
Data Incompleteness May form a Hard-to-Overcome Barrier to Decoding Life’s Mechanism


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Incompleteness of Genomic Data
3. E. coli Data Incompleteness
4. One of the Best-Studied Multicellular Models, C. elegans, Is Still Very Far from “n = all”
5. The Unsolved Mysteries of the Fully Synthetic JCVI-syn3 Genome
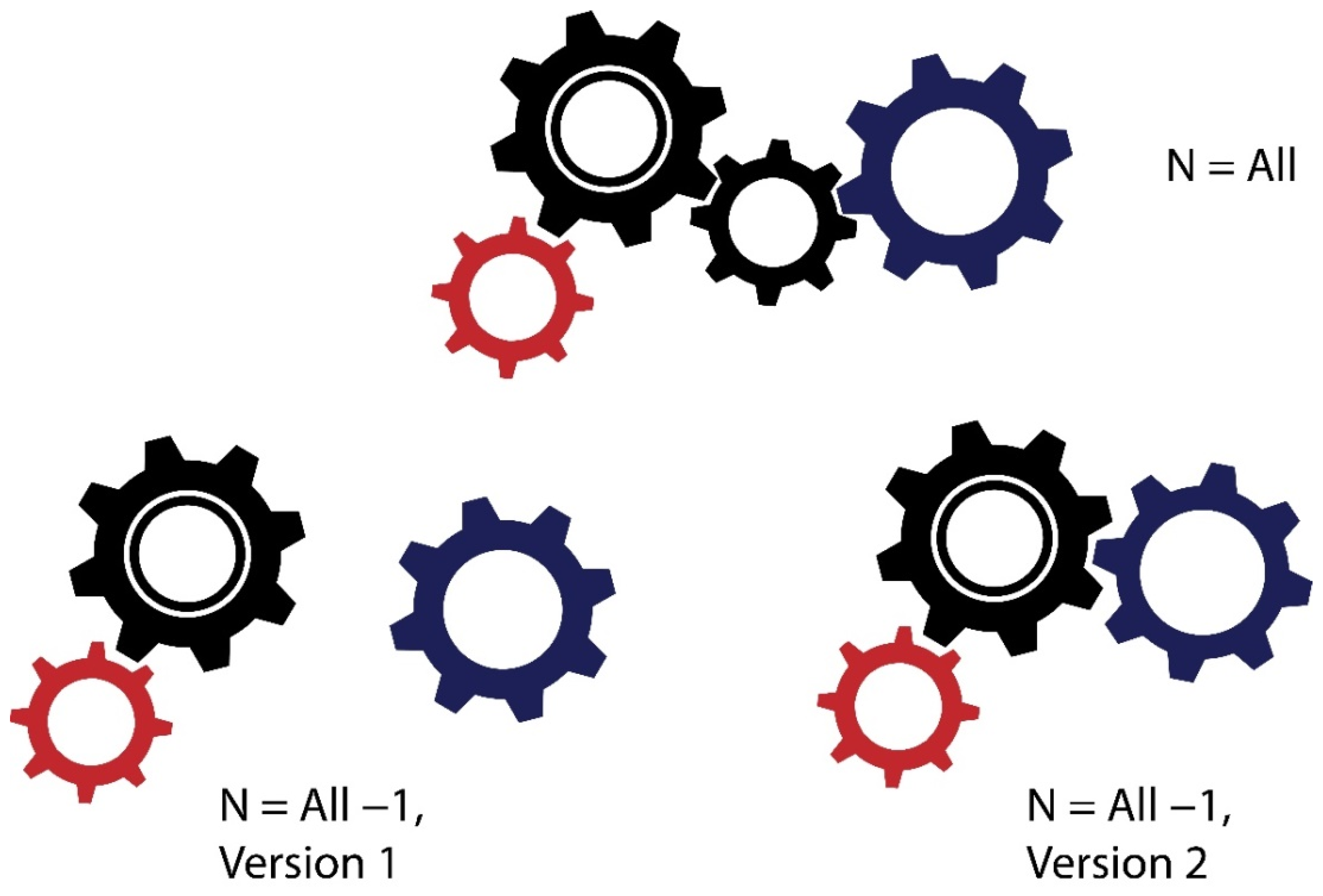
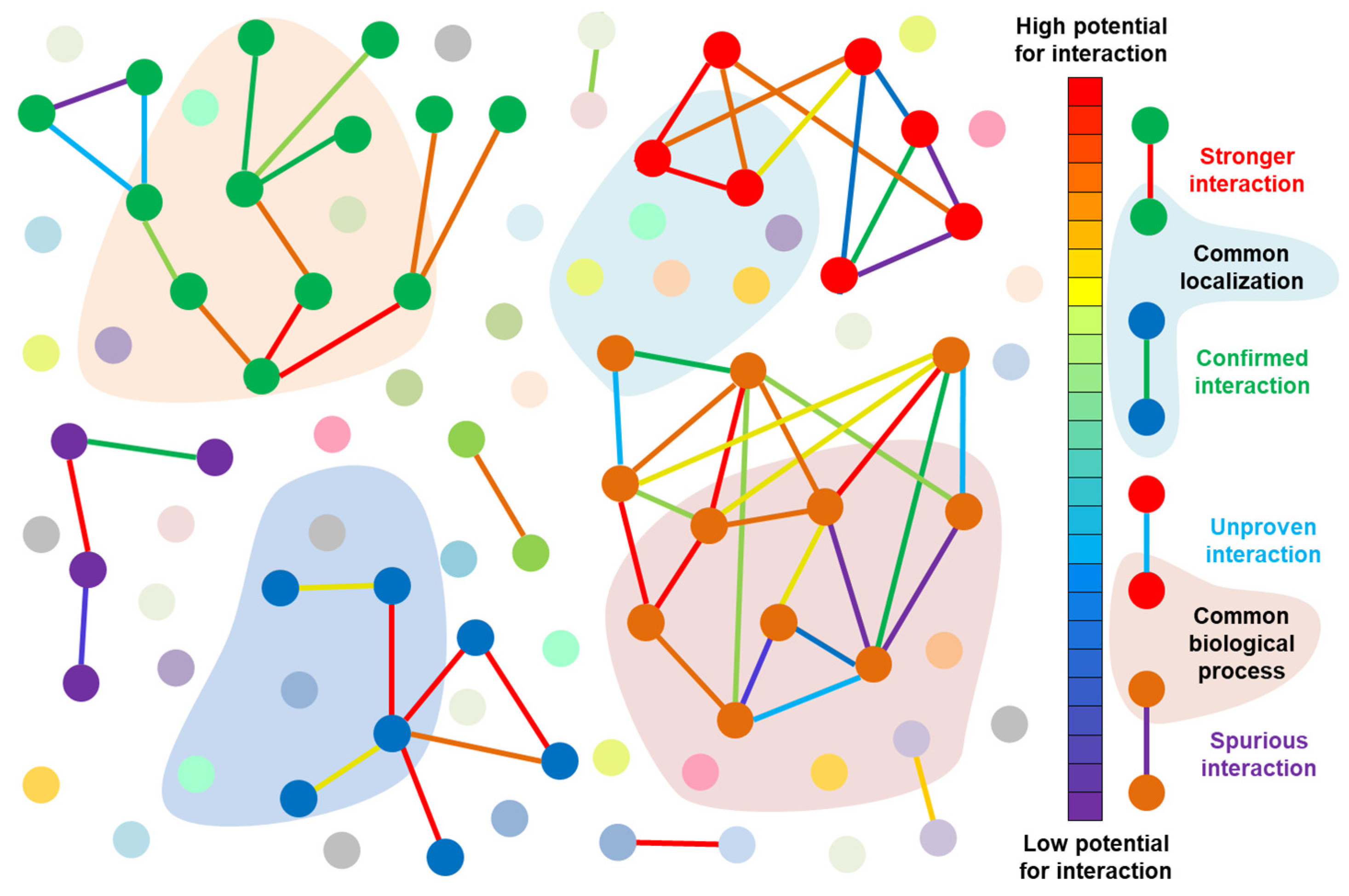
6. Complete Interactomes—An (Unreachable?) Dream of Systems Biologists
7. Bio-Databases and Ontologies for Biomedical Literature: The Inherent Incompleteness of Gene Ontology
8. Conclusions: Is Data Completeness One More Unsolvable Problem of Biology?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Errington, T.M.; Mathur, M.; Soderberg, C.K.; Denis, A.; Perfito, N.; Iorns, E.; Nosek, B.A. Investigating the replicability of preclinical cancer biology. eLife 2021, 10, e71601. [Google Scholar] [CrossRef]
- Errington, T.M.; Denis, A.; Perfito, N.; Iorns, E.; Nosek, B.A. Reproducibility in Cancer Biology: Challenges for assessing replicability in preclinical cancer biology. eLife 2021, 10, e67995. [Google Scholar] [CrossRef] [PubMed]
- Errington, T.M.; Denis, A.; Allison, A.B.; Araiza, R.; Aza-Blanc, P.; Bower, L.R.; Campos, J.; Chu, H.; Denson, S.; Donham, C. Experiments from unfinished Registered Reports in the Reproducibility Project: Cancer Biology. eLife 2021, 10, e73430. [Google Scholar] [CrossRef]
- Rodgers, P.; Collings, A. Reproducibility in Cancer Biology: What have we learned? eLife 2021, 10, e75830. [Google Scholar] [CrossRef]
- Hannun, Y.A. Build a registry of results that students can replicate. Nature 2021, 600, 571. [Google Scholar] [CrossRef]
- Begley, C.G.; Ioannidis, J.P. Reproducibility in science: Improving the standard for basic and preclinical research. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Helzlsouer, K.; Meerzaman, D.; Taplin, S.; Dunn, B.K. Humanizing Big Data: Recognizing the Human Aspect of Big Data. Front. Oncol. 2020, 10, 186. [Google Scholar] [CrossRef]
- Stevens, M.; Wehrens, R.; de Bont, A. Conceptualizations of Big Data and their epistemological claims in healthcare: A discourse analysis. Big Data Soc. 2018, 5, 2053951718816727. [Google Scholar] [CrossRef]
