
Is Brain-Derived Neurotrophic Factor a Metabolic Hormone in Peripheral Tissues?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
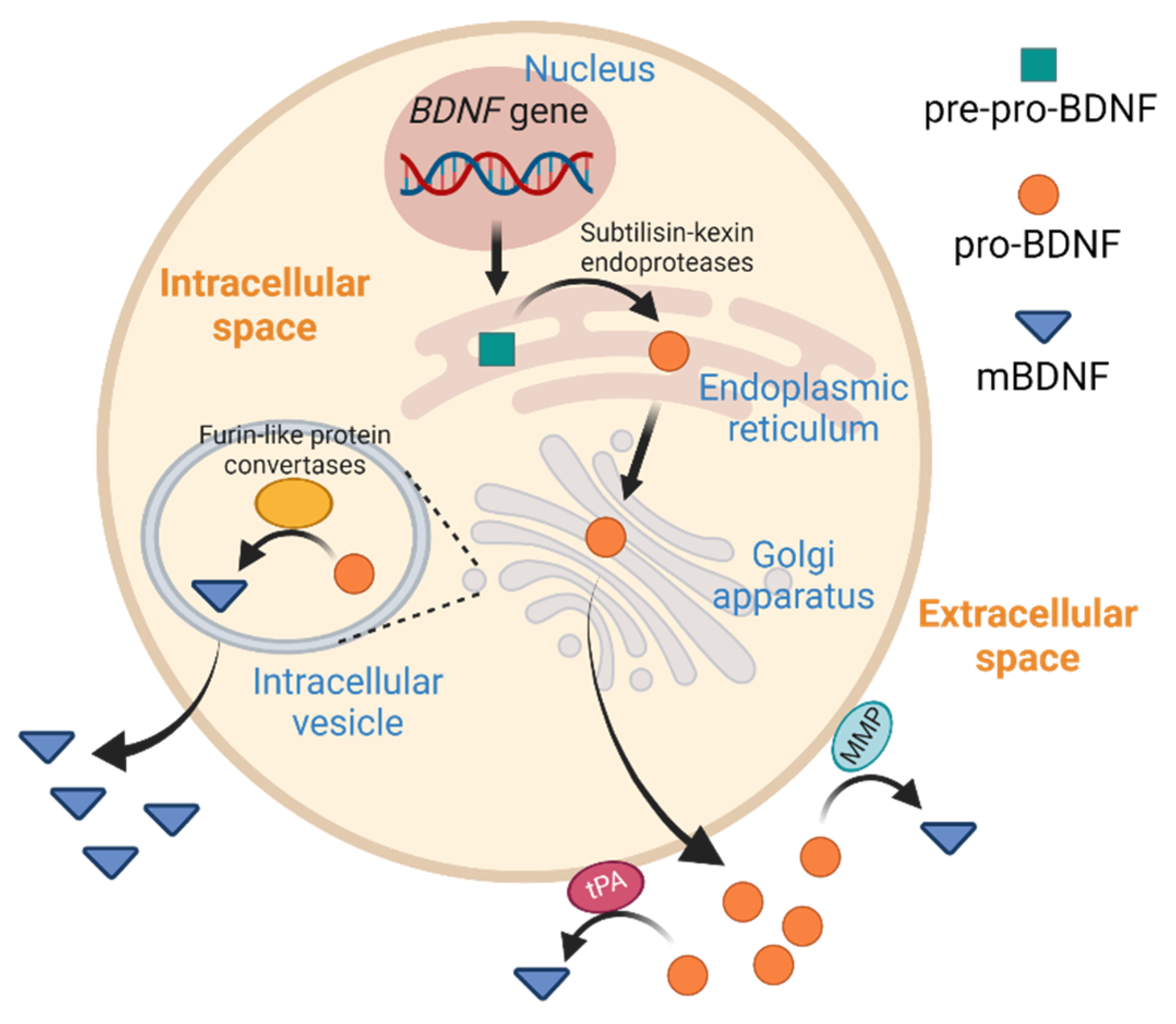
1. Introduction
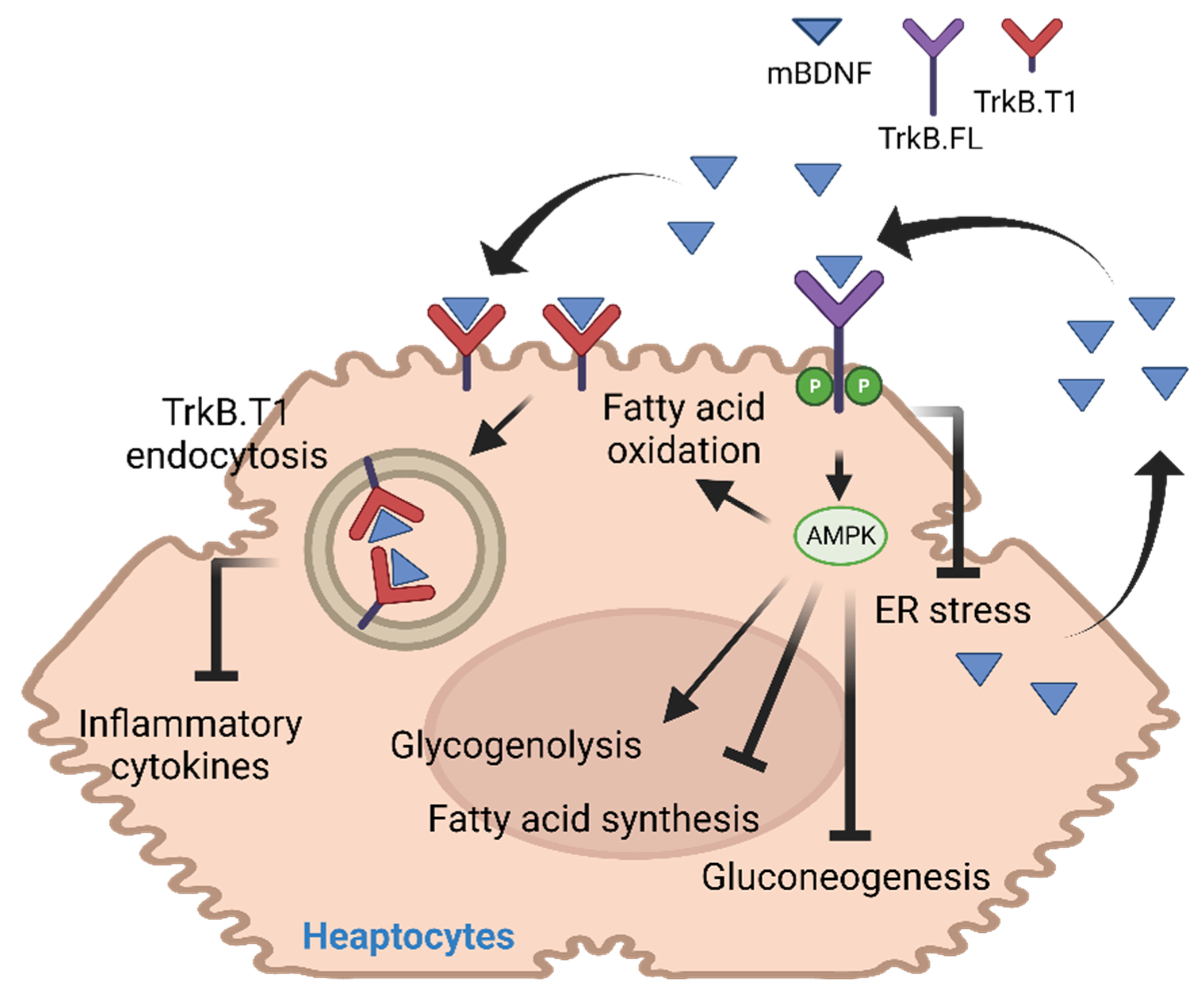
2. BDNF in Liver
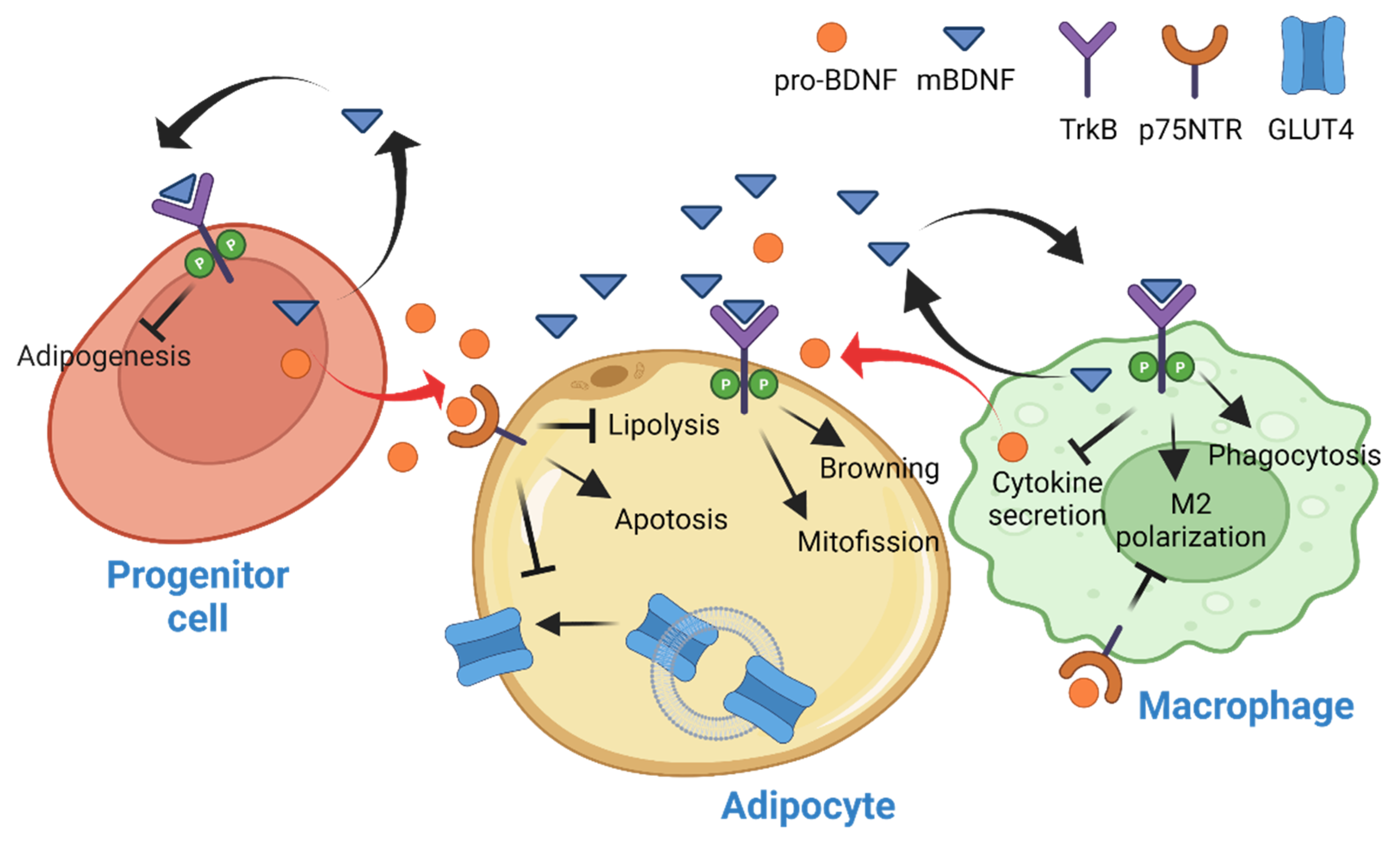
3. BDNF in Adipose Tissue
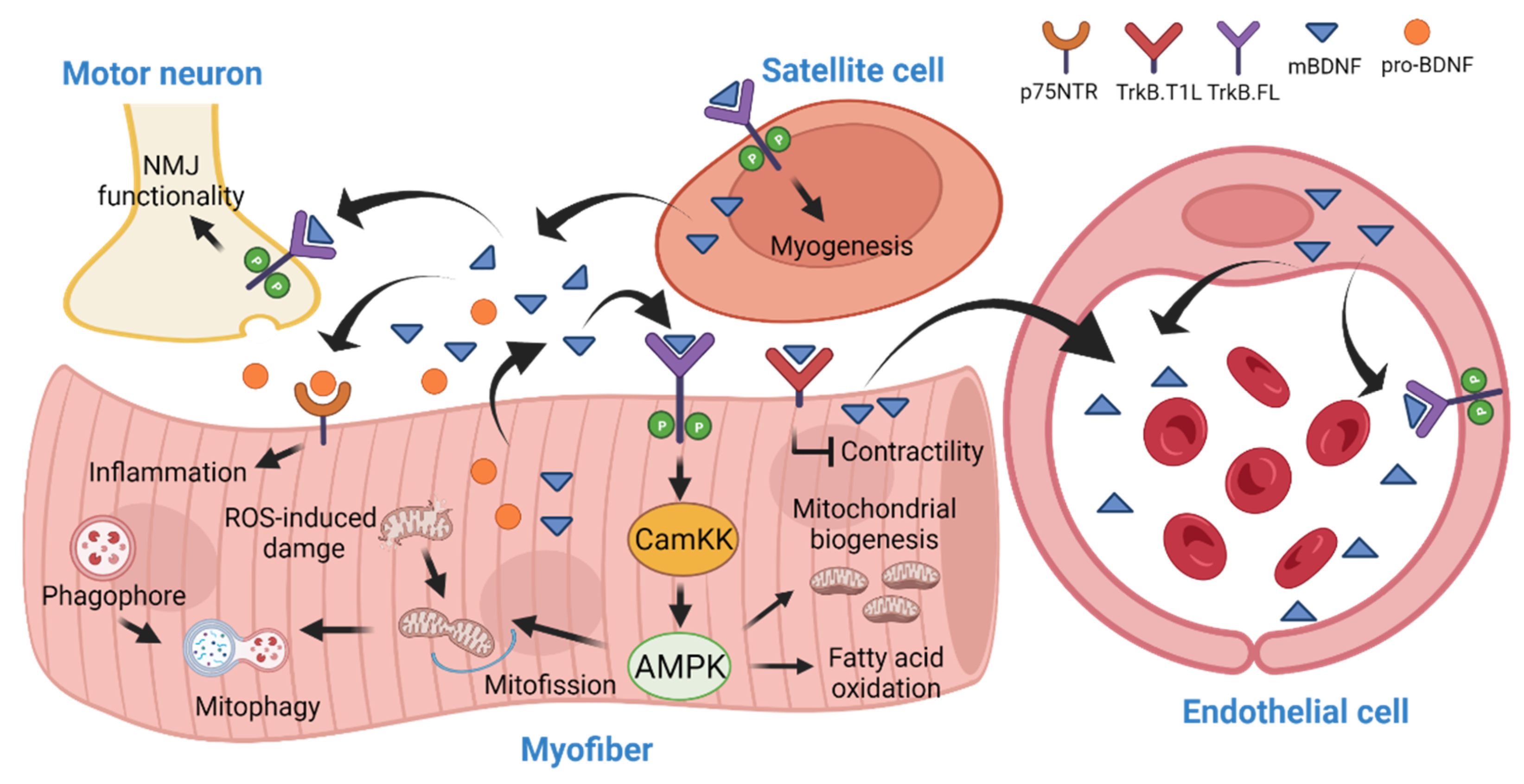
4. BDNF in Skeletal Muscle
5. Unresolved Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kowianski, P.; Lietzau, G.; Czuba, E.; Waskow, M.; Steliga, A.; Morys, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Je, H.S.; Yang, F.; Ji, Y.; Potluri, S.; Fu, X.Q.; Luo, Z.G.; Nagappan, G.; Chan, J.P.; Hempstead, B.; Son, Y.J.; et al. ProBDNF and mature BDNF as punishment and reward signals for synapse elimination at mouse neuromuscular junctions. J. Neurosci. 2013, 33, 9957–9962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marosi, K.; Mattson, M.P. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 2014, 25, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Kernie, S.G.; Liebl, D.J.; Parada, L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000, 19, 1290–1300. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Goulding, E.H.; Zang, K.; Cepoi, D.; Cone, R.D.; Jones, K.R.; Tecott, L.H.; Reichardt, L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003, 6, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Di Rosa, M.C.; Zimbone, S.; Saab, M.W.; Tomasello, M.F. The Pleiotropic Potential of BDNF beyond Neurons: Implication for a Healthy Mind in a Healthy Body. Life 2021, 11, 1256. [Google Scholar] [CrossRef] [PubMed]
- Bariohay, B.; Lebrun, B.; Moyse, E.; Jean, A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology 2005, 146, 5612–5620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsh, G.S.; Schwartz, M.W. Genetic approaches to studying energy balance: Perception and integration. Nat. Rev. Genet. 2002, 3, 589–600. [Google Scholar] [CrossRef]
- An, J.J.; Liao, G.Y.; Kinney, C.E.; Sahibzada, N.; Xu, B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab. 2015, 22, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.S.; Connie Hung, C.C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O’Rahilly, S.; Farooqi, I.S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.C.P.; Abreu, G.M.; Palhinha, L.; Zembrzuski, V.M.; Campos Junior, M.; Carneiro, J.R.I.; Nogueira Neto, J.F.; Magno, F.; Rosado, E.L.; Maya-Monteiro, C.M.; et al. A Rare Potential Pathogenic Variant in the BDNF Gene is Found in a Brazilian Patient with Severe Childhood-Onset Obesity. Diabetes Metab. Syndr. Obes. 2021, 14, 11–22. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tsuchida, A.; Itakura, Y.; Nonomura, T.; Ono, M.; Hirota, F.; Inoue, T.; Nakayama, C.; Taiji, M.; Noguchi, H. Brain-derived neurotrophic factor regulates glucose metabolism by modulating energy balance in diabetic mice. Diabetes 2000, 49, 436–444. [Google Scholar] [CrossRef] [Green Version]
- Otani, K.; Okada, M.; Yamawaki, H. Diverse distribution of tyrosine receptor kinase B isoforms in rat multiple tissues. J. Vet. Med. Sci. 2017, 79, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Camerino, C.; Conte, E.; Cannone, M.; Caloiero, R.; Fonzino, A.; Tricarico, D. Nerve Growth Factor, Brain-Derived Neurotrophic Factor and Osteocalcin Gene Relationship in Energy Regulation, Bone Homeostasis and Reproductive Organs Analyzed by mRNA Quantitative Evaluation and Linear Correlation Analysis. Front. Physiol. 2016, 7, 456. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, A.; Nakagawa, T.; Itakura, Y.; Ichihara, J.; Ogawa, W.; Kasuga, M.; Taiji, M.; Noguchi, H. The effects of brain-derived neurotrophic factor on insulin signal transduction in the liver of diabetic mice. Diabetologia 2001, 44, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassiman, D.; Denef, C.; Desmet, V.J.; Roskams, T. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001, 33, 148–158. [Google Scholar] [CrossRef]
