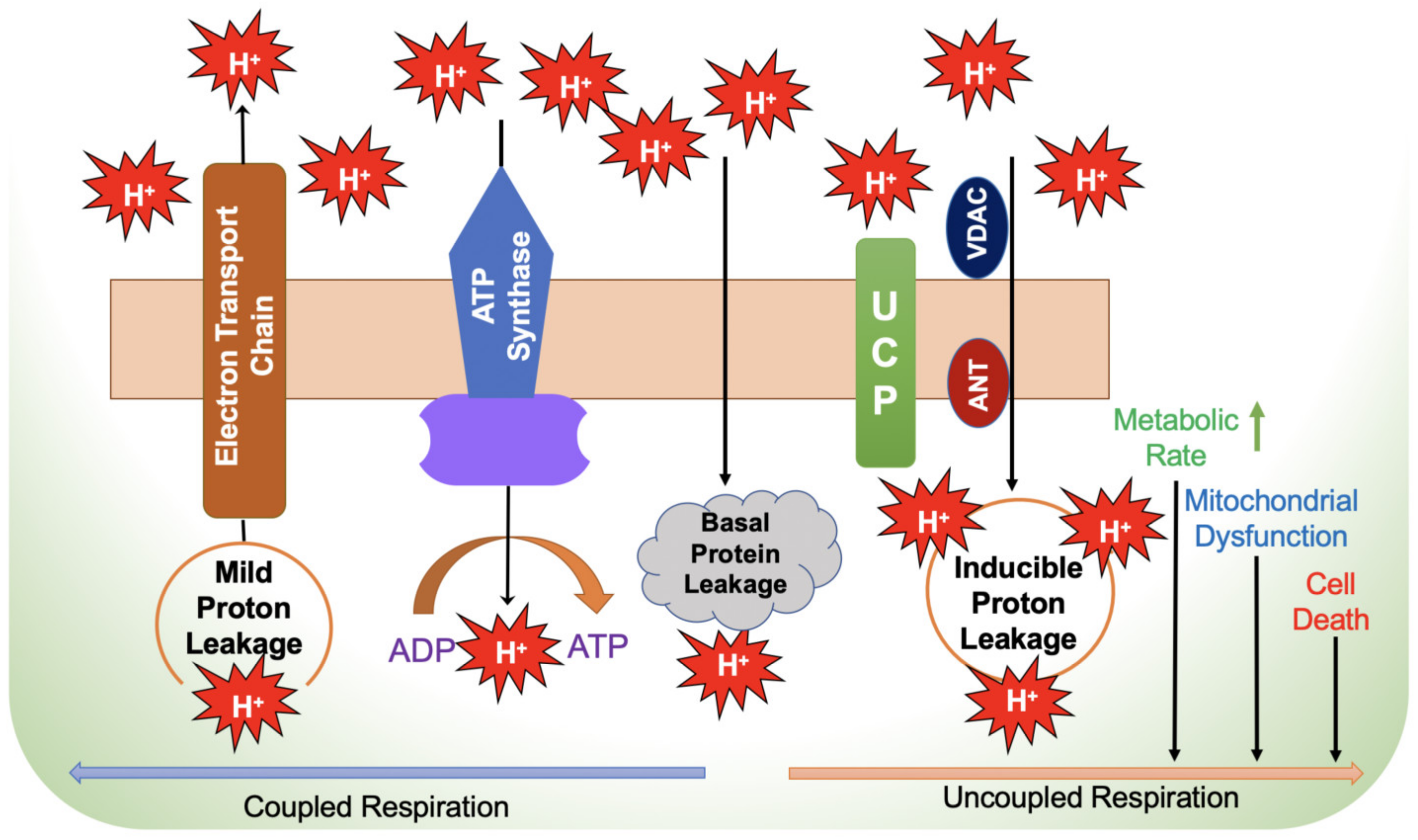
2.4. Electron Flux in the Electron Transport Chain, Electron Leakage and ROS Production
2.4.1. Electron Leakage and ROS Production
2.4.2. Blockade of ETC Complexes Suppresses ROS Generation
2.5. Failure of Liver ROS Clearance Capacity
2.5.1. Deregulation of ROS Homeostasis Causes Progressive Liver Injury
2.5.2. Imbalance of ROS Homeostasis Aggravates Inflammation in ALD
2.6. Mechanisms of Inefficient Respiratory Chain
2.7. Hepatic Tricarboxylic Acid (TCA) Cycle and Mitochondrial Respiratory Efficiency
2.8. Hepatic Inflammation
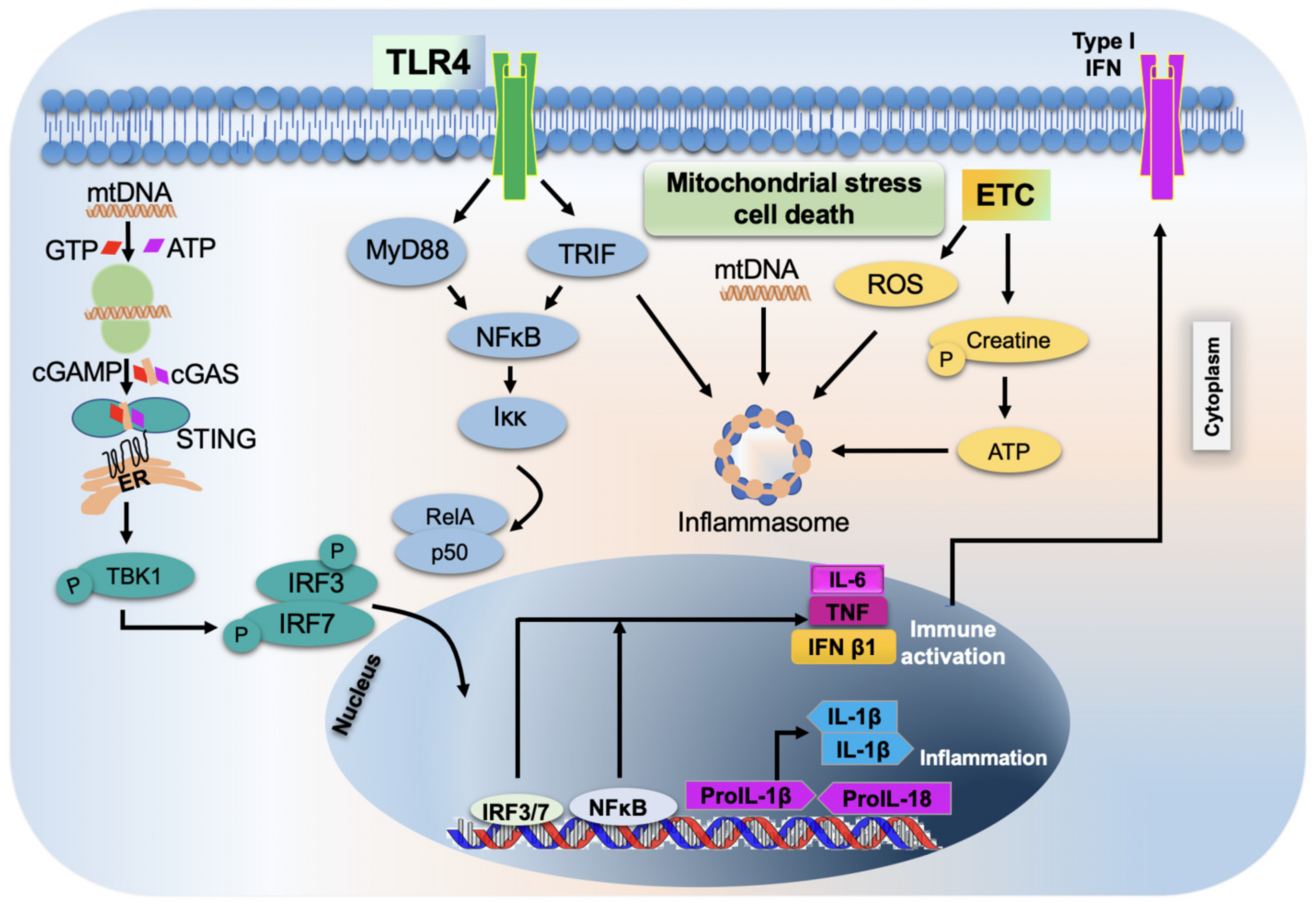
2.8.1. Role of Mitochondria in the Inflammatory Response
2.8.2. Activation of TLR9
2.8.3. Activation of Inflammasome by Mitochondria
2.8.4. Role of Mitochondria in the Activation and Release of Pro-Inflammatory Cytokines
2.8.5. Mitochondrial-Targeted Treatments for Inflammatory Responses
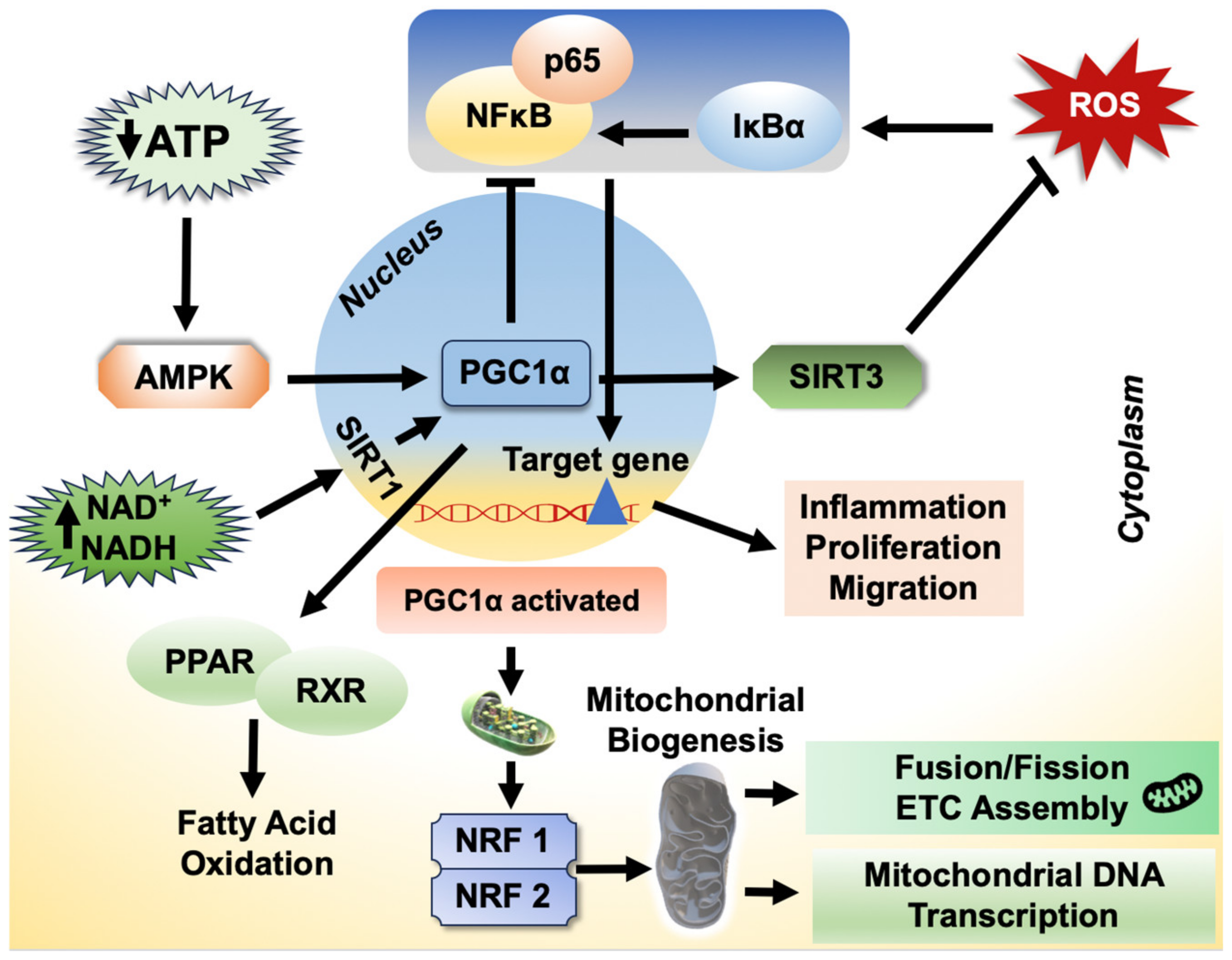
2.8.6. Role of PGC-1α on Inflammatory Response
2.9. Mitochondrial Protein Methylation—Fusion and Fission
2.10. The Insufficiency of Antioxidants and Impaired ROS Clearance
2.11. Potential Mitochondrial Mechanisms of More Conventional Liver Disease Treatments
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dave, D.T.; Patel, B.M. Mitochondrial metabolism in cancer cachexia: Novel drug target. Curr. Drug Metab. 2019, 20, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, G.K.; Pahwa, P.; Gupta, A.; Navik, U.S.; Reddy, P.H.; Bhatti, J.S. Targeting Mitochondria as a Novel Disease-Modifying Therapeutic Strategy in Cancer. In Handbook of Oxidative Stress in Cancer: Mechanistic Aspects; Springer: Berlin/Heidelberg, Germany, 2022; pp. 241–262. [Google Scholar]
- Mitochondrial Toxicity & Stunting Our Energy Production: An Epidemic of Mass Proportions? 2023. Available online: https://drjessmd.com/mitochondrial-toxicity-stunting-our-energy-production-an-epidemic-of-mass-proportions/ (accessed on 20 June 2023).
- Onishi, M.; Okamoto, K. Mitochondrial clearance: Mechanisms and roles in cellular fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. ITPRs/inositol 1,4,5-trisphosphate receptors in autophagy: From enemy to ally. Autophagy 2015, 11, 1944–1948. [Google Scholar] [CrossRef]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular nature and physiological role of the mitochondrial calcium uniporter channel. Am. J. Physiol. Cell Physiol. 2021, 320, C465–C482. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Sheu, S.S. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim. Biophys. Acta 2009, 1787, 1291–1308. [Google Scholar] [CrossRef]
- Tomar, D.; Jaña, F.; Dong, Z.; Quinn, W.J., 3rd; Jadiya, P.; Breves, S.L.; Daw, C.C.; Srikantan, S.; Shanmughapriya, S.; Nemani, N.; et al. Blockade of MCU-mediated Ca(2+) uptake perturbs lipid metabolism via PP4-dependent AMPK dephosphorylation. Cell Rep. 2019, 26, 3709–3725.e3707. [Google Scholar] [CrossRef]
- Oliva-Vilarnau, N.; Hankeova, S.; Vorrink, S.U.; Mkrtchian, S.; Andersson, E.R.; Lauschke, V.M. Calcium signaling in liver injury and regeneration. Front. Med. 2018, 5, 192. [Google Scholar] [CrossRef]
- Chernikov, A.; Krapivin, A.; Khazanov, V.; Kuz’menko, D.; VIu, S.; Udut, V. Effect of the energy metabolism regulator Yantar-Antitox on the system of energy production in rat liver during experimental pathology of beta-oxidation. Eksperimental’naia I Klin. Farmakol. 2012, 75, 24–27. [Google Scholar]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondrial regulation of cell death: A phylogenetically conserved control. Microb. Cell 2016, 3, 101. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Torpey, J.; Madine, J.; Wood, A.; Lian, L.Y. Cyclophilin D binds to the acidic C-terminus region of α-Synuclein and affects its aggregation characteristics. Sci. Rep. 2020, 10, 10159. