

Genetic Signature of Pinctada fucata Inferred from Population Genomics: Source Tracking of the Invasion in Mischief Reef of Nansha Islands
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
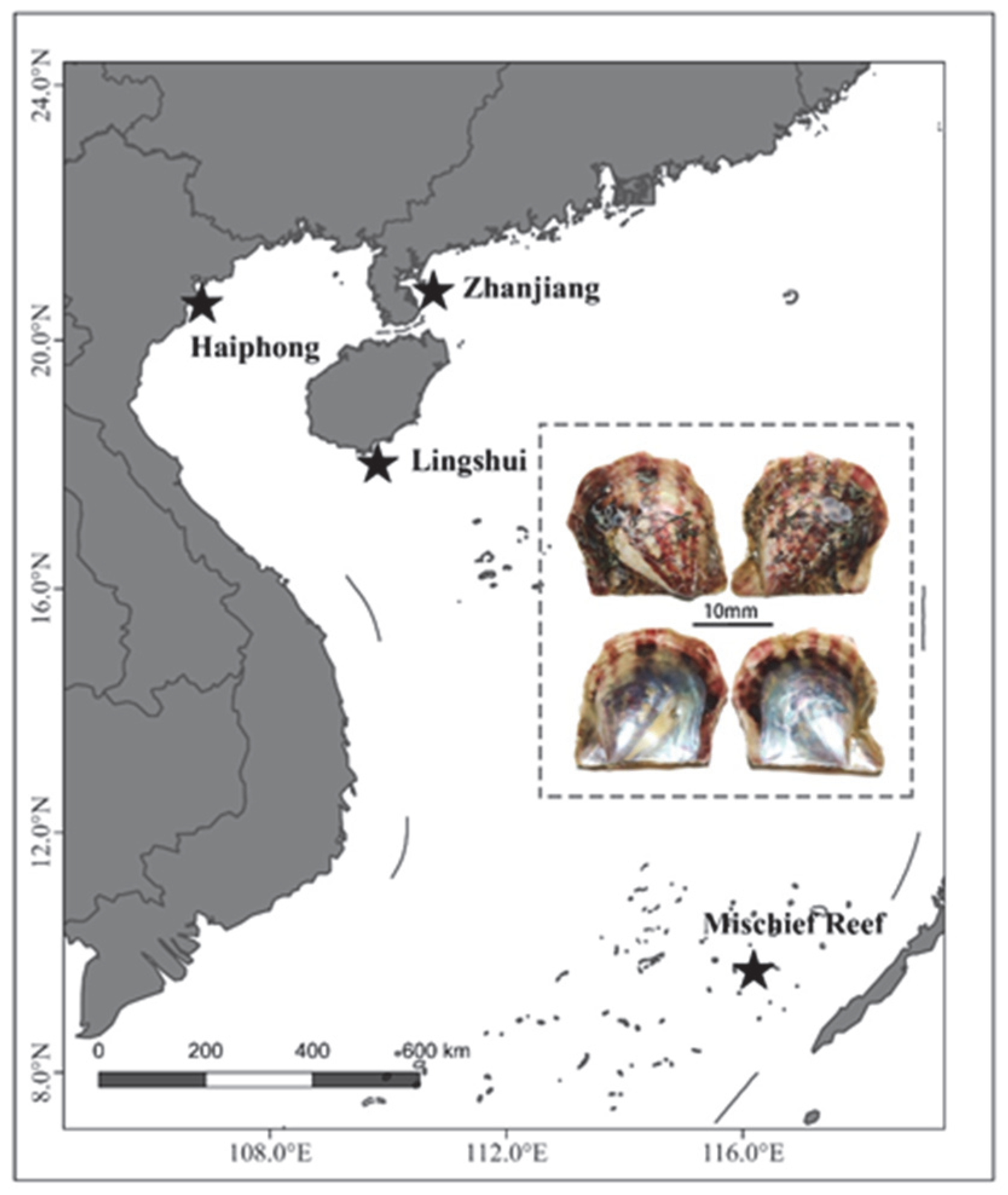
2. Materials and Methods
2.1. Sampling and Sequencing
2.2. Alignment and Variation Calling
2.3. Population Genetics Analysis
2.4. Potential Selected Regions in the Introduced Population
3. Results
3.1. Variant Calling and Population Genetic Diversity
3.2. Population Genetic Structure
3.3. Population Genetic Structure
4. Discussion
4.1. Population Genetic Structure and the Potential Origin of the Mischief Reef Population
4.2. Population Genetic Diversity and LD in P. fucata Populations
4.3. Potential Selected Regions in the Introduced Population
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kappel, C.V. Losing pieces of the puzzle: Threats to marine, estuarine, and diadromous species. Front. Ecol. Environ. 2005, 3, 275–282. [Google Scholar] [CrossRef]
- Costello, M.J.; Coll, M.; Danovaro, R.; Halpin, P.; Ojaveer, H.; Miloslavich, P. A Census of Marine Biodiversity Knowledge, Resources, and Future Challenges. PLoS ONE 2010, 5, e12110. [Google Scholar] [CrossRef] [Green Version]
- Ojaveer, H.; Galil, B.S.; Carlton, J.T.; Alleway, H.; Goulletquer, P.; Lehtiniemi, M.; Marchini, A.; Miller, W.; Occhipinti-Ambrogi, A.; Peharda, M.; et al. Historical baselines in marine bioinvasions: Implications for policy and management. PLoS ONE 2018, 13, e0202383. [Google Scholar] [CrossRef] [Green Version]
- Kolar, C.S.; Lodge, D.M. Progress in invasion biology: Predicting invaders. Trends Ecol. Evol. 2001, 16, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Katsanevakis, S.; Moustakas, A. Uncertainty in Marine Invasion Science. Front. Mar. Sci. 2018, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Seebens, H.; Blackburn, T.M.; Dyer, E.E.; Genovesi, P.; Hulme, P.E.; Jeschke, J.M.; Pagad, S.; Pyšek, P.; Winter, M.; Arianoutsou, M.; et al. No saturation in the accumulation of alien species worldwide. Nat. Commun. 2017, 8, 14435. [Google Scholar] [CrossRef]
- Werschkun, B.; Banerji, S.; Basurko, O.C.; David, M.; Fuhr, F.; Gollasch, S.; Grummt, T.; Haarich, M.; Jha, A.N.; Kacan, S.; et al. Emerging risks from ballast water treatment: The run-up to the International Ballast Water Management Convention. Chemosphere 2014, 112, 256–266. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.T. The Pearl Oyster Pinctada fucata (Gould) (Family Pteriidae)//Estuarine and Marine Bivalve Mollusk Culture; CRC Press: Boca Raton, FL, USA, 1991; pp. 245–260. [Google Scholar]
- Cunha, R.L.; Blanc, F.; Bonhomme, F.; Arnaud-Haond, S. Evolutionary Patterns in Pearl Oysters of the Genus Pinctada (Bivalvia: Pteriidae). Mar. Biotechnol. 2010, 13, 181–192. [Google Scholar] [CrossRef]