- Meek, T.H.; Wisse, B.E.; Thaler, J.P.; Guyenet, S.J.; Matsen, M.E.; Fischer, J.D.; Taborsky, G.J., Jr.; Schwartz, M.W.; Morton, G.J. BDNF action in the brain attenuates diabetic hyperglycemia via insulin-independent inhibition of hepatic glucose production. Diabetes 2013, 62, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Liu, T.; Mi, L.; Kuang, H.; Xiong, X.; Chen, Z.; Li, S.; Lin, J.D. hnRNPU/TrkB Defines a Chromatin Accessibility Checkpoint for Liver Injury and Nonalcoholic Steatohepatitis Pathogenesis. Hepatology 2020, 71, 1228–1246. [Google Scholar] [CrossRef]
- Genzer, Y.; Chapnik, N.; Froy, O. Effect of brain-derived neurotrophic factor (BDNF) on hepatocyte metabolism. Int. J. Biochem. Cell Biol. 2017, 88, 69–74. [Google Scholar] [CrossRef]
- Cirrik, S.; Hacioglu, G.; Abidin, I.; Aydin-Abidin, S.; Noyan, T. Endoplasmic reticulum stress in the livers of BDNF heterozygous knockout mice. Arch. Physiol. Biochem. 2019, 125, 378–386. [Google Scholar] [CrossRef]
- Ahuja, P.; Ng, C.F.; Pang, B.P.S.; Chan, W.S.; Tse, M.C.L.; Bi, X.; Kwan, H.R.; Brobst, D.; Herlea-Pana, O.; Yang, X.; et al. Muscle-generated BDNF (brain derived neurotrophic factor) maintains mitochondrial quality control in female mice. Autophagy 2021, 18, 1367–1384. [Google Scholar] [CrossRef]
- Stoilov, P.; Castren, E.; Stamm, S. Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem. Biophys. Res. Commun. 2002, 290, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Ren, Q.; Zhang, J.C.; Chen, Q.X.; Hashimoto, K. Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: Rethinking the brain-liver axis. Transl. Psychiatry 2017, 7, e1128. [Google Scholar] [CrossRef]
- Teillon, S.; Calderon, G.A.; Rios, M. Diminished diet-induced hyperglycemia and dyslipidemia and enhanced expression of PPARalpha and FGF21 in mice with hepatic ablation of brain-derived neurotropic factor. J. Endocrinol. 2010, 205, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; Endotext [Internet]: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bernhard, F.; Landgraf, K.; Kloting, N.; Berthold, A.; Buttner, P.; Friebe, D.; Kiess, W.; Kovacs, P.; Bluher, M.; Korner, A. Functional relevance of genes implicated by obesity genome-wide association study signals for human adipocyte biology. Diabetologia 2013, 56, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Sornelli, F.; Fiore, M.; Chaldakov, G.N.; Aloe, L. Adipose tissue-derived nerve growth factor and brain-derived neurotrophic factor: Results from experimental stress and diabetes. Gen. Physiol. Biophys. 2009, 28, 179–183. [Google Scholar] [PubMed]
- Nakagomi, A.; Okada, S.; Yokoyama, M.; Yoshida, Y.; Shimizu, I.; Miki, T.; Kobayashi, Y.; Minamino, T. Role of the central nervous system and adipose tissue BDNF/TrkB axes in metabolic regulation. NPJ Aging Mech. Dis. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay, J.; Gauthier, M.F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.; et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2020, 2, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Blaszkiewicz, M.; Wood, E.; Koizar, S.; Willows, J.; Anderson, R.; Tseng, Y.H.; Godwin, J.; Townsend, K.L. The involvement of neuroimmune cells in adipose innervation. Mol. Med. 2020, 26, 126. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Lee, C.W.; Lee, J.; Choi, D.J.; Sohng, J.K.; Park, Y.I. 7,8-Dihydroxyflavone inhibits adipocyte differentiation via antioxidant activity and induces apoptosis in 3T3-L1 preadipocyte cells. Life Sci. 2016, 144, 103–112. [Google Scholar] [CrossRef]