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. mPTP opening caused by Cdk5 loss is due to increased mitochondrial Ca(2+) uptake. Oncogene 2020, 39, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Yumnam, S.; Hong, G.E.; Raha, S.; Saralamma, V.V.; Lee, H.J.; Lee, W.S.; Kim, E.H.; Kim, G.S. Mitochondrial dysfunction and Ca(2+) Overload contributes to hesperidin induced paraptosis in hepatoblastoma cells, HepG2. J. Cell Physiol. 2016, 231, 1261–1268. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis regulators Bcl-2 and caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Castilho, R.F.; Meinicke, A.R.; Almeida, A.M.; Hermes-Lima, M.; Vercesi, A.E. Oxidative damage of mitochondria induced by Fe(II)citrate is potentiated by Ca2+ and includes lipid peroxidation and alterations in membrane proteins. Arch. Biochem. Biophys. 1994, 308, 158–163. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Nasr, P.; Bottai, M.; Ekstedt, M.; Kechagias, S.; Hultcrantz, R.; Stål, P. Elevated serum ferritin is associated with increased mortality in non-alcoholic fatty liver disease after 16 years of follow-up. Liver Int. 2016, 36, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Petta, S.; Manuguerra, R.; Luong, T.V.; Cabibi, D.; Corradini, E.; Craxì, A.; Pinzani, M.; Tsochatzis, E.; Pietrangelo, A. Evaluating the association of serum ferritin and hepatic iron with disease severity in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, R.; Yang, S.; Ma, X.; Yu, C. Association between serum ferritin level and the various stages of non-alcoholic fatty liver disease: A systematic review. Front. Med. 2022, 9, 934989. [Google Scholar] [CrossRef] [PubMed]
- Imeryuz, N.; Tahan, V.; Sonsuz, A.; Eren, F.; Uraz, S.; Yuksel, M.; Akpulat, S.; Ozcelik, D.; Haklar, G.; Celikel, C.; et al. Iron preloading aggravates nutritional steatohepatitis in rats by increasing apoptotic cell death. J. Hepatol. 2007, 47, 851–859. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Motta, B.M.; Canavesi, E.; Pelusi, S.; Pulixi, E.A.; Fracanzani, A.L.; Fargion, S. The A736V TMPRSS6 polymorphism influences hepatic iron overload in nonalcoholic fatty liver disease. PLoS ONE 2012, 7, e48804. [Google Scholar] [CrossRef]
- Cornejo, P.; Varela, P.; Videla, L.A.; Fernández, V. Chronic iron overload enhances inducible nitric oxide synthase expression in rat liver. Nitric Oxide 2005, 13, 54–61. [Google Scholar] [CrossRef]
- Cornejo, P.; Fernández, V.; Vial, M.T.; Videla, L.A. Hepatoprotective role of nitric oxide in an experimental model of chronic iron overload. Nitric Oxide 2007, 16, 143–149. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Ge, C.; Min, J.; Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022, 29, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Pratte, J.; Giardina, C. Ferroptosis and its potential as a therapeutic target. Biochem. Pharmacol. 2021, 186, 114486. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, L.; Iorga, A.; Dara, L. Cell death in liver diseases: A review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.; Wu, Q.; Fang, X.; Duan, L.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef]
- Lőrincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef]
- Niu, B.; Lei, X.; Xu, Q.; Ju, Y.; Xu, D.; Mao, L.; Li, J.; Zheng, Y.; Sun, N.; Zhang, X.; et al. Protecting mitochondria via inhibiting VDAC1 oligomerization alleviates ferroptosis in acetaminophen-induced acute liver injury. Cell Biol. Toxicol. 2022, 38, 505–530. [Google Scholar] [CrossRef]
- Tomah, S.; Alkhouri, N.; Hamdy, O. Nonalcoholic fatty liver disease and type 2 diabetes: Where do Diabetologists stand? Clin. Diabetes Endocrinol. 2020, 6, 9. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Eapen, C.E.; Pullimood, A.B.; Balasubramanian, K.A. Oxidative stress in experimental liver microvesicular steatosis: Role of mitochondria and peroxisomes. J. Gastroenterol. Hepatol. 2006, 21, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, J.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Lago, N.R.; Pirola, C.J.; Sookoian, S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J. Cell Mol. Med. 2011, 15, 1329–1338. [Google Scholar] [CrossRef]