- Tomaru, Y.; Udaka, N.; Kawabata, Z.; Nakano, S.-I. Seasonal change of seston size distribution and phytoplankton composition in bivalve pearl oyster Pinctada fucata martensii culture farm. Hydrobiologia 2002, 481, 181–185. [Google Scholar] [CrossRef]
- Choi, Y.H.; Chang, Y.J. Gametogenic cycle of the transplanted-cultured pearl oyster, Pinctada fucata martensii (Bivalvia: Pteriidae) in Korea. Aquaculture 2003, 220, 781–790. [Google Scholar] [CrossRef]
- Yu, D.; Chu, K.; Jia, X. Preliminary analysis on genetic relationship of common pearl oysters of Pinctada in china. Oceanol. Et Limnol. Sin. 2006, 37, 211. [Google Scholar]
- Wang, Z.R.; Chen, R.Q. The species of the Pterioda from the Nansha Islands waters. In Studies on Marine Species of the Nasha Islands and Neighboring Waters; Chen, Q.C., Ed.; China Ocean Press: Beijing, China, 1991; pp. 150–151. [Google Scholar]
- Chen, Q.C. Studies on Marine Biodiversity of the Nasha Islands and Neighboring Waters II; China Ocean Press: Beijing, China, 1996; p. 101. [Google Scholar]
- Edelist, D.; Rilov, G.; Golani, D.; Carlton, J.T.; Spanier, E. Restructuring the Sea: Profound shifts in the world’s most invaded marine ecosystem. Divers. Distrib. 2013, 19, 69–77. [Google Scholar] [CrossRef]
- Cristescu, M.E. Genetic reconstructions of invasion history. Mol. Ecol. 2015, 24, 2212–2225. [Google Scholar] [CrossRef] [PubMed]
- Handley, L.J.L.; Estoup, A.; Evans, D.M.; Thomas, C.E.; Lombaert, E.; Facon, B.; Aebi, A.; Roy, H.E. Ecological genetics of invasive alien species. Entomophaga 2011, 56, 409–428. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, X.; Zhang, G.; Zhang, F. Classification of Jinjiang oysters Crassostrea rivularis (Gould, 1861) from China, based on morphology and phylogenetic analysis. Aquaculture 2004, 242, 137–155. [Google Scholar] [CrossRef]
- Muirhead, J.R.; Gray, D.K.; Kelly, D.W.; Ellis, S.M.; Heath, D.D.; Macisaac, H.J. Identifying the source of species invasions: Sampling intensity vs. genetic diversity. Mol. Ecol. 2008, 17, 1020–1035. [Google Scholar] [CrossRef]
- Darling, J.A.; Bagley, M.J.; Roman, J.; Tepolt, C.K.; Geller, J.B. Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol. Ecol. 2008, 17, 4992–5007. [Google Scholar] [CrossRef]
- Rius, M.; Turon, X.; Bernardi, G.; Volckaert, F.A.M.; Viard, F. Marine invasion genetics: From spatio-temporal patterns to evolutionary outcomes. Biol. Invasions 2014, 17, 869–885. [Google Scholar] [CrossRef] [Green Version]
- Troost, K. Causes and effects of a highly successful marine invasion: Case-study of the introduced Pacific oyster Crassostrea gigas in continental NW European estuaries. J. Sea Res. 2010, 64, 145–165. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todesco, M.; Owens, G.L.; Bercovich, N.; Légaré, J.-S.; Soudi, S.; Burge, D.O.; Huang, K.; Ostevik, K.L.; Drummond, E.B.M.; Imerovski, I.; et al. Massive haplotypes underlie ecotypic differentiation in sunflowers. Nature 2020, 584, 602–607. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watterson, G. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 3, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P.W. A standardized genetic differentiation measure. Evolution 2005, 59, 1633–1638. [Google Scholar]