- Jo, D.; Son, Y.; Yoon, G.; Song, J.; Kim, O.Y. Role of Adiponectin and Brain Derived Neurotrophic Factor in Metabolic Regulation Involved in Adiposity and Body Fat Browning. J. Clin. Med. 2020, 10, 56. [Google Scholar] [CrossRef]
- Song, H.D.; Kim, S.N.; Saha, A.; Ahn, S.Y.; Akindehin, S.; Son, Y.; Cho, Y.K.; Kim, M.; Park, J.H.; Jung, Y.S.; et al. Aging-Induced Brain-Derived Neurotrophic Factor in Adipocyte Progenitors Contributes to Adipose Tissue Dysfunction. Aging Dis. 2020, 11, 575–587. [Google Scholar] [CrossRef]
- Baeza-Raja, B.; Sachs, B.D.; Li, P.; Christian, F.; Vagena, E.; Davalos, D.; Le Moan, N.; Ryu, J.K.; Sikorski, S.L.; Chan, J.P.; et al. p75 Neurotrophin Receptor Regulates Energy Balance in Obesity. Cell Rep. 2016, 14, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Colitti, M.; Montanari, T. Brain-derived neurotrophic factor modulates mitochondrial dynamics and thermogenic phenotype on 3T3-L1 adipocytes. Tissue Cell 2020, 66, 101388. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.; Youm, Y.H.; Ravussin, A.; Dixit, V.D. Quantification of adipose tissue leukocytosis in obesity. Methods Mol. Biol. 2013, 1040, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Barouch, R.; Appel, E.; Kazimirsky, G.; Brodie, C. Macrophages express neurotrophins and neurotrophin receptors. Regulation of nitric oxide production by NT-3. J. Neuroimmunol. 2001, 112, 72–77. [Google Scholar] [CrossRef]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.L.; Bartke, I.; et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef]
- Kruse, N.; Cetin, S.; Chan, A.; Gold, R.; Luhder, F. Differential expression of BDNF mRNA splice variants in mouse brain and immune cells. J. Neuroimmunol. 2007, 182, 13–21. [Google Scholar] [CrossRef]
- Wong, I.; Liao, H.; Bai, X.; Zaknic, A.; Zhong, J.; Guan, Y.; Li, H.Y.; Wang, Y.J.; Zhou, X.F. ProBDNF inhibits infiltration of ED1+ macrophages after spinal cord injury. Brain Behav. Immun. 2010, 24, 585–597. [Google Scholar] [CrossRef]
- Ji, X.C.; Dang, Y.Y.; Gao, H.Y.; Wang, Z.T.; Gao, M.; Yang, Y.; Zhang, H.T.; Xu, R.X. Local Injection of Lenti-BDNF at the Lesion Site Promotes M2 Macrophage Polarization and Inhibits Inflammatory Response After Spinal Cord Injury in Mice. Cell Mol. Neurobiol. 2015, 35, 881–890. [Google Scholar] [CrossRef]
- Asami, T.; Ito, T.; Fukumitsu, H.; Nomoto, H.; Furukawa, Y.; Furukawa, S. Autocrine activation of cultured macrophages by brain-derived neurotrophic factor. Biochem. Biophys. Res. Commun. 2006, 344, 941–947. [Google Scholar] [CrossRef]
- Sasaki, S.; Takeda, K.; Ouhara, K.; Shirawachi, S.; Kajiya, M.; Matsuda, S.; Kono, S.; Shiba, H.; Kurihara, H.; Mizuno, N. Involvement of Rac1 in macrophage activation by brain-derived neurotrophic factor. Mol. Biol. Rep. 2021, 48, 5249–5257. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Huang, H.B.; Huang Tseng, H.Y.; Lu, M.C. Brain-Derived Neurotrophic Factor Suppressed Proinflammatory Cytokines Secretion and Enhanced MicroRNA(miR)-3168 Expression in Macrophages. Int. J. Mol. Sci. 2022, 23, 570. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Herbruggen, O.; Nassenstein, C.; Lommatzsch, M.; Quarcoo, D.; Renz, H.; Braun, A. Tumor necrosis factor-alpha and interleukin-6 regulate secretion of brain-derived neurotrophic factor in human monocytes. J. Neuroimmunol. 2005, 160, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, N.; Vatterott, P.; Egwiekhor, A.; Rochlin, M.W. Brain-derived neurotrophic factor attracts geniculate ganglion neurites during embryonic targeting. Dev. Neurosci. 2010, 32, 184–196. [Google Scholar] [CrossRef] [Green Version]