- Garcia, J.; Decker, C.W.; Sanchez, S.J.; Ouk, J.M.; Siu, K.M.; Han, D. Obesity and steatosis promotes mitochondrial remodeling that enhances respiratory capacity in the liver of ob/ob mice. FEBS Lett. 2018, 592, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E311–E319. [Google Scholar] [CrossRef]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustín, P.; Ramos, E.; Navarro, E.; Parada, E.; Sánchez-López, N.; Peláez-Aguado, L.; Cabrera-García, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, A.S.; Bayley, J.P. The role of complex II in disease. Biochim. Biophys. Acta 2013, 1827, 543–551. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Siebels, I.; Dröse, S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta 2013, 1827, 1156–1164. [Google Scholar] [CrossRef]
- Turrens, F.T.; Alexandre, A.; Lehninger, L.L. Reprint of: Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 2022, 726, 109232. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J. Reactive oxygen species and alpha,beta-unsaturated aldehydes as second messengers in signal transduction. Ann. NY Acad. Sci. 2010, 1203, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 1, 1–13. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic liver disease: Pathogenesis and current management. Alcohol. Res. 2017, 38, 147–161. [Google Scholar] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Grunhut, J.; Wang, W.; Aykut, B.; Gakhal, I.; Torres-Hernandez, A.; Miller, G. Macrophages in nonalcoholic steatohepatitis: Friend or foe? Eur. Med. J. Hepatol. 2018, 6, 100–109. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular pathogenesis of nonalcoholic steatohepatitis- (NASH-) Related hepatocellular carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef]
- Perez, M.J.; Cederbaum, A.I. Proteasome inhibition potentiates CYP2E1-mediated toxicity in HepG2 cells. Hepatology 2003, 37, 1395–1404. [Google Scholar] [CrossRef]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in alcoholic and non-alcoholic liver injury. Roles of ROS, reactive intermediates and lipid overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Mansouri, A.; Fromenty, B.; Berson, A.; Robin, M.A.; Grimbert, S.; Beaugrand, M.; Erlinger, S.; Pessayre, D. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J. Hepatol. 1997, 27, 96–102. [Google Scholar] [CrossRef]
- Mansouri, A.; Gaou, I.; De Kerguenec, C.; Amsellem, S.; Haouzi, D.; Berson, A.; Moreau, A.; Feldmann, G.; Lettéron, P.; Pessayre, D.; et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999, 117, 181–190. [Google Scholar] [CrossRef]
- Demeilliers, C.; Maisonneuve, C.; Grodet, A.; Mansouri, A.; Nguyen, R.; Tinel, M.; Lettéron, P.; Degott, C.; Feldmann, G.; Pessayre, D.; et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology 2002, 123, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- Léveillé, M.; Estall, J.L. Mitochondrial dysfunction in the transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The efficiency and plasticity of mitochondrial energy transduction. Biochem. Soc. Trans. 2005, 33, 897–904. [Google Scholar] [CrossRef]
- Eyenga, P.; Rey, B.; Eyenga, L.; Sheu, S.S. Regulation of oxidative phosphorylation of liver mitochondria in Sepsis. Cells 2022, 11, 1598. [Google Scholar] [CrossRef] [PubMed]
