
Development of a Rapid Reverse Transcription-Recombinase Polymerase Amplification Couple Nucleic Acid Lateral Flow Method for Detecting Porcine Epidemic Diarrhoea Virus

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Clinical Samples and Nucleic Acid Preparation
2.2. Primers and Probe Design
2.3. RT-qPCR
2.4. RPA Primer Screening
2.5. RT-RPA and RT-RPA-NALF
2.6. Evaluation of the RT-RPA-NALF
3. Results
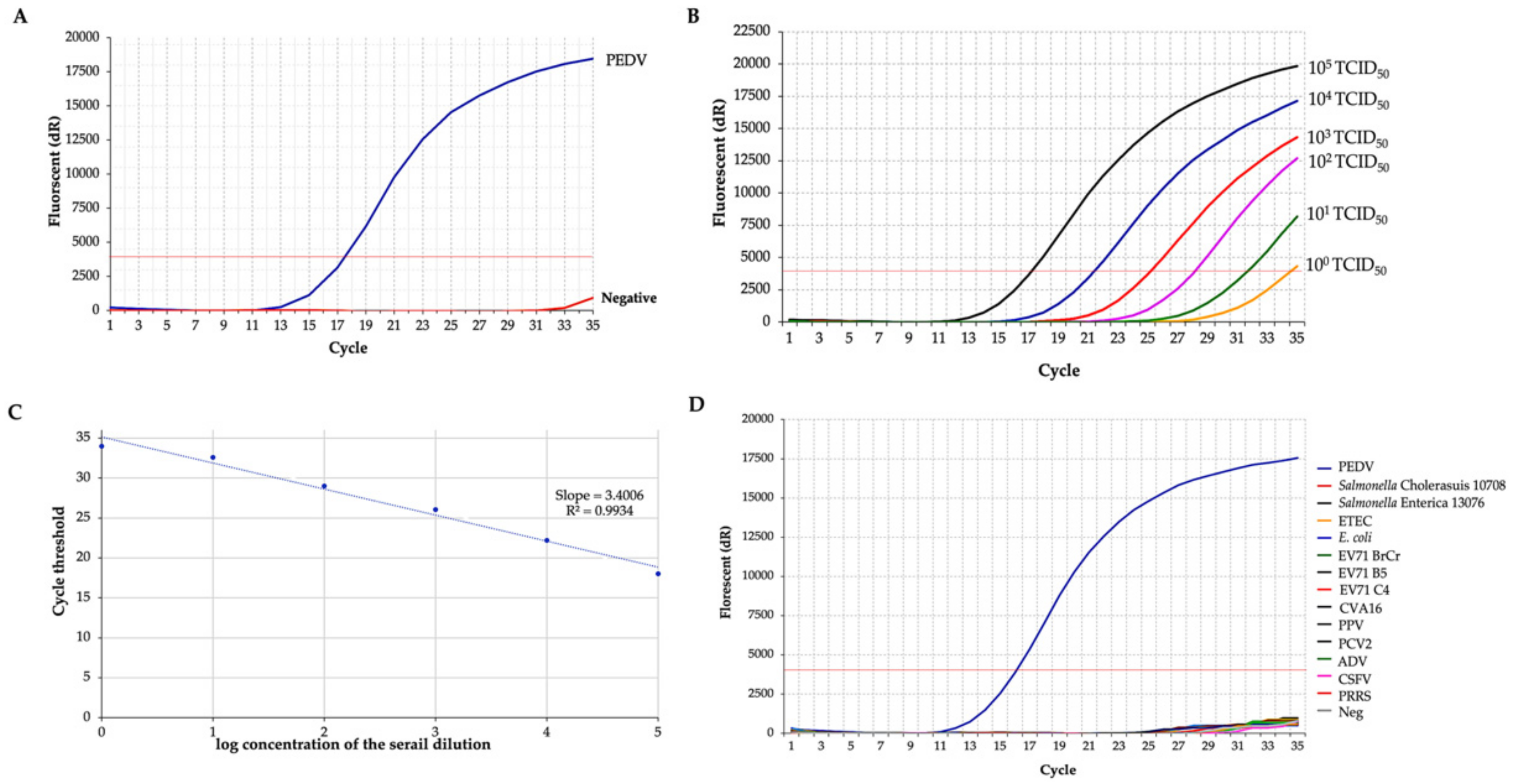
3.1. Establishment of the PEDV RT-qPCR
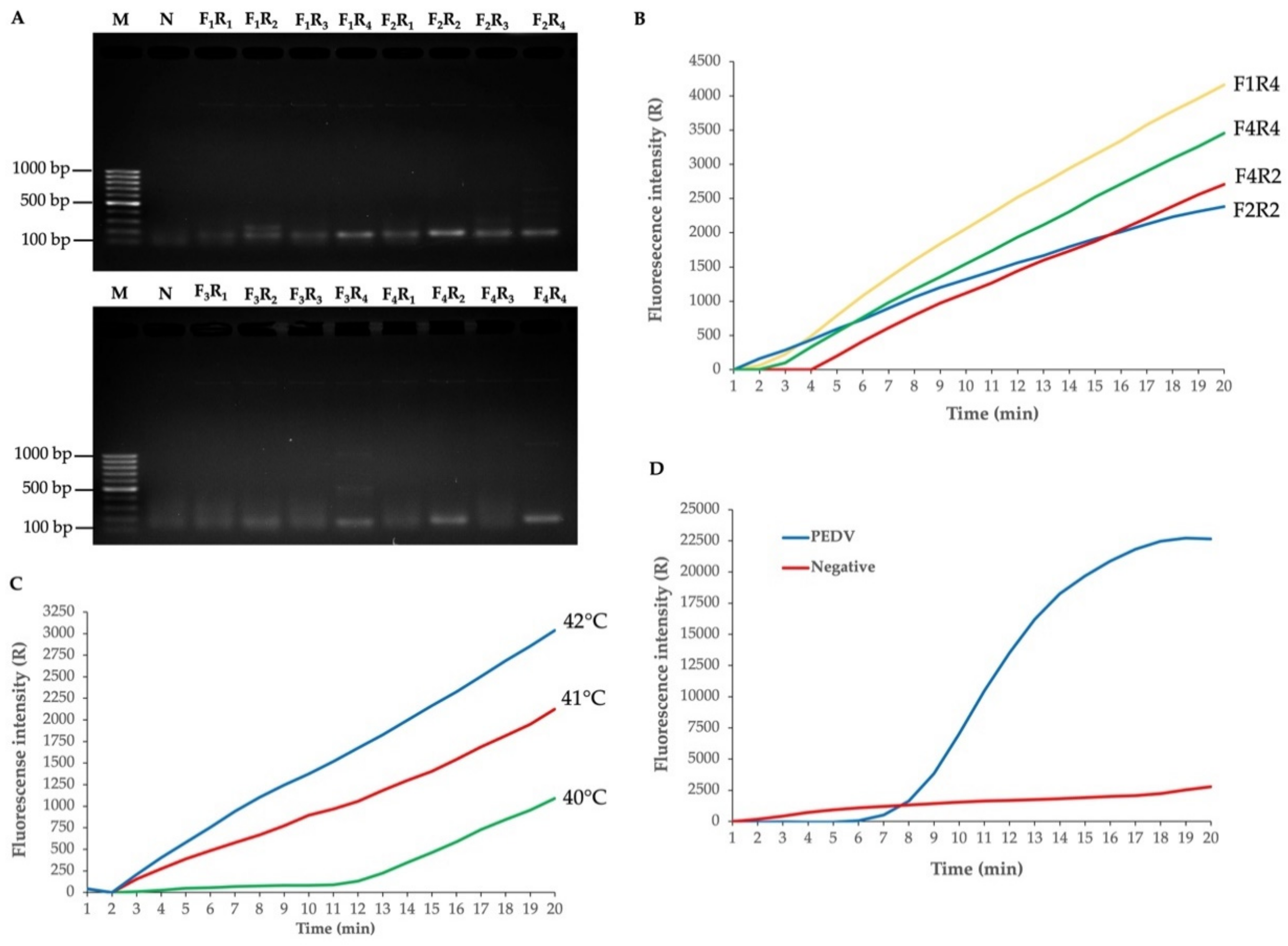
3.2. Establishment of the PEDV RT-RPA-NALF
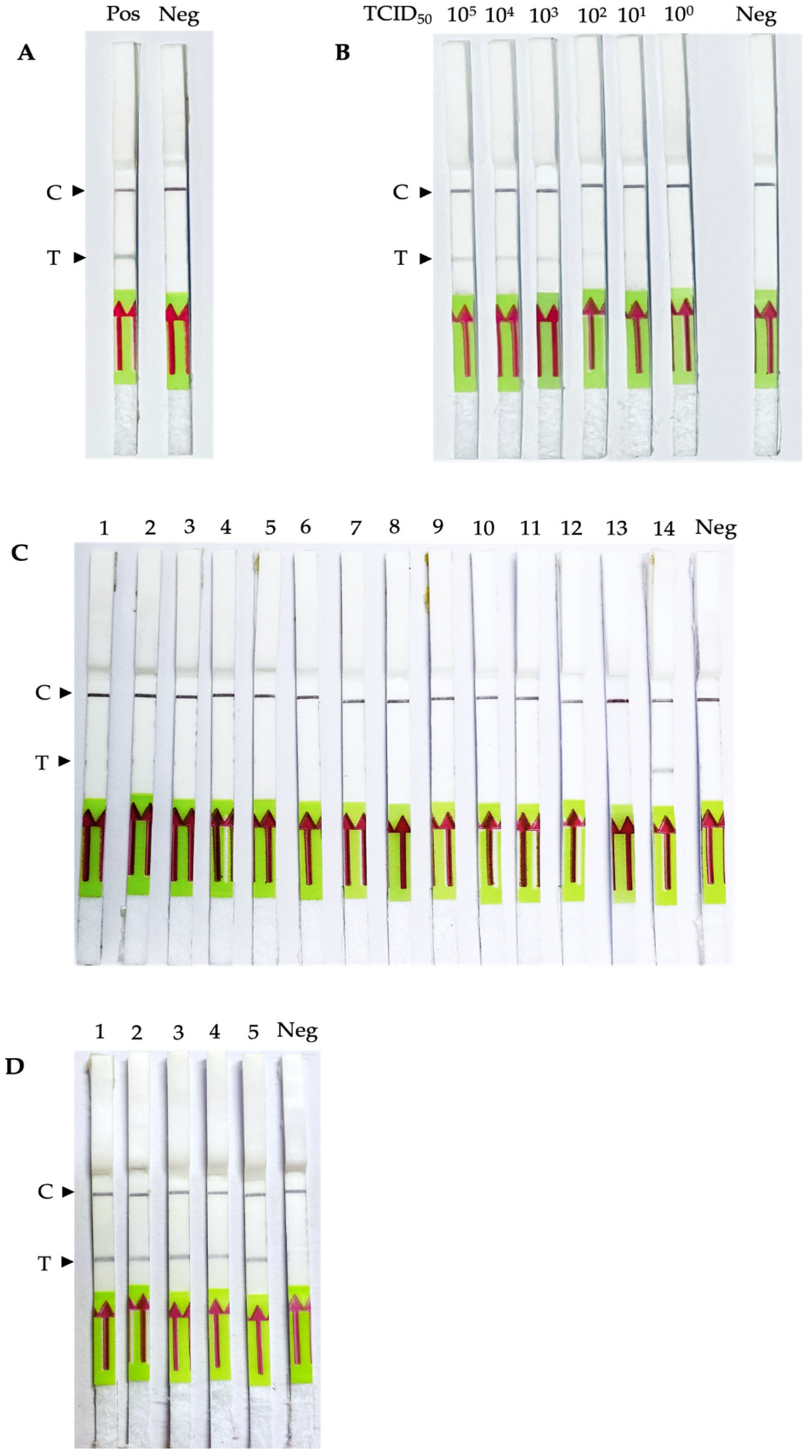
3.3. Sensitivity, Specificity and Usability of the PEDV RT-RPA-NALF
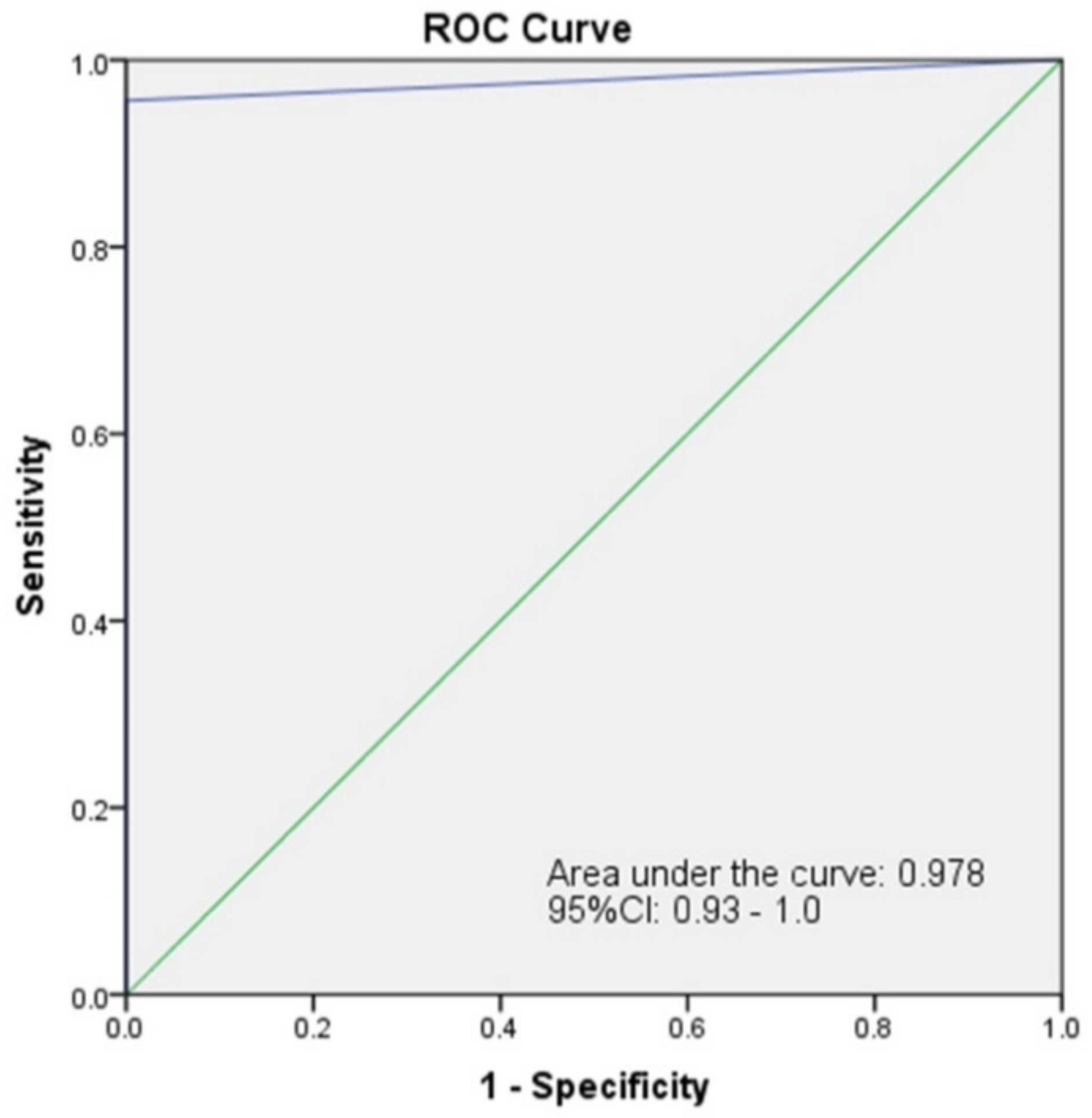
3.4. Validation of Simulated and Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Suda, Y.; Miyazaki, A.; Miyazawa, K.; Shibahara, T.; Ohashi, S. Systemic and Intestinal Porcine Epidemic Diarrhea Virus-specific Antibody Response and Distribution of Antibody-Secreting Cells in Experimentally Infected Conventional Pigs. Vet. Res. 2021, 52, 2. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Porcine Epidemic Diarrhea Virus: An Emerging and Re-emerging Epizootic Swine Virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Ma, Z.; Li, Y.; Gao, S.; Xiao, S. Porcine Epidemic Diarrhea Virus: Molecular Mechanisms of Attenuation And Vaccines. Microb. Pathog. 2020, 149, 104553. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Lv, C.; Xiao, Y.; Li, X. Porcine Epidemic Diarrhea Virus: Current Insights. Virus Adapt. Treat. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sritun, J.; Inthong, N.; Jala, S.; Phatthanakunanan, S.; Sirinarumitr, K.; Lertwatcharasarakul, P.; Sirinarumitr, T. Expression of Recombinant S2 Domain of Spike Protein of Porcine Epidemic Diarrhea Virus. Thai J. Vet. Med. 2021, 51, 221–230. [Google Scholar]