- Baeza-Raja, B.; Li, P.; Le Moan, N.; Sachs, B.D.; Schachtrup, C.; Davalos, D.; Vagena, E.; Bridges, D.; Kim, C.; Saltiel, A.R.; et al. p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2012, 109, 5838–5843. [Google Scholar] [CrossRef] [Green Version]
- Gaidhu, M.P.; Anthony, N.M.; Patel, P.; Hawke, T.J.; Ceddia, R.B. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: Role of ATGL, HSL, and AMPK. Am. J. Physiol. Cell Physiol. 2010, 298, C961–C971. [Google Scholar] [CrossRef] [Green Version]
- Saka, Y.; Yoshimura, O.; Tahara, H.; Takeda, Y.; Moriyama, H.; Maejima, H.; Tobimatsu, Y. The mRNA expression of neurotrophins in different skeletal muscles of young rats. Hiroshima J. Med. Sci. 2007, 56, 23–28. [Google Scholar]
- Yang, X.; Brobst, D.; Chan, W.S.; Tse, M.C.L.; Herlea-Pana, O.; Ahuja, P.; Bi, X.; Zaw, A.M.; Kwong, Z.S.W.; Jia, W.H.; et al. Muscle-generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 2019, 12, eaau1468. [Google Scholar] [CrossRef]
- Mousavi, K.; Jasmin, B.J. BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J. Neurosci. 2006, 26, 5739–5749. [Google Scholar] [CrossRef] [Green Version]
- Cefis, M.; Chaney, R.; Quirie, A.; Santini, C.; Marie, C.; Garnier, P.; Prigent-Tessier, A. Endothelial cells are an important source of BDNF in rat skeletal muscle. Sci. Rep. 2022, 12, 311. [Google Scholar] [CrossRef] [PubMed]
- Delezie, J.; Weihrauch, M.; Maier, G.; Tejero, R.; Ham, D.J.; Gill, J.F.; Karrer-Cardel, B.; Rüegg, M.A.; Tabares, L.; Handschin, C. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 16111–16120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maderova, D.; Krumpolec, P.; Slobodova, L.; Schon, M.; Tirpakova, V.; Kovanicova, Z.; Klepochova, R.; Vajda, M.; Sutovsky, S.; Cvecka, J.; et al. Acute and regular exercise distinctly modulate serum, plasma and skeletal muscle BDNF in the elderly. Neuropeptides 2019, 78, 101961. [Google Scholar] [CrossRef] [PubMed]
- Cuppini, R.; Sartini, S.; Agostini, D.; Guescini, M.; Ambrogini, P.; Betti, M.; Bertini, L.; Vallasciani, M.; Stocchi, V. Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch. Ital. Biol. 2007, 145, 99–110. [Google Scholar] [PubMed]
- Matthews, V.B.; Astrom, M.B.; Chan, M.H.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Akerstrom, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Kaneko, F.; Iwamoto, E.; Saitoh, S.; Yamada, T. Neuromuscular electrical stimulation increases serum brain-derived neurotrophic factor in humans. Exp. Brain Res. 2019, 237, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef]
- Chan, C.B.; Tse, M.C.; Liu, X.; Zhang, S.; Schmidt, R.; Otten, R.; Liu, L.; Ye, K. Activation of muscular TrkB by its small molecular agonist 7,8-dihydroxyflavone sex-dependently regulates energy metabolism in diet-induced obese mice. Chem. Biol. 2015, 22, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Carbone, J.W.; McClung, J.P.; Pasiakos, S.M. Skeletal muscle responses to negative energy balance: Effects of dietary protein. Adv. Nutr. 2012, 3, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K.; Pedersen, M.; Krabbe, K.S.; Bruunsgaard, H.; Matthews, V.B.; Febbraio, M.A. Role of exercise-induced brain-derived neurotrophic factor production in the regulation of energy homeostasis in mammals. Exp. Physiol. 2009, 94, 1153–1160. [Google Scholar] [CrossRef]