- Eyenga, P.; Roussel, D.; Morel, J.; Rey, B.; Romestaing, C.; Gueguen-Chaignon, V.; Sheu, S.S.; Viale, J.P. Time course of liver mitochondrial function and intrinsic changes in oxidative phosphorylation in a rat model of sepsis. Intensive Care Med. Exp. 2018, 6, 31. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging role of hepatic ketogenesis in fatty liver disease. Front. Physiol. 2022, 13, 946474. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- d’Avignon, D.A.; Puchalska, P.; Ercal, B.; Chang, Y.; Martin, S.E.; Graham, M.J.; Patti, G.J.; Han, X.; Crawford, P.A. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight 2018, 3, e99762. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E163–E173. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Basso, P.J.; Andrade-Oliveira, V.; Câmara, N.O.S. Targeting immune cell metabolism in kidney diseases. Nat. Rev. Nephrol. 2021, 17, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate immunity, inflammation and tumour progression: Double-edged swords. J. Intern. Med. 2019, 285, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Zengeler, K.E.; Lukens, J.R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 2021, 21, 454–468. [Google Scholar] [CrossRef]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Hoffmann, J.A.; Galluzzi, L. Pharmacological modulation of nucleic acid sensors—Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2019, 18, 845–867. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging role of mitochondrial dna as a major driver of inflammation and disease progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef]
- Cardon, L.R.; Burge, C.; Clayton, D.A.; Karlin, S. Pervasive CpG suppression in animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 1994, 91, 3799–3803. [Google Scholar] [CrossRef] [PubMed]
- Pollack, Y.; Kasir, J.; Shemer, R.; Metzger, S.; Szyf, M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984, 12, 4811–4824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Imajo, M.; Tsuchiya, Y.; Nishida, E. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life 2006, 58, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, L.; Gornati, L.; Artuso, I.; Zanoni, I.; Granucci, F. Below the surface: The inner lives of TLR4 and TLR9. J. Leukoc. Biol. 2019, 106, 147–160. [Google Scholar] [CrossRef]
- Kopp, E.; Medzhitov, R. Recognition of microbial infection by Toll-like receptors. Curr. Opin. Immunol. 2003, 15, 396–401. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Tumurkhuu, G.; Shimada, K.; Dagvadorj, J.; Crother, T.R.; Zhang, W.; Luthringer, D.; Gottlieb, R.A.; Chen, S.; Arditi, M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res. 2016, 119, e76–e90. [Google Scholar] [CrossRef]
- Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The role of mitochondrial dysfunction in the progression of alzheimer’s disease. Curr. Med. Chem. 2018, 25, 5578–5587. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Dhanwani, R.; Takahashi, M.; Sharma, S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 2018, 50, 82–87. [Google Scholar] [CrossRef]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Miao, E.A.; Ting, J.P. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 2013, 39, 432–441. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef]
- Schmidts, A.; Wehrli, M.; Maus, M.V. Toward better understanding and management of CAR-T cell-associated toxicity. Annu. Rev. Med. 2021, 72, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Glab, J.A.; Cao, Z.; Puthalakath, H. Bcl-2 family proteins, beyond the veil. Int. Rev. Cell Mol. Biol. 2020, 351, 1–22. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Lopez, A.; Reyna, D.E.; Gitego, N.; Kopp, F.; Zhou, H.; Miranda-Roman, M.A.; Nordstrøm, L.U.; Narayanagari, S.R.; Chi, P.; Vilar, E.; et al. Co-targeting of BAX and BCL-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat. Commun. 2022, 13, 1199. [Google Scholar] [CrossRef]
