
Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and the Mutations
2.2. Expression and Molecular Biology of Nav1.4 Constructs
2.3. Preparation of Cell Lines for Transfection
2.4. Electrophysiological Recordings
2.5. Construction of Activation and Inactivation Curves
2.6. Data Analysis and Statistics
3. Results
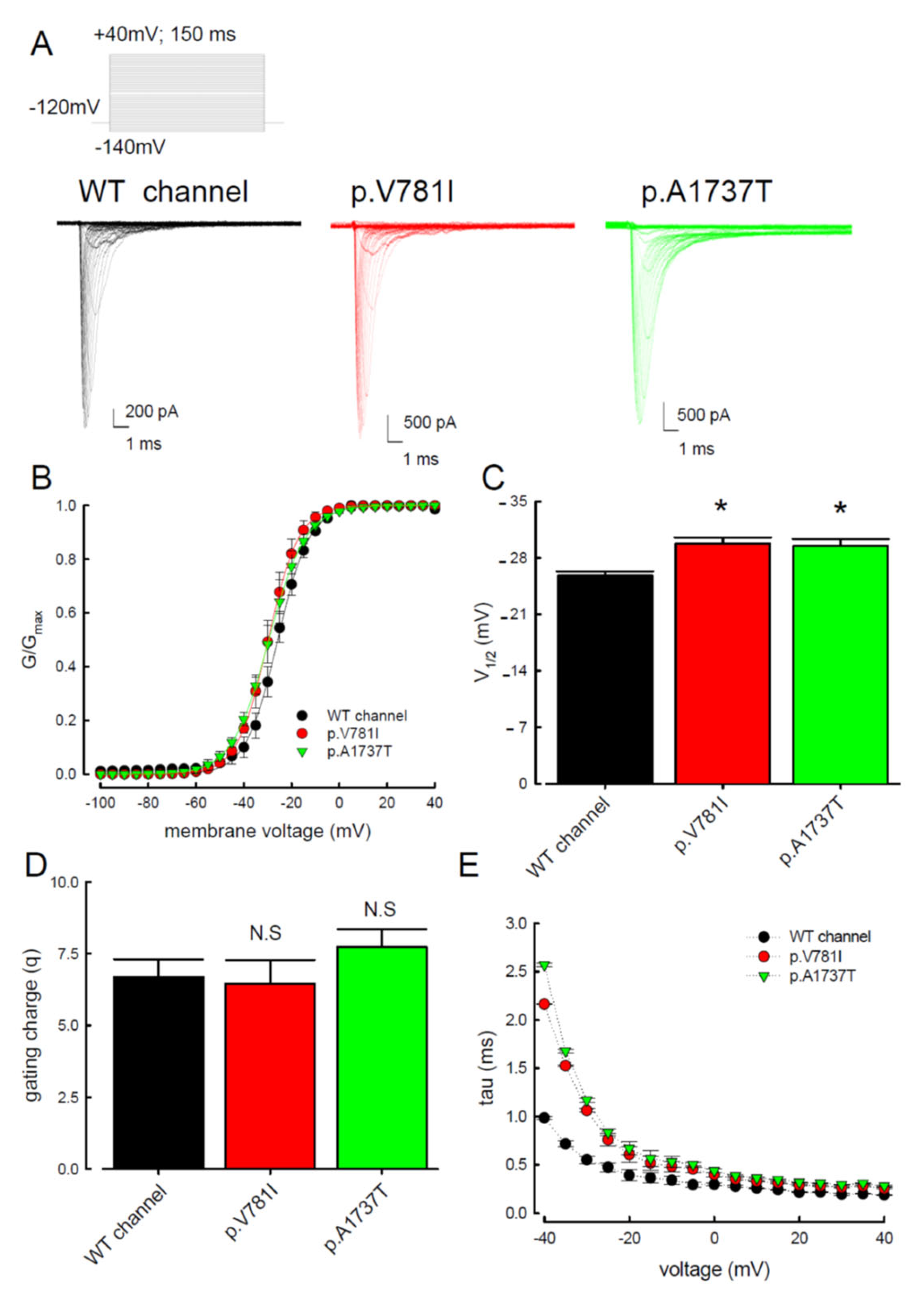
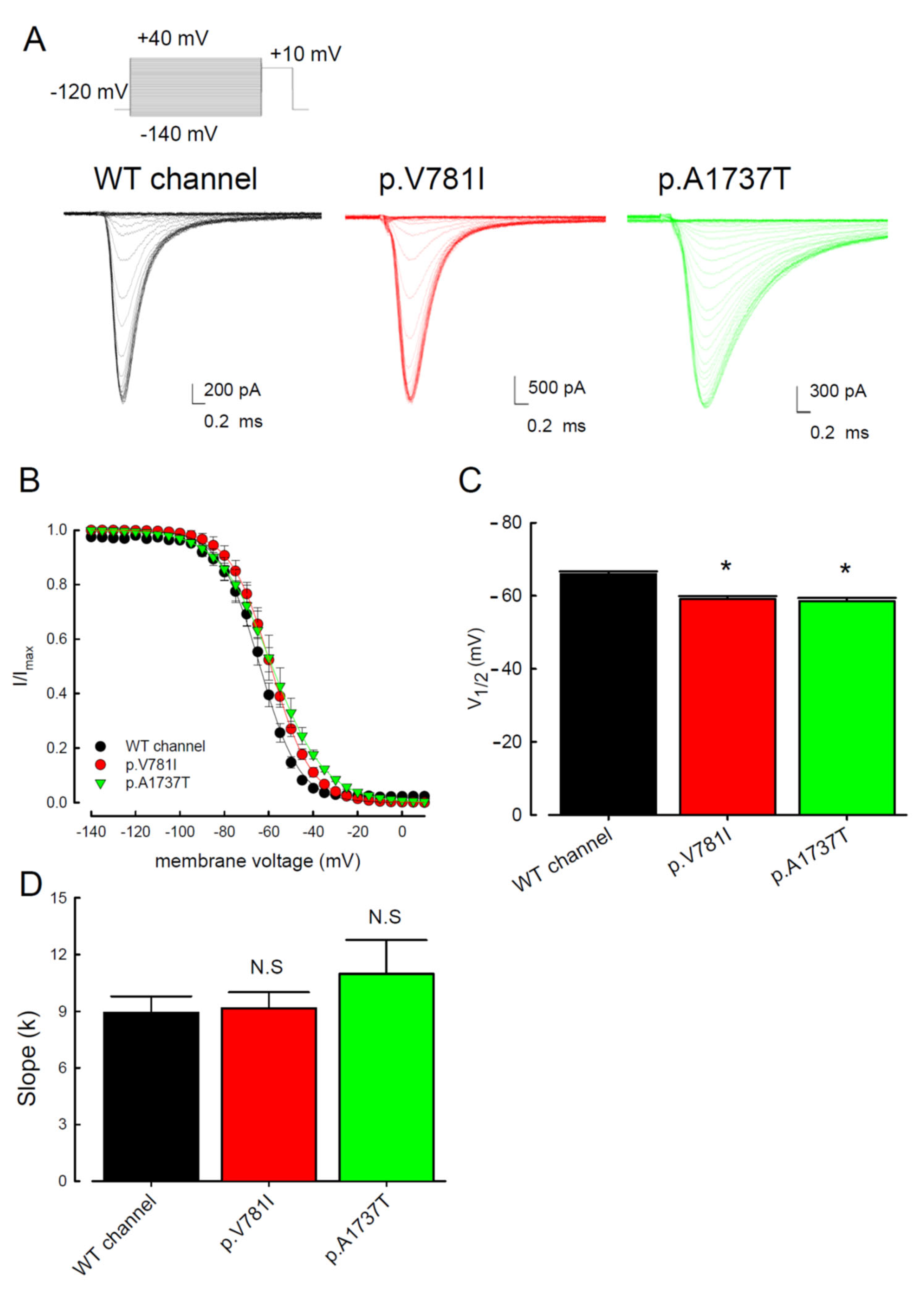