- Dzamko, N.; Schertzer, J.D.; Ryall, J.G.; Steel, R.; Macaulay, S.L.; Wee, S.; Chen, Z.P.; Michell, B.J.; Oakhill, J.S.; Watt, M.J.; et al. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J. Physiol. 2008, 586, 5819–5831. [Google Scholar] [CrossRef] [PubMed]
- Hingst, J.R.; Kjobsted, R.; Birk, J.B.; Jorgensen, N.O.; Larsen, M.R.; Kido, K.; Larsen, J.K.; Kjeldsen, S.A.S.; Fentz, J.; Frosig, C.; et al. Inducible deletion of skeletal muscle AMPKalpha reveals that AMPK is required for nucleotide balance but dispensable for muscle glucose uptake and fat oxidation during exercise. Mol. Metab. 2020, 40, 101028. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jorgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.E.; Villadiego, A.; Morse, D.P. Fatty acid oxidation intermediates and the effect of fasting on oxidation in red and white skeletal muscle. Muscle Nerve 1983, 6, 367–373. [Google Scholar] [CrossRef]
- de Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential changes in the signal transduction responses of skeletal muscle following food deprivation. FASEB J. 2006, 20, 2579–2581. [Google Scholar] [CrossRef] [Green Version]
- Brockhoff, M.; Rion, N.; Chojnowska, K.; Wiktorowicz, T.; Eickhorst, C.; Erne, B.; Frank, S.; Angelini, C.; Furling, D.; Ruegg, M.A.; et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J. Clin. Investig. 2017, 127, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [Green Version]
- de Lange, P.; Moreno, M.; Silvestri, E.; Lombardi, A.; Goglia, F.; Lanni, A. Fuel economy in food-deprived skeletal muscle: Signaling pathways and regulatory mechanisms. FASEB J. 2007, 21, 3431–3441. [Google Scholar] [CrossRef]
- Antony, R.; Li, Y. BDNF secretion from C2C12 cells is enhanced by methionine restriction. Biochem. Biophys. Res. Commun. 2020, 533, 1347–1351. [Google Scholar] [CrossRef]
- Giacco, A.; Cioffi, F.; Cuomo, A.; Simiele, R.; Senese, R.; Silvestri, E.; Amoresano, A.; Fontanarosa, C.; Petito, G.; Moreno, M.; et al. Mild Endurance Exercise during Fasting Increases Gastrocnemius Muscle and Prefrontal Cortex Thyroid Hormone Levels through Differential BHB and BCAA-Mediated BDNF-mTOR Signaling in Rats. Nutrients 2022, 14, 1166. [Google Scholar] [CrossRef]
- Dolcet, X.; Egea, J.; Soler, R.M.; Martin-Zanca, D.; Comella, J.X. Activation of phosphatidylinositol 3-kinase, but not extracellular-regulated kinases, is necessary to mediate brain-derived neurotrophic factor-induced motoneuron survival. J. Neurochem. 1999, 73, 521–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, S.B.; Treebak, J.T.; Viollet, B.; Schjerling, P.; Vaulont, S.; Wojtaszewski, J.F.; Richter, E.A. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E331–E339. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Roves, P.M.; Osler, M.E.; Holmstrom, M.H.; Zierath, J.R. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J. Biol. Chem. 2008, 283, 35724–35734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.; Tse, M.C.L.; Yang, X.; Brobst, D.; Liu, Z.; Pang, B.P.S.; Chan, W.S.; Zaw, A.M.; Chow, B.K.C.; Ye, K.; et al. BDNF mimetic alleviates body weight gain in obese mice by enhancing mitochondrial biogenesis in skeletal muscle. Metabolism 2018, 87, 113–122. [Google Scholar] [CrossRef]
- Matsumoto, J.; Takada, S.; Furihata, T.; Nambu, H.; Kakutani, N.; Maekawa, S.; Mizushima, W.; Nakano, I.; Fukushima, A.; Yokota, T.; et al. Brain-Derived Neurotrophic Factor Improves Impaired Fatty Acid Oxidation Via the Activation of Adenosine Monophosphate-Activated Protein Kinase-a—Proliferator-Activated Receptor-r Coactivator-1a Signaling in Skeletal Muscle of Mice With Heart Failure. Circ. Heart Fail. 2021, 14, e005890. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [Green Version]