- Kim, S.-J.; Nguyen, V.-G.; Huynh, T.-M.; Park, Y.-H.; Park, B.-K.; Chung, H.-C. Molecular Characterization of Porcine Epidemic Diarrhea Virus and Its New Genetic Classification Based on the Nucleocapsid Gene. Viruses 2020, 12, 790. [Google Scholar] [CrossRef]
- Jung, K.; Saif, L.J. Porcine Epidemic Diarrhea Virus Infection: Etiology, Epidemiology, Pathogenesis and Immunoprophylaxis. Vet. J. 2015, 204, 134–143. [Google Scholar] [CrossRef]
- Stott, C.J.; Temeeyasen, G.; Tripipat, T.; Kaewprommal, P.; Tantituvanont, A.; Piriyapongsa, J.; Nilubol, D. Evolutionary and Epidemiological Analyses Based on Spike Genes of Porcine Epidemic Diarrhea Virus Circulating in Thailand in 2008–2015. Infect. Genet. Evol. 2017, 50, 70–76. [Google Scholar] [CrossRef]
- Temeeyasen, G.; Srijanwad, A.; Tripipat, T.; Tantituvanont, A.; Nilubol, D. Emergence of a Classical Variant of Porcine Epidemic Diarrhea Virus Novel to Thailand Responsible for The Milder Clinical Disease in a Herd Previously Infected with a Pandemic Variant. Thai J. Vet. Med. 2018, 48, 663–669. [Google Scholar]
- Diel, D.; Lawson, S.; Okda, F.; Singrey, A.; Clement, T.; Fernandes, M.; Christopher-Hennings, J.; Nelson, E. Porcine Epidemic Diarrhea Virus: An Overview of Current Virological and Serological Diagnostic Methods. Virus Res. 2016, 226, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Fu, Y.; Li, B.; Chen, J.; Wang, J.; Yin, B.; Sha, W.; Liu, G. Development of a Multiplex RT-PCR for the Detection of Major Diarrhoeal Viruses in Pig Herds in China. Transbound. Emerg. Dis. 