- Mayer-Schonberger, V.; Cukier, K. Big Data: A Revolution that will Transform How We Live, Work, and Think; Mariner Books; Houghton Mifflin Harcourt: Boston, MA, USA, 2014. [Google Scholar]
- Hutter, H.; Moerman, D. Big Data in Caenorhabditis elegans: Quo vadis? Mol. Biol. Cell 2015, 26, 3909–3914. [Google Scholar] [CrossRef]
- Aggarwal, S.; Raj, A.; Kumar, D.; Dash, D.; Yadav, A.K. False discovery rate: The Achilles’ heel of proteogenomics. Brief. Bioinform. 2022, bbac163. [Google Scholar] [CrossRef] [PubMed]
- Elouataoui, W.; Alaoui, I.E.; Gahi, Y. Data Quality in the Era of Big Data: A Global Review. Big Data Intell. Smart Appl. 2022, 994, 1–25. [Google Scholar] [CrossRef]
- Kasif, S.; Roberts, R.J. We need to keep a reproducible trace of facts, predictions, and hypotheses from gene to function in the era of big data. PLoS Biol. 2020, 18, e3000999. [Google Scholar] [CrossRef]
- Danchin, A.; Ouzounis, C.; Tokuyasu, T.; Zucker, J.D. No wisdom in the crowd: Genome annotation in the era of big data—Current status and future prospects. Microb. Biotechnol. 2018, 11, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.; Leeuwenberg, A.M.; Beekers, I.; Verkouter, I.; Jacobs, J.; Bots, M.L.; Asselbergs, F.W.; Moons, K.; Debray, T. Missing data is poorly handled and reported in prediction model studies using machine learning: A literature review. J. Clin. Epidemiol. 2022, 142, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
- Miao, Z.; Humphreys, B.D.; McMahon, A.P.; Kim, J. Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 2021, 17, 710–724. [Google Scholar] [CrossRef]
- Wu, S.; Chen, D.; Snyder, M.P. Network biology bridges the gaps between quantitative genetics and multi-omics to map complex diseases. Curr. Opin. Chem. Biol. 2022, 66, 102101. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, N.; Michailidis, G. Unsupervised Multi-Omics Data Integration Methods: A Comprehensive Review. Front. Genet. 2022, 13, 854752. [Google Scholar] [CrossRef]
- Kurokawa, M.; Ying, B.W. Experimental Challenges for Reduced Genomes: The Cell Model Escherichia coli. Microorganisms 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; King, Z.A.; Sastry, A.; Palsson, B.O. The y-ome defines the 35% of Escherichia coli genes that lack experimental evidence of function. Nucleic Acids Res. 2019, 47, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Dellomonaco, C.; Clomburg, J.M.; Miller, E.N.; Gonzalez, R. Engineered reversal of