- Lejri, I.; Grimm, A.; Eckert, A. Mitochondria, Estrogen and Female Brain Aging. Front. Aging Neurosci. 2018, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.M.; Lombardo, P.S.; Malik, N.; Brun, S.N.; Hellberg, K.; Van Nostrand, J.L.; Garcia, D.; Baumgart, J.; Diffenderfer, K.; Asara, J.M.; et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 2021, 7, eabg4544. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 2019, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, B.; Li, T.; Zhu, Y.; Luo, G.; Jiang, Y.; Tang, F.; Jian, Z.; Xiao, Y. AMPK activation serves a critical role in mitochondria quality control via modulating mitophagy in the heart under chronic hypoxia. Int. J. Mol. Med. 2018, 41, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, S.P.; Shao, Q.; Li, P.F.; Sun, Y.; Luo, L.Z.; Yan, X.Q.; Fan, Z.Y.; Hu, J.; Zhao, J.; et al. Brain-derived neurotrophic factor mimetic, 7,8-dihydroxyflavone, protects against myocardial ischemia by rebalancing optic atrophy 1 processing. Free Radic. Biol. Med. 2019, 145, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhu, Y.; Li, Y.; Ding, X.; Ma, W.; Han, X.; Wang, B. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 2019, 24, 511–528. [Google Scholar] [CrossRef]
- Shim, M.S.; Kim, K.Y.; Noh, M.; Ko, J.Y.; Ahn, S.; An, M.A.; Iwata, T.; Perkins, G.A.; Weinreb, R.N.; Ju, W.K. Optineurin E50K triggers BDNF deficiency-mediated mitochondrial dysfunction in retinal photoreceptor cell line. Biochem. Biophys. Res. Commun. 2018, 503, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Takada, S.; Kinugawa, S.; Furihata, T.; Nambu, H.; Kakutani, N.; Tsuda, M.; Fukushima, A.; Yokota, T.; Tanaka, S.; et al. Brain-Derived Neurotrophic Factor Improves Limited Exercise Capacity in Mice With Heart Failure. Circulation 2018, 138, 2064–2066. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Cameron, I.; Franklin, P.; Spedding, M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur. J. Neurosci. 2004, 20, 1189–1196. [Google Scholar] [CrossRef]
- Nguyen, H.T.N.; Kato, H.; Masuda, K.; Yamaza, H.; Hirofuji, Y.; Sato, H.; Pham, T.T.M.; Takayama, F.; Sakai, Y.; Ohga, S.; et al. Impaired neurite development associated with mitochondrial dysfunction in dopaminergic neurons differentiated from exfoliated deciduous tooth-derived pulp stem cells of children with autism spectrum disorder. Biochem. Biophys. Rep. 2018, 16, 24–31. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Vergnes, L.; Reue, K.; Gomez-Pinilla, F. Coupling energy homeostasis with a mechanism to support plasticity in brain trauma. Biochim. Biophys. Acta 2014, 1842, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Du, J.; Pan, Y.; Chen, T.; Zhao, L.; Zhu, Y.; Chen, Y.; Zheng, Y.; Liu, Y.; Sun, L.; et al. Activation of cardiac TrkB receptor by its small molecular agonist 7,8-dihydroxyflavone inhibits doxorubicin-induced cardiotoxicity via enhancing mitochondrial oxidative phosphorylation. Free Radic. Biol. Med. 2019, 130, 557–567. [Google Scholar] [CrossRef]
- Prince, C.S.; Maloyan, A.; Myatt, L. Tropomyosin Receptor Kinase B Agonist, 7,8-Dihydroxyflavone, Improves Mitochondrial Respiration in Placentas From Obese Women. Reprod. Sci. 2018, 25, 452–462. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Siemen, D.; Mawrin, C.; Horn, T.F.; Dietzmann, K. The neurotrophin receptor TrkB is colocalized to mitochondrial membranes. Int. J. Biochem. Cell Biol. 2006, 38, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Seidl, K.; Erck, C.; Buchberger, A. Evidence for the participation of nerve growth factor and its low-affinity receptor (p75NTR) in the regulation of the myogenic program. J. Cell Physiol. 1998, 176, 10–21. [Google Scholar] [CrossRef]