- Jost, L.O.U. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [PubMed]
- Sundqvist, L.; Keenan, K.; Zackrisson, M.; Prodöhl, P.; Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016, 6, 3461–3475. [Google Scholar] [CrossRef] [PubMed]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.B. Population Genetics; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- McMahon, R.F. Evolutionary and physiological adaptations of aquatic invasive animals: R selection versus resistance. Can. J. Fish. Aquat. Sci. 2002, 59, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Sousa, R.; Mendes, T.; Borges, R.; Vilares, P.; Vasconcelos, V.; Guilhermino, L.; Antunes, A. Low Genetic Diversity and High Invasion Success of Corbicula fluminea (Bivalvia, Corbiculidae) (Müller, 1774) in Portugal. PLoS ONE 2016, 11, e0158108. [Google Scholar] [CrossRef] [Green Version]
- Sousa, R.; Gutiérrez, J.; Aldridge, D. Non-indigenous invasive bivalves as ecosystem engineers. Biol. Invasions 2009, 11, 2367–2385. [Google Scholar] [CrossRef]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The Population Biology of Invasive Species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef] [Green Version]
- Colautti, R.I.; MacIsaac, H.J. A neutral terminology to define ‘invasive’ species. Divers. Distrib. 2004, 10, 135–141. [Google Scholar] [CrossRef]
- Takeuchi, T.; Masaoka, T.; Aoki, H.; Koyanagi, R.; Fujie, M.; Satoh, N. Divergent northern and southern populations and demographic history of the pearl oyster in the western Pacific revealed with genomic SNPs. Evol. Appl. 2019, 13, 837–853. [Google Scholar] [CrossRef] [Green Version]
- Ficetola, G.F.; Bonin, A.; Miaud, C. Population genetics reveals origin and number of founders in a biological invasion. Mol. Ecol. 2008, 17, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.; Penone, C.; Pennino, M.G.; Courchamp, F. Predicting future invaders and future invasions. Proc. Natl. Acad. Sci. USA 2019, 116, 7905–7910. [Google Scholar] [CrossRef] [PubMed]
- Gwak, W.S.; Nakayama, K. Genetic variation of hatchery and wild stocks of the pearl oyster Pinctada fucata martensii (Dunker, 1872), assessed by mitochondrial DNA analysis. Aquac. Int. 2011, 19, 585–591. [Google Scholar] [CrossRef]
- Huang, J.; Xu, M.; He, M. Discriminant analysis of four cultured populations of the pearl oyster, Pinctada fucata martensii, using morphology and single-nucleotide polymorphism markers. J. World Aquac. Soc. 2019, 51, 1373–1385. [Google Scholar] [CrossRef]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, B.A.; Krueger-Hadfield, S.A.; Murren, C.J.; Nice, C.C.; Strand, A.E.; Sotka, E.E. Founder effects shape linkage disequilibrium and genomic diversity of a partially clonal invader. Mol. Ecol. 2021, 30, 1962–1978. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Hou, Z.; Lin, Y.; Lan, W.; Tan, F.; Xing, F.; Li, G.; Guo, W.; Huang, Y. Going with the flow: Analysis of population structure reveals high gene flow shaping invasion pattern and inducing range expansion of Mikania micrantha in Asia. Ann. Bot. 2020, 125, 1113–1126. [Google Scholar] [CrossRef]