2019, 67, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, B.; Sun, J.; Zhu, L.; Zhou, J.; Zhao, Y.; Yu, Z.; Sun, B.; Guo, R.; He, K.; Li, B. Development of a Novel Double Antibody Sandwich Quantitative Enzyme-Linked Immunosorbent Assay for Detection of Porcine Epidemic Diarrhea Virus Antigen. Front. Vet. Sci. 2020, 7, 540248. [Google Scholar] [CrossRef] [PubMed]
- Craw, P.; Balachandran, W. Isothermal Nucleic Acid Amplification Technologies for Point-of-Care Diagnostics: A Critical Review. Lab Chip 2012, 12, 2469–2486. [Google Scholar] [CrossRef] [PubMed]
- Olaf, P.; Colin, H.S.; Derek, L.; Niall, A.A. DNA Detection using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar]
- Euler, M.; Wang, Y.; Nentwich, O.; Piepenburg, O.; Hufert, F.T.; Weidmann, M. Recombinase Polymerase Amplification Assay for Rapid Detection of Rift Valley Fever Virus. J. Clin. Virol. 2012, 54, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Amer, H.; El Wahed, A.A.; Shalaby, M.; Almajhdi, F.; Hufert, F.; Weidmann, M. A New Approach for Diagnosis of Bovine Coronavirus using a Reverse Transcription Recombinase Polymerase Amplification Assay. J. Virol. Methods 2013, 193, 337–340. [Google Scholar] [CrossRef]
- El Wahed, A.A.; El-Deeb, A.; El-Tholoth, M.; El Kader, H.A.; Ahmed, A.; Hassan, S.; Hoffmann, B.; Haas, B.; Shalaby, M.A.; Hufert, F.T.; et al. A Portable Reverse Transcription Recombinase Polymerase Amplification Assay for Rapid Detection of Foot-and-Mouth Disease Virus. PLoS ONE 2013, 8, e71642. [Google Scholar] [CrossRef] [Green Version]