the beta-oxidation cycle for the synthesis of fuels and chemicals. Nature 2011, 476, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, T.E.; Pedersen, M.; LaCroix, R.A.; Ebrahim, A.; Bonde, M.; Herrgard, M.J.; Palsson, B.O.; Sommer, M.; Feist, A.M. Evolution of Escherichia coli to 42 degrees C and subsequent genetic engineering reveals adaptive mechanisms and novel mutations. Mol. Biol. Evol. 2014, 31, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Hufnagel, D.A.; DePas, W.H.; Chapman, M.R. The disulfide bonding system suppresses CsgD-independent cellulose production in Escherichia coli. J. Bacteriol. 2014, 196, 3690–3699. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martinez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muniz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- Urtecho, G.; Tripp, A.D.; Insigne, K.D.; Kim, H.; Kosuri, S. Systematic Dissection of Sequence Elements Controlling sigma70 Promoters Using a Genomically Encoded Multiplexed Reporter Assay in Escherichia coli. Biochemistry 2019, 58, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.I.; Ross, C.; Siegele, D.A.; Hu, J.C. Insights from the reanalysis of high-throughput chemical genomics data for Escherichia coli K-12. G3 2021, 11, jkaa035. [Google Scholar] [CrossRef] [PubMed]
- Glenwinkel, L.; Taylor, S.R.; Langebeck-Jensen, K.; Pereira, L.; Reilly, M.B.; Basavaraju, M.; Rafi, I.; Yemini, E.; Pocock, R.; Sestan, N.; et al. In silico analysis of the transcriptional regulatory logic of neuronal identity specification throughout the C. elegans nervous system. eLife 2021, 10, e64906. [Google Scholar] [CrossRef]
- Godini, R.; Handley, A.; Pocock, R. Transcription Factors That Control Behavior-Lessons From C. elegans. Front. Neurosci. 2021, 15, 745376. [Google Scholar] [CrossRef]
- Hillier, L.W.; Coulson, A.; Murray, J.I.; Bao, Z.; Sulston, J.E.; Waterston, R.H. Genomics in C. elegans: So many genes, such a little worm. Genome Res. 2005, 15, 1651–1660. [Google Scholar] [CrossRef]
- Li-Leger, E.; Feichtinger, R.; Flibotte, S.; Holzkamp, H.; Schnabel, R.; Moerman, D.G. Identification of essential genes in Caenorhabditis elegans through whole-genome sequencing of legacy mutant collections. G3 2021, 11, jkab328. [Google Scholar] [CrossRef] [PubMed]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, C.; Chu, J.S. Identification of Essential Genes in Caenorhabditis elegans with Lethal Mutations Maintained by Genetic Balancers. Methods Mol. Biol. 2022, 2377, 345–362. [Google Scholar] [CrossRef]