- Duk, M.; Kanapin, A.; Rozhmina, T.; Bankin, M.; Surkova, S.; Samsonova, A.; Samsonova, M. The Genetic Landscape of Fiber Flax. Front. Plant Sci. 2021, 12, 764612. [Google Scholar] [CrossRef]
- Hill, W.G. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Bruen, T.C.; Philippe, H.; Bryant, D. A Simple and Robust Statistical Test for Detecting the Presence of Recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Juric, I.; Cosgrove, E.J.; Bowman, R.; Fitzpatrick, J.W.; Schoech, S.J.; Clark, A.G.; Coop, G. Allele frequency dynamics in a pedigreed natural population. Proc. Natl. Acad. Sci. USA 2018, 116, 2158–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, M.A.; Wahlberg, N.; van Eijk, M.; Brakefield, P.M.; Zwaan, B.J. Mitochondrial DNA Signature for Range-Wide Populations of Bicyclus anynana Suggests a Rapid Expansion from Recent Refugia. PLoS ONE 2011, 6, e21385. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, M.; Fu, Z.; Yang, J.; Zhou, S.; Yu, G.; Zhou, W.; Ma, Z. A Comparative Study on Low and High Salinity Tolerance of Two Strains of Pinctada fucata. Front. Mar. Sci. 2021, 8, 1039. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| He | Ho | θw | θπ | Tajima’s D | |
|---|---|---|---|---|---|
| Mischief | 0.2377 | 0.0942 | 0.0037 | 0.0034 | −0.2932 |
| Zhanjiang | 0.2403 | 0.0967 | 0.0044 | 0.0045 | 0.0347 |
| Lingshui | 0.2502 | 0.1233 | 0.0049 | 0.0045 | −0.3231 |
| Haiphong | 0.2622 | 0.1556 | 0.0048 | 0.0051 | 0.2054 |
| Hainan | Mischief Reef | Zhanjiang | |
|---|---|---|---|
| Lingshui | |||
| Mischief | 0.048 | ||
| Zhanjiang | 0.069 | 0.095 | |
| Haiphong | 0.087 | 0.118 | 0.102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, B.; Yu, G.; Wang, L.; Liu, Y.; Yang, C.; Liu, M.; Sun, D. Genetic Signature of Pinctada fucata Inferred from Population Genomics: Source Tracking of the Invasion in Mischief Reef of Nansha Islands. Biology 2023, 12, 97. https://doi.org/10.3390/biology12010097
Shan B, Yu G, Wang L, Liu Y, Yang C, Liu M, Sun D. Genetic Signature of Pinctada fucata Inferred from Population Genomics: Source Tracking of the Invasion in Mischief Reef of Nansha Islands. Biology. 2023; 12(1):97. https://doi.org/10.3390/biology12010097
Chicago/Turabian StyleShan, Binbin, Gang Yu, Liangming Wang, Yan Liu, Changping Yang, Manting Liu, and Dianrong Sun. 2023. "Genetic Signature of Pinctada fucata Inferred from Population Genomics: Source Tracking of the Invasion in Mischief Reef of Nansha Islands" Biology 12, no. 1: 97. https://doi.org/10.3390/biology12010097