- Boyle, D.S.; Lehman, D.A.; Lillis, L.; Peterson, D.; Singhal, M.; Armes, N.; Parker, M.; Piepenburg, O.; Overbaugh, J. Rapid Detection of HIV-1 Proviral DNA for Early Infant Diagnosis Using Recombinase Polymerase Amplification. mBio 2013, 4, e00135-13. [Google Scholar] [CrossRef] [Green Version]
- Euler, M.; Wang, Y.; Otto, P.; Tomaso, H.; Escudero, R.; Anda, P.; Hufert, F.T.; Weidmann, M. Recombinase Polymerase Amplification Assay for Rapid Detection of Francisella tularensis. J. Clin. Microbiol. 2012, 50, 2234–2238. [Google Scholar] [CrossRef] [Green Version]
- Euler, M.; Wang, Y.; Heidenreich, D.; Patel, P.; Strohmeier, O.; Hakenberg, S.; Niedrig, M.; Hufert, F.T.; Weidmann, M. Development of a Panel of Recombinase Polymerase Amplification Assays for Detection of Biothreat Agents. J. Clin. Microbiol. 2013, 51, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Kersting, S.; Rausch, V.; Bier, F.F.; Von Nickisch-Rosenegk, M. Multiplex Isothermal Solid-Phase Recombinase Polymerase Amplification for the Specific and Fast DNA-based Detection of Three Bacterial Pathogens. Mikrochim. Acta 2014, 181, 1715–1723. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Felipe, S.; Tortajada-Genaro, L.A.; Morais, S.; Puchades, R.; Maquieira, Á. Isothermal DNA Amplification Strategies for Duplex Microorganism Detection. Food Chem. 2015, 174, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Powell, M.L.; Bowler, F.R.; Martinez, A.J.; Greenwood, C.J.; Armes, N.; Piepenburg, O. New Fpg Probe Chemistry for Direct Detection of Recombinase Polymerase Amplification on Lateral Flow Strips. Anal. Biochem. 