- Reyna, D.E.; Garner, T.P.; Lopez, A.; Kopp, F.; Choudhary, G.S.; Sridharan, A.; Narayanagari, S.R.; Mitchell, K.; Dong, B.; Bartholdy, B.A.; et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 2017, 32, 490–505.e410. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Peña-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e939. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Galluzzi, L. BAX and BAK dynamics control mitochondrial DNA release during apoptosis. Cell Death Differ. 2022, 29, 1296–1298. [Google Scholar] [CrossRef]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1α as a pivotal factor in lipid and metabolic regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Léveillé, M.; Besse-Patin, A.; Jouvet, N.; Gunes, A.; Sczelecki, S.; Jeromson, S.; Khan, N.P.; Baldwin, C.; Dumouchel, A.; Correia, J.C.; et al. PGC-1α isoforms coordinate to balance hepatic metabolism and apoptosis in inflammatory environments. Mol. Metab. 2020, 34, 72–84. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega Á, L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, K.; Liu, C.; Hu, X.; Que, W.; Ito, H.; Takahashi, K.; Nakajima, M.; Tanaka, T.; Ren, K.; et al. 5-Aminolevulinic acid combined with sodium ferrous citrate (5-ALA/SFC) ameliorated liver injury in a murine acute graft-versus-host disease model by reducing inflammation responses through PGC1-α activation. Drug Discov. Ther. 2021, 14, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Sheng, H.; Bai, Y.F.; Weng, Y.Y.; Fan, X.Y.; Lou, L.J.; Zhang, F. Neohesperidin enhances PGC-1α-mediated mitochondrial biogenesis and alleviates hepatic steatosis in high fat diet fed mice. Nutr. Diabetes 2020, 10, 27. [Google Scholar] [CrossRef]
- Wan, X.; Zhu, X.; Wang, H.; Feng, Y.; Zhou, W.; Liu, P.; Shen, W.; Zhang, L.; Liu, L.; Li, T.; et al. PGC1α protects against hepatic steatosis and insulin resistance via enhancing IL10-mediated anti-inflammatory response. FASEB J. 2020, 34, 10751–10761. [Google Scholar] [CrossRef]
- Moreno, R.; Sobotzik, J.M.; Schultz, C.; Schmitz, M.L. Specification of the NF-kappaB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res. 2010, 38, 6029–6044. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 12, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, K.G.; Yoo, E.K.; Kim, Y.H.; Kim, Y.N.; Kim, H.S.; Kim, H.T.; Park, J.Y.; Lee, K.U.; Jang, W.G.; et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid. Redox Signal. 2007, 9, 301–307. [Google Scholar] [CrossRef]
- Brault, J.J.; Jespersen, J.G.; Goldberg, A.L. Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J. Biol. Chem. 2010, 285, 19460–19471. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T. Arginine methylation at a glance. J. Cell Sci. 2007, 120, 4243–4246. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Drozak, J. Protein histidine methylation. Curr. Protein Pept. Sci. 2020, 21, 675–689. [Google Scholar] [CrossRef]
- Małecki, J.M.; Davydova, E.; Falnes, P. Protein methylation in mitochondria. J. Biol. Chem. 2022, 298, 101791. [Google Scholar] [CrossRef]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Human METTL12 is a mitochondrial methyltransferase that modifies citrate synthase. FEBS Lett. 2017, 591, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Orekhov, A.N. Mitochondrial lipid homeostasis at the crossroads of liver and heart diseases. Int. J. Mol. Sci. 2021, 22, 6949. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Zhang, I.W.; López-Vicario, C.; Duran-Güell, M.; Clària, J. Mitochondrial dysfunction in advanced liver disease: Emerging concepts. Front. Mol. Biosci. 2021, 8, 772174. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Zhou, Y.; Long, D.; Zhao, Y.; Li, S.; Liang, Y.; Wan, L.; Zhang, J.; Xue, F.; Feng, L. Oxidative stress-mediated mitochondrial fission promotes hepatic stellate cell activation via stimulating oxidative phosphorylation. Cell Death Dis. 2022, 13, 689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, T.E.; Chen, M.; Xu, D.; Zhu, Y.; Hu, B.Y.; Lin, Z.F.; Pan, J.J.; Wang, X.; Wu, C.; et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer 2020, 122, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Umbaugh, D.S.; Jaeschke, H. Mitochondrial dynamics in drug-induced liver injury. Livers 2021, 1, 102–115. [Google Scholar] [CrossRef]