- Miura, P.; Amirouche, A.; Clow, C.; Belanger, G.; Jasmin, B.J. Brain-derived neurotrophic factor expression is repressed during myogenic differentiation by miR-206. J. Neurochem. 2012, 120, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Jin, X.A. Brain-derived neurotrophic factor inhibits neuromuscular junction maturation in a cAMP-PKA-dependent way. Neurosci. Lett. 2015, 591, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xie, K.; Lu, B. Neurotrophins promote maturation of developing neuromuscular synapses. J. Neurosci. 1995, 15, 4796–4805. [Google Scholar] [CrossRef] [Green Version]
- Garcia, N.; Tomas, M.; Santafe, M.M.; Besalduch, N.; Lanuza, M.A.; Tomas, J. The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J. Neurosci. 2010, 30, 16514–16522. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.; Ruggiero, F.P.; Chang, Q.; Shi, Y.J.; Rich, M.M.; Kraner, S.; Balice-Gordon, R.J. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron 1999, 24, 567–583. [Google Scholar] [CrossRef] [Green Version]
- McKay, B.R.; Nederveen, J.P.; Fortino, S.A.; Snijders, T.; Joanisse, S.; Kumbhare, D.A.; Parise, G. Brain-derived neurotrophic factor is associated with human muscle satellite cell differentiation in response to muscle-damaging exercise. Appl. Physiol. Nutr. Metab. 2020, 45, 581–590. [Google Scholar] [CrossRef]
- Yu, T.; Chang, Y.; Gao, X.L.; Li, H.; Zhao, P. Dynamic Expression and the Role of BDNF in Exercise-induced Skeletal Muscle Regeneration. Int. J. Sports Med. 2017, 38, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Clow, C.; Jasmin, B.J. Brain-derived neurotrophic factor regulates satellite cell differentiation and skeltal muscle regeneration. Mol. Biol. Cell 2010, 21, 2182–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aby, K.; Antony, R.; Eichholz, M.; Srinivasan, R.; Li, Y. Enhanced pro-BDNF-p75NTR pathway activity in denervated skeletal muscle. Life Sci. 2021, 286, 120067. [Google Scholar] [CrossRef] [PubMed]
- Halievski, K.; Xu, Y.; Haddad, Y.W.; Tang, Y.P.; Yamada, S.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Muscle BDNF improves synaptic and contractile muscle strength in Kennedy’s disease mice in a muscle-type specific manner. J. Physiol. 2020, 598, 2719–2739. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Kwatra, M.; Dwivedi, D.K.; Choubey, P.; Lahkar, M.; Jangra, A. 7,8-Dihydroxyflavone alleviated the high-fat diet and alcohol-induced memory impairment: Behavioral, biochemical and molecular evidence. Psychopharmacology 2020, 237, 1827–1840. [Google Scholar] [CrossRef]
- Krabbe, K.S.; Nielsen, A.R.; Krogh-Madsen, R.; Plomgaard, P.; Rasmussen, P.; Erikstrup, C.; Fischer, C.P.; Lindegaard, B.; Petersen, A.M.; Taudorf, S.; et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 2007, 50, 431–438. [Google Scholar] [CrossRef]
- Glud, M.; Christiansen, T.; Larsen, L.H.; Richelsen, B.; Bruun, J.M. Changes in Circulating BDNF in relation to Sex, Diet, and Exercise: A 12-Week Randomized Controlled Study in Overweight and Obese Participants. J. Obes. 2019, 2019, 4537274. [Google Scholar] [CrossRef]
- Polyakova, M.; Stuke, K.; Schuemberg, K.; Mueller, K.; Schoenknecht, P.; Schroeter, M.L. BDNF as a biomarker for successful treatment of mood disorders: A systematic & quantitative meta-analysis. J. Affect. Disord. 2015, 174, 432–440. [Google Scholar] [CrossRef]