2018, 543, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Posthuma-Trumpie, G.A.; Korf, J.; van Amerongen, A. Lateral Flow (Immuno)Assay: Its Strengths, Weaknesses, Opportunities and Threats. A Literature Survey. Anal. Bioanal. Chem. 2008, 393, 569–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Enríquez, A.; Herrera-Camacho, I.; Millán-Pérez-Peña, L.; Reyes-Leyva, J.; Santos-López, G.; Rivera-Benítez, J.F.; Rosas-Murrieta, N.H. Predicted 3D Model of the M Protein of Porcine Epidemic Diarrhea Virus and Analysis of its Immunogenic Potential. PLoS ONE 2022, 17, e0263582. [Google Scholar] [CrossRef]
- Duarte, M.; Tobler, K.; Bridgen, A.; Rasschaert, D.; Ackermann, M.; Laude, H. Sequence Analysis of the Porcine Epidemic Diarrhea Virus Genome between the Nucleocapsid and Spike Protein Genes Reveals a Polymorphic ORF. Virology 1994, 198, 466–476. [Google Scholar] [CrossRef]
- Kailasa, S.K.; Mehta, V.N.; Koduru, J.R.; Basu, H.; Singhal, R.K.; Murthy, Z.V.P.; Park, T.J. An Overview of Molecular Biology and Nanotechnology based Analytical Methods for the Detection of Sars-Cov-2: Promising Biotools for the Rapid Diagnosis of Covid-19. Analyst 2021, 146, 1489–1513. [Google Scholar] [CrossRef]
- Alvarez, J.; Sarradell, J.; Morrison, R.; Perez, A. Impact of Porcine Epidemic Diarrhea on Performance of Growing Pigs. PLoS ONE 2015, 10, e0120532. [Google Scholar] [CrossRef]
- Song, D.; Huang, D.; Peng, Q.; Huang, T.; Chen, Y.; Zhang, T.; Nie, X.; He, H.; Wang, P.; Liu, Q.; et al. Molecular Characterization and Phylogenetic Analysis of Porcine Epidemic Diarrhea Viruses Associated with Outbreaks of Severe Diarrhea in Piglets in Jiangxi, China 2013. PLoS ONE 2015, 10, e0120310. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Xiao, S. Porcine Epidemic Diarrhea in China. Virus Res. 2016, 226, 7–13. [Google Scholar] [CrossRef]