- Wei, Y.; Clark, S.E.; Thyfault, J.P.; Uptergrove, G.M.; Li, W.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am. J. Pathol. 2009, 174, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Casas-Grajales, S.; Muriel, P. Antioxidants in liver health. World J. Gastrointest. Pharmacol. Ther. 2015, 6, 59–72. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Jee, J.P.; Lim, S.J.; Park, J.S.; Kim, C.K. Stabilization of all-trans retinol by loading lipophilic antioxidants in solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2006, 63, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Jang, S.Y.; Kang, H.T.; Wei, B.; Jeoun, U.W.; Yoon, G.S.; Hwang, E.S. Modulation of mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and mitophagy. Mol. Cells. 2017, 40, 503–514. [Google Scholar] [CrossRef]
- Zozina, V.I.; Covantev, S.; Goroshko, O.A.; Krasnykh, L.M.; Kukes, V.G. Coenzyme Q10 in cardiovascular and metabolic diseases: Current state of the problem. Curr. Cardiol. Rev. 2018, 14, 164–174. [Google Scholar] [CrossRef]
- Barros, A.I.; Nunes, F.M.; Gonçalves, B.; Bennett, R.N.; Silva, A.P. Effect of cooking on total vitamin C contents and antioxidant activity of sweet chestnuts (Castanea sativa Mill.). Food Chem. 2011, 128, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Minich, D.M.; Brown, B.I. A review of dietary (phyto)nutrients for glutathione support. Nutrients 2019, 11, 2073. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Schenker, S.; Henderson, G.I. 4-hydroxynonenal detoxification by mitochondrial glutathione S-transferase is compromised by short-term ethanol consumption in rats. Alcohol. Clin. Exp. Res. 2002, 26, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Checa, J.C.; Hirano, T.; Tsukamoto, H.; Kaplowitz, N. Mitochondrial glutathione depletion in alcoholic liver disease. Alcohol 1993, 10, 469–475. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Amorim, R.; Teixeira, J.; Karkucinska-Wieckowska, A.; Carvalho, A.; Pereira, S.P.; Simões, R.F.; Szymanska, S.; Dąbrowski, M.; Janikiewicz, J.; et al. The alterations of mitochondrial function during NAFLD progression-An independent effect of mitochondrial ROS production. Int. J. Mol. Sci. 2021, 22, 6848. [Google Scholar] [CrossRef] [PubMed]
- García-Berumen, C.I.; Vargas-Vargas, M.A.; Ortiz-Avila, O.; Piña-Zentella, R.M.; Ramos-Gómez, M.; Figueroa-García, M.D.C.; Mejía-Zepeda, R.; Rodríguez-Orozco, A.R.; Saavedra-Molina, A.; Cortés-Rojo, C. Avocado oil alleviates non-alcoholic fatty liver disease by improving mitochondrial function, oxidative stress and inflammation in rats fed a high fat-High fructose diet. Front. Pharmacol. 2022, 13, 1089130. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Avila, O.; Gallegos-Corona, M.A.; Sánchez-Briones, L.A.; Calderón-Cortés, E.; Montoya-Pérez, R.; Rodriguez-Orozco, A.R.; Campos-García, J.; Saavedra-Molina, A.; Mejía-Zepeda, R.; Cortés-Rojo, C. Protective effects of dietary avocado oil on impaired electron transport chain function and exacerbated oxidative stress in liver mitochondria from diabetic rats. J. Bioenerg. Biomembr. 2015, 47, 337–353. [Google Scholar] [CrossRef]
- Anfuso, B.; Giraudi, P.J.; Tiribelli, C.; Rosso, N. Silybin modulates collagen turnover in an in vitro model of NASH. Molecules 2019, 24, 1280. [Google Scholar] [CrossRef]
- Vecchione, G.; Grasselli, E.; Cioffi, F.; Baldini, F.; Oliveira, P.J.; Sardão, V.A.; Cortese, K.; Lanni, A.; Voci, A.; Portincasa, P.; et al. The nutraceutic silybin counteracts excess lipid accumulation and ongoing oxidative stress in an in vitro model of non-alcoholic fatty liver disease progression. Front. Nutr. 2017, 4, 42. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.J.; Shang, L.C.; Zhang, Y.H.; Zhou, Y.; Shi, X.L. Puerarin protects against high-fat high-sucrose diet-induced non-alcoholic fatty liver disease by modulating PARP-1/PI3K/AKT signaling pathway and facilitating mitochondrial homeostasis. Phytother. Res. 2019, 33, 2347–2359. [Google Scholar] [CrossRef]