- Edgley, M.; Baillie, D.; Riddle, D.; Rose, A. Genetic Balancers. WormBook: The Online Review of C. elegans Biology. Nucleic Acids Res. 2007, 35, D472-5. [Google Scholar] [CrossRef]
- Consortium, C.e.D.M. large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 2012, 2, 1415–1425. [Google Scholar] [CrossRef]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Hatje, K.; Muhlhausen, S.; Simm, D.; Kollmar, M. The Protein-Coding Human Genome: Annotating High-Hanging Fruits. Bioessays 2019, 41, e1900066. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Harnessing model organism genomics to underpin the machine learning-based prediction of essential genes in eukaryotes—Biotechnological implications. Biotechnol. Adv. 2021, 54, 107822. [Google Scholar] [CrossRef] [PubMed]
- Vallin, E.; Gallagher, J.; Granger, L.; Martin, E.; Belougne, J.; Maurizio, J.; Duverger, Y.; Scaglione, S.; Borrel, C.; Cortier, E.; et al. A genome-wide collection of Mos1 transposon insertion mutants for the C. elegans research community. PLoS ONE 2012, 7, e30482. [Google Scholar] [CrossRef] [PubMed]
- Simonis, N.; Rual, J.F.; Carvunis, A.R.; Tasan, M.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Sahalie, J.M.; Venkatesan, K.; Gebreab, F.; et al. Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 2009, 6, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Remmelzwaal, S.; Boxem, M. Protein interactome mapping in Caenorhabditis elegans. Curr. Opin. Syst. Biol. 2019, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.A., 3rd; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, aad6253. [Google Scholar] [CrossRef]
- Antczak, M.; Michaelis, M.; Wass, M.N. Environmental conditions shape the nature of a minimal bacterial genome. Nat. Commun. 2019, 10, 3100. [Google Scholar] [CrossRef]
- Coyle, M.; Hu, J.; Gartner, Z. Mysteries in a Minimal Genome. ACS Cent. Sci. 2016, 2, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Breuer, M.; Earnest, T.M.; Merryman, C.; Wise, K.S.; Sun, L.; Lynott, M.R.; Hutchison, C.A.; Smith, H.O.; Lapek, J.D.; Gonzalez, D.J.; et al. Essential metabolism for a minimal cell. eLife 2019, 8, e36842. [Google Scholar] [CrossRef]