- Nilsson, J.; Ekblom, O.; Ekblom, M.; Lebedev, A.; Tarassova, O.; Moberg, M.; Lovden, M. Acute increases in brain-derived neurotrophic factor in plasma following physical exercise relates to subsequent learning in older adults. Sci. Rep. 2020, 10, 4395. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jakobsen, A.H.; Pilegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release from the human brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef] [Green Version]
- Bejot, Y.; Mossiat, C.; Giroud, M.; Prigent-Tessier, A.; Marie, C. Circulating and brain BDNF levels in stroke rats. Relevance to clinical studies. PLoS ONE 2011, 6, e29405. [Google Scholar] [CrossRef] [PubMed]
- Podyma, B.; Parekh, K.; Guler, A.D.; Deppmann, C.D. Metabolic homeostasis via BDNF and its receptors. Trends Endocrinol. Metab. 2021, 32, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Blum, R.; Pichler, B.; Lepier, A.; Kafitz, K.W.; Konnerth, A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 2003, 426, 74–78. [Google Scholar] [CrossRef]
- Biffo, S.; Offenhauser, N.; Carter, B.D.; Barde, Y.A. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development 1995, 121, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Eide, F.F.; Vining, E.R.; Eide, B.L.; Zang, K.; Wang, X.Y.; Reichardt, L.F. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J. Neurosci. 1996, 16, 3123–3129. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Tomassoni-Ardori, F.; Babini, L.; Becker, J.; Barrick, C.; Puverel, S.; Tessarollo, L. BDNF modulates heart contraction force and long-term homeostasis through truncated TrkB.T1 receptor activation. J. Cell Biol. 2015, 210, 1003–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, S.G.; Lovering, R.M.; Renn, C.L.; Leitch, C.C.; Liu, X.; Tallon, L.J.; Sadzewicz, L.D.; Pratap, A.; Ott, S.; Sengamalay, N.; et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Physiol. Cell Physiol. 2012, 302, C141–C153. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L. Transport of human recombinant brain-derived neurotrophic factor (BDNF) through the rat blood-brain barrier in vivo using vector-mediated peptide drug delivery. Pharm. Res. 1994, 11, 738–746. [Google Scholar] [CrossRef]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Alcala-Barraza, S.R.; Lee, M.S.; Hanson, L.R.; McDonald, A.A.; Frey, W.H., 2nd; McLoon, L.K. Intranasal delivery of neurotrophic factors BDNF, CNTF, EPO, and NT-4 to the CNS. J. Drug. Target. 2010, 18, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Duman, R.; Sanacora, G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cai, Z.D.; Jiang, W.T.; Fang, Y.Y.; Sun, W.X.; Wang, X. Systematic review and meta-analysis of the effects of exercise on depression in adolescents. Child. Adolesc. Psychiatry Ment. Health 2022, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.Y.; Kelliny, S.; Liu, L.C.; Al-Hawwas, M.; Zhou, X.F.; Bobrovskaya, L. Peripheral ProBDNF Delivered by an AAV Vector to the Muscle Triggers Depression-Like Behaviours in Mice. Neurotox. Res. 2020, 38, 626–639. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iu, E.C.Y.; Chan, C.B. Is Brain-Derived Neurotrophic Factor a Metabolic Hormone in Peripheral Tissues? Biology 2022, 11, 1063. https://doi.org/10.3390/biology11071063
Iu ECY, Chan CB. Is Brain-Derived Neurotrophic Factor a Metabolic Hormone in Peripheral Tissues? Biology. 2022; 11(7):1063. https://doi.org/10.3390/biology11071063
Chicago/Turabian StyleIu, Elsie Chit Yu, and Chi Bun Chan. 2022. "Is Brain-Derived Neurotrophic Factor a Metabolic Hormone in Peripheral Tissues?" Biology 11, no. 7: 1063. https://doi.org/10.3390/biology11071063