- Fang, K.; Wu, F.; Chen, G.; Dong, H.; Li, J.; Zhao, Y.; Xu, L.; Zou, X.; Lu, F. Diosgenin ameliorates palmitic acid-induced lipid accumulation via AMPK/ACC/CPT-1A and SREBP-1c/FAS signaling pathways in LO2 cells. BMC Complement. Altern. Med. 2019, 19, 255. [Google Scholar] [CrossRef]
- Guo, X.L.; Liang, B.; Wang, X.W.; Fan, F.G.; Jin, J.; Lan, R.; Yang, J.H.; Wang, X.C.; Jin, L.; Cao, Q. Glycyrrhizic acid attenuates CCl₄-induced hepatocyte apoptosis in rats via a p53-mediated pathway. World J. Gastroenterol. 2013, 19, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Gumpricht, E.; Dahl, R.; Devereaux, M.W.; Sokol, R.J. Licorice compounds glycyrrhizin and 18beta-glycyrrhetinic acid are potent modulators of bile acid-induced cytotoxicity in rat hepatocytes. J. Biol. Chem. 2005, 280, 10556–10563. [Google Scholar] [CrossRef]
- Jin, X.; Li, L.; Peng, Q.; Gan, C.; Gao, L.; He, S.; Tan, S.; Pu, W.; Liu, Y.; Gong, Y.; et al. Glycyrrhetinic acid restricts mitochondrial energy metabolism by targeting SHMT2. iScience 2022, 25, 104349. [Google Scholar] [CrossRef]
- Arumugam, M.K.; Paal, M.C.; Donohue, T.M., Jr.; Ganesan, M.; Osna, N.A.; Kharbanda, K.K. Beneficial effects of betaine: A comprehensive review. Biology 2021, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.K.; Chava, S.; Perumal, S.K.; Paal, M.C.; Rasineni, K.; Ganesan, M.; Donohue, T.M., Jr.; Osna, N.A.; Kharbanda, K.K. Acute ethanol-induced liver injury is prevented by betaine administration. Front. Physiol. 2022, 13, 940148. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.K.; Perumal, S.K.; Rasineni, K.; Donohue, T.M., Jr.; Osna, N.A.; Kharbanda, K.K. Lipidomic analysis of liver lipid droplets after chronic alcohol consumption with and without betaine supplementation. Biology 2023, 12, 462. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Angulo, P.; Jorgensen, R.A.; Sylvestre, P.B.; Lindor, K.D. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: Results of a pilot study. Am. J. Gastroenterol. 2001, 96, 2711–2717. [Google Scholar] [CrossRef]
- Kawakami, S.; Han, K.H.; Nakamura, Y.; Shimada, K.; Kitano, T.; Aritsuka, T.; Nagura, T.; Ohba, K.; Nakamura, K.; Fukushima, M. Effects of dietary supplementation with betaine on a nonalcoholic steatohepatitis (NASH) mouse model. J. Nutr. Sci. Vitaminol. 2012, 58, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Heidari, R.; Niknahad, H.; Sadeghi, A.; Mohammadi, H.; Ghanbarinejad, V.; Ommati, M.M.; Hosseini, A.; Azarpira, N.; Khodaei, F.; Farshad, O.; et al. Betaine treatment protects liver through regulating mitochondrial function and counteracting oxidative stress in acute and chronic animal models of hepatic injury. Biomed. Pharmacother. 2018, 103, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; King, A.L.; Osna, N.A.; McVicker, B.L.; Tuma, D.J.; Wisecarver, J.L.; Bailey, S.M. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int. J. Hepatol. 2012, 2012, 962183. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Mailliard, M.E.; Baldwin, C.R.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 2007, 46, 314–321. [Google Scholar] [CrossRef]
- Varatharajalu, R.; Garige, M.; Leckey, L.C.; Arellanes-Robledo, J.; Reyes-Gordillo, K.; Shah, R.; Lakshman, M.R. Adverse signaling of scavenger receptor class B1 and PGC1s in alcoholic hepatosteatosis and steatohepatitis and protection by betaine in rat. Am. J. Pathol. 2014, 184, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J. Betaine enhances the cellular survival via mitochondrial fusion and fission factors, MFN2 and DRP1. Anim. Cells Syst. 2018, 22, 289–298. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Characterization | Regulation | Abnormalities | References |