- Pelletier, J.F.; Glass, J.I.; Strychalski, E.A. Cellular mechanics during division of a genomically minimal cell. Trends Cell Biol. 2022; preprint. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, W.; Cheng, M.; Omenn, G.S.; Freddolino, P.L.; Zhang, Y. Functions of Essential Genes and a Scale-Free Protein Interaction Network Revealed by Structure-Based Function and Interaction Prediction for a Minimal Genome. J. Proteome Res. 2021, 20, 1178–1189. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Marcotte, E.M.; Xenarios, I.; Yeates, T.O. Protein function in the post-genomic era. Nature 2000, 405, 823–826. [Google Scholar] [CrossRef]
- Brehme, M.; Vidal, M. A global protein-lipid interactome map. Mol. Syst. Biol. 2010, 6, 443. [Google Scholar] [CrossRef]
- Kunowska, N.; Stelzl, U. Decoding the cellular effects of genetic variation through interaction proteomics. Curr. Opin. Chem. Biol. 2021, 66, 102100. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-Scale Human Interactomics. Trends Biochem. Sci. 2017, 42, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Yook, S.H.; Oltvai, Z.N.; Barabasi, A.L. Functional and topological characterization of protein interaction networks. Proteomics 2004, 4, 928–942. [Google Scholar] [CrossRef]
- Snider, J.; Kotlyar, M.; Saraon, P.; Yao, Z.; Jurisica, I.; Stagljar, I. Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 2015, 11, 848. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef]
- Cusick, M.E.; Klitgord, N.; Vidal, M.; Hill, D.E. Interactome: Gateway into systems biology. Hum. Mol. Genet. 2005, 14, R171–R181. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Long, L.; Falcon, S.; Gentleman, R. Graphs in molecular biology. BMC Bioinform. 2007, 8 (Suppl. 6), S8. [Google Scholar] [CrossRef]
- Koh, G.C.; Porras, P.; Aranda, B.; Hermjakob, H.; Orchard, S.E. Analyzing protein-protein interaction networks. J. Proteome Res. 2012, 11, 2014–2031. [Google Scholar] [CrossRef]
- Mason, O.; Verwoerd, M. Graph theory and networks in Biology. IET Syst. Biol. 2007, 1, 89–119. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Zhao, Y.; Cai, L.; Xue, H.; Zhu, X.; Lu, H.; Zhang, J.; Sun, S.; Ling, L.; Zhang, N.; et al. Topological structure analysis of the protein-protein interaction network in budding yeast. Nucleic Acids Res. 2003, 31, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Wuchty, S.; Oltvai, Z.N.; Barabasi, A.L. Evolutionary conservation of motif constituents in the yeast protein interaction network. Nat. Genet. 2003, 35, 176–179. [Google Scholar] [CrossRef]