- Krõlov, K.; Frolova, J.; Tudoran, O.; Suhorutsenko, J.; Lehto, T.; Sibul, H.; Mäger, I.; Laanpere, M.; Tulp, I.; Langel, Ü. Sensitive and Rapid Detection of Chlamydia trachomatis by Recombinase Polymerase Amplification Directly from Urine Samples. J. Mol. Diagn. 2014, 16, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.D.; Jaykus, L.-A. Development of a Recombinase Polymerase Amplification Assay for Detection of Epidemic Human Noroviruses. Sci. Rep. 2017, 7, 40244. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lian, K.; Zhu, M.; Tang, Y.; Zhang, M. Visual Detection of Porcine Epidemic Diarrhea Virus by Recombinase Polymerase Amplification Combined with Lateral Flow Dipstrip. BMC Vet. Res. 2022, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, R.; Wang, J.; Han, Q.; Liu, L.; Li, Y.; Yuan, W. Real-time Reverse Transcription Recombinase Polymerase Amplification Assay for Rapid Detection of Porcine Epidemic Diarrhea Virus. J. Virol. Methods 2018, 253, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, M.; Li, J.; Cai, W.; Xie, Y.; Si, G.; Xiao, L.; Cong, F.; He, D. Rapid Detection of Porcine Deltacoronavirus and Porcine Epidemic Diarrhea Virus using the Duplex Recombinase Polymerase Amplification Method. J. Virol. Methods 2021, 292, 114096. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Primers and Probe | Sequence 5′–3′ | Product Size (bp) |
|---|---|---|---|
| RT-qPCR | MPEDVF | ATGTCTAACGGTTCTATTCCC | 113 |
| MPEDVR | TAATGGCCATACTGAAGCAC | ||
| RT-RPA-NALF | F1 | CTGTGATGGGCCGACAGGTCTGCATTCCAG | 170 |
| F2 | CTGTGATGGGCCGACAGGTCTGCATTCCAGTG | ||
| F3 | CTGTGATGGGCCGACAGGTCTGCATTCCAGTGC | ||
| F4 | CTGTGATGGGCCGACAGGTCTGCATTCCAGTGCTTG | ||
| R1 | GACAATTGTTGTAGTGGCCTTGGCGACTG | ||
| R2 | GACAATTGTTGTAGTGGCCTTGGCGACTGTG | ||