|---|---|---|---|---|
| Hepatic iron overload | Influences the conversion of hydrogen peroxide (H2O2) to highly toxic hydroxyl radicals (HO•). | Hepcidin is feedback-regulated by iron concentrations in plasma and the liver and by erythropoietic demand for iron. | ROS accumulation, cytotoxicity and oxidative stress. | [25] |
| Ferritin overload | Primary intracellular iron storage protein in both prokaryotes and eukaryotes. | Participates in oxidation–reduction, iron ion transport across membranes and cellular iron ion homeostasis. | Hemochromatosis or hemosiderosis. | [26,27] |
| Chronic iron overload | Enhances iNOS synthase. | Activation of extracellular signal-regulated kinase (ERK1/2) and nuclear transcription factor (NFκB) in the liver. | Liver steatosis and fibrosis. | [28,29] |
| Ferroptosis | Intracellular iron-dependent form of cell death that is distinct from necrosis and autophagy. | Accumulation of lipid peroxides. | Neurological dysfunction and cell death. | [36] |
| Voltage-dependent anion channel 1 (VDAC) in outer mitochondrial membrane | Cellular Ca2+ homeostasis by mediating the transport of Ca2+ in and out of mitochondria. VDAC1 is highly Ca2+-permeable and modulates Ca2+ access to the mitochondrial intermembrane space. | Mitochondria-mediated apoptosis by the release of apoptotic proteins. | Increase in calcium into the mitochondria leads to apoptosis. | [5,6] |
| VDAC oligomerization | VDAC oligomerization inducing mitochondrial outer membrane permeabilization causing mtDNA release. | Mitochondrial stress releases mtDNA into the cytosol, thereby triggering the type Ι interferon (IFN) response. | Regulates Ca2+ influx, metabolism, inflammasome activation and cell death. | [43] |
| Mitochondrial calcium uniporter | Transmembrane protein that allows for the passage of calcium ions from cytosol into mitochondria. | Regulated by MICU1 and MICU2 and plays a fundamental role in the shaping of global calcium signaling and in the control of aerobic metabolism, as well as apoptosis. | Oxidative stress-elevated mitochondrial calcium and its function in neurodegenerative disorders. Hepatic lipid accumulation through MCU/PP4/AMPK molecular signaling mechanism. | [8] |
| Depolarization of the inner mitochondrial membrane mediated | Allows for antioxidant molecules to exit mitochondria, reducing the organelles’ ability to neutralize ROS. | Caspase-mediated apoptosis. | Increased ROS production. | [13] |
| Paraptosis, a non-apoptotic type of programmed cell death | Non-apoptotic type of programmed cell death. | Fragmentation of DNA and caspase activation, cell death occurring by cytoplasmic vacuolation. | Mitochondrial swelling. | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arumugam, M.K.; Gopal, T.; Kalari Kandy, R.R.; Boopathy, L.K.; Perumal, S.K.; Ganesan, M.; Rasineni, K.; Donohue, T.M., Jr.; Osna, N.A.; Kharbanda, K.K. Mitochondrial Dysfunction-Associated Mechanisms in the Development of Chronic Liver Diseases. Biology 2023, 12, 1311. https://doi.org/10.3390/biology12101311
Arumugam MK, Gopal T, Kalari Kandy RR, Boopathy LK, Perumal SK, Ganesan M, Rasineni K, Donohue TM Jr., Osna NA, Kharbanda KK. Mitochondrial Dysfunction-Associated Mechanisms in the Development of Chronic Liver Diseases. Biology. 2023; 12(10):1311. https://doi.org/10.3390/biology12101311
Chicago/Turabian StyleArumugam, Madan Kumar, Thiyagarajan Gopal, Rakhee Rathnam Kalari Kandy, Lokesh Kumar Boopathy, Sathish Kumar Perumal, Murali Ganesan, Karuna Rasineni, Terrence M. Donohue, Jr., Natalia A. Osna, and Kusum K. Kharbanda. 2023. "Mitochondrial Dysfunction-Associated Mechanisms in the Development of Chronic Liver Diseases" Biology 12, no. 10: 1311. https://doi.org/10.3390/biology12101311