- James, K.; Wipat, A.; Cockell, S. Expanding Interactome Analyses beyond Model Eukaryotes. Brief Funct. Genom. 2021, 21, 243–269. [Google Scholar] [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current Challenges and Opportunities in Designing Protein-Protein Interaction Targeted Drugs. Adv. Appl. Bioinform. Chem. AABC 2020, 13, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, G.N.; Klapa, M.I.; Moschonas, N.K. How Far Are We from the Completion of the Human Protein Interactome Reconstruction? Biomolecules 2022, 12, 140. [Google Scholar] [CrossRef]
- Alberts, B. The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 1998, 92, 291–294. [Google Scholar] [CrossRef]
- von Hippel, P.H. From “simple” DNA-protein interactions to the macromolecular machines of gene expression. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Ginalski, K. The interactome: Predicting the protein-protein interactions in cells. Cell. Mol. Biol. Lett. 2009, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, I.A.; Luck, K.; Spirohn, K.; Wang, Y.; Pollis, C.; Schlabach, S.; Bian, W.; Kim, D.K.; Kishore, N.; Hao, T.; et al. Network-based prediction of protein interactions. Nat. Commun. 2019, 10, 1240. [Google Scholar] [CrossRef]
- Johnson, K.L.; Qi, Z.; Yan, Z.; Wen, X.; Nguyen, T.C.; Zaleta-Rivera, K.; Chen, C.J.; Fan, X.; Sriram, K.; Wan, X.; et al. Revealing protein-protein interactions at the transcriptome scale by sequencing. Mol. Cell 2021, 81, 3877. [Google Scholar] [CrossRef]
- Wuchty, S.; Muller, S.A.; Caufield, J.H.; Hauser, R.; Aloy, P.; Kalkhof, S.; Uetz, P. Proteome Data Improves Protein Function Prediction in the Interactome of Helicobacter pylori. Mol. Cell. Proteom. MCP 2018, 17, 961–973. [Google Scholar] [CrossRef]
- Dilucca, M.; Cimini, G.; Giansanti, A. Bacterial Protein Interaction Networks: Connectivity is Ruled by Gene Conservation, Essentiality and Function. Curr. Genom. 2021, 22, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Gao, F.; Lin, Y. Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes. Sci. Rep. 2015, 5, 13210. [Google Scholar] [CrossRef] [PubMed]
- Huxley, T. Thomas Huxley Quotes. Available online: https://www.quotes.net/quote/56043 (accessed on 10 March 2022).