| R3 | ACAATTGTTGTAGTGGCCTTGGCGACTGTGAC | ||
| R4 | GACAATTGTTGTAGTGGCCTTGGCGACTGTGACG | ||
| Probe | CTGGTGTAACGCTAACACTCCTTAGTGG [FAM-dT] A [THF] A [BHQ-1-DT] TGCTTGTAGAGCG [3PHOS] |
| RT-qPCR | Total | 1 Coincident Rate | 2 Sensitivity | 3 Specificity | ||
|---|---|---|---|---|---|---|
| Positive | Negative | |||||
| RT-RPA-NALF | 98.33% | 95.65% | 100% | |||
| RT-RPA-NALF Positive | 22 | 0 | 22 | |||
| Negative | 1 | 37 | 38 | |||
| Total | 23 | 37 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pewlaoo, S.; Phanthong, S.; Kong-Ngoen, T.; Santajit, S.; Tunyong, W.; Buranasinsup, S.; Kaeoket, K.; Thavorasak, T.; Pumirat, P.; Sookrung, N.; et al. Development of a Rapid Reverse Transcription-Recombinase Polymerase Amplification Couple Nucleic Acid Lateral Flow Method for Detecting Porcine Epidemic Diarrhoea Virus. Biology 2022, 11, 1018. https://doi.org/10.3390/biology11071018
Pewlaoo S, Phanthong S, Kong-Ngoen T, Santajit S, Tunyong W, Buranasinsup S, Kaeoket K, Thavorasak T, Pumirat P, Sookrung N, et al. Development of a Rapid Reverse Transcription-Recombinase Polymerase Amplification Couple Nucleic Acid Lateral Flow Method for Detecting Porcine Epidemic Diarrhoea Virus. Biology. 2022; 11(7):1018. https://doi.org/10.3390/biology11071018
Chicago/Turabian StylePewlaoo, Seatthanan, Siratcha Phanthong, Thida Kong-Ngoen, Sirijan Santajit, Witawat Tunyong, Shutipen Buranasinsup, Kampon Kaeoket, Techit Thavorasak, Pornpan Pumirat, Nitat Sookrung, and et al. 2022. "Development of a Rapid Reverse Transcription-Recombinase Polymerase Amplification Couple Nucleic Acid Lateral Flow Method for Detecting Porcine Epidemic Diarrhoea Virus" Biology 11, no. 7: 1018. https://doi.org/10.3390/biology11071018