- Attwood, T.K.; Kell, D.B.; McDermott, P.; Marsh, J.; Pettifer, S.R.; Thorne, D. Calling International Rescue: Knowledge lost in literature and data landslide! Biochem. J. 2009, 424, 317–333. [Google Scholar] [CrossRef]
- Gaudet, P.; Dessimoz, C. Gene Ontology: Pitfalls, Biases, and Remedies. In The Gene Ontology Handbook, Methods in Molecular Biology; Dessimoz, C., Škunca, N., Eds.; Springer Open Humana Press: Berlin/Heidelberg, Germany, 2017; Volume 1446. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Chen, J.; Zhang, X.; Guo, M.; Yu, G. A Literature Review of Gene Function Prediction by Modeling Gene Ontology. Front. Genet. 2020, 11, 400. [Google Scholar] [CrossRef]
- Zhang, D.; Guelfi, S.; Garcia-Ruiz, S.; Costa, B.; Reynolds, R.H.; D’Sa, K.; Liu, W.; Courtin, T.; Peterson, A.; Jaffe, A.E.; et al. Incomplete annotation has a disproportionate impact on our understanding of Mendelian and complex neurogenetic disorders. Sci. Adv. 2020, 6, eaay8299. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Biology Past and Biology Future: Where have we been and where are we going. Neural Regener. Res. 2013, 8, 2309–2316. [Google Scholar]
- Brenner, S. Loose ends. Curr. Biol. 1995, 5, 1328. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Brenner, S. Sequences and consequences. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2010, 365, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Unsolvable Problems of Biology: It Is Impossible to Create Two Identical Organisms, to Defeat Cancer, or to Map Organisms onto Their Genomes. Biochemistry 2018, 83, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. Nobel lecture: Nature’s gift to science. Biosci. Rep. 2003, 23, 225–237. [Google Scholar] [CrossRef]
- Weinberg, R. Point: Hypotheses first. Nature 2010, 464, 678. [Google Scholar] [CrossRef] [PubMed]
- Golub, T. Counterpoint: Data first. Nature 2010, 464, 679. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From Big Data to Precision Medicine. Front. Med. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Voit, E.O. Perspective: Dimensions of the scientific method. PLoS Comput. Biol. 2019, 15, e1007279. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Chowdhury, S. Big data quality dimensions: A systematic literature review. JISTEM-J. Inf. Syst. Technol. Manag. 2020, 17, e202017003. [Google Scholar] [CrossRef]
- Hassenstein, M.J.; Vanella, P. Data Quality—Concepts and Problems. Encyclopedia 2022, 2, 498–510. [Google Scholar] [CrossRef]
- Wang, R.Y.; Strong, D.M. Beyond accuracy: What data quality means to data consumers. J. Manag. Inf. Syst. 1996, 12, 5–33. [Google Scholar] [CrossRef]
- Taran, S.; Adhikari, N.K.; Fan, E. Falsifiability in medicine: What clinicians can learn from Karl Popper. Intensive Care Med. 2021, 47, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Popper, K. The Logic of Scientific Discovery; Routledge: Oxfordshire, UK, 2005. [Google Scholar]
- Johnston, M.; Fields, S. Grass-roots genomics. Nat. Genet. 2000, 24, 5–6. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondratyeva, L.; Alekseenko, I.; Chernov, I.; Sverdlov, E. Data Incompleteness May form a Hard-to-Overcome Barrier to Decoding Life’s Mechanism. Biology 2022, 11, 1208. https://doi.org/10.3390/biology11081208
Kondratyeva L, Alekseenko I, Chernov I, Sverdlov E. Data Incompleteness May form a Hard-to-Overcome Barrier to Decoding Life’s Mechanism. Biology. 2022; 11(8):1208. https://doi.org/10.3390/biology11081208
Chicago/Turabian StyleKondratyeva, Liya, Irina Alekseenko, Igor Chernov, and Eugene Sverdlov. 2022. "Data Incompleteness May form a Hard-to-Overcome Barrier to Decoding Life’s Mechanism" Biology 11, no. 8: 1208. https://doi.org/10.3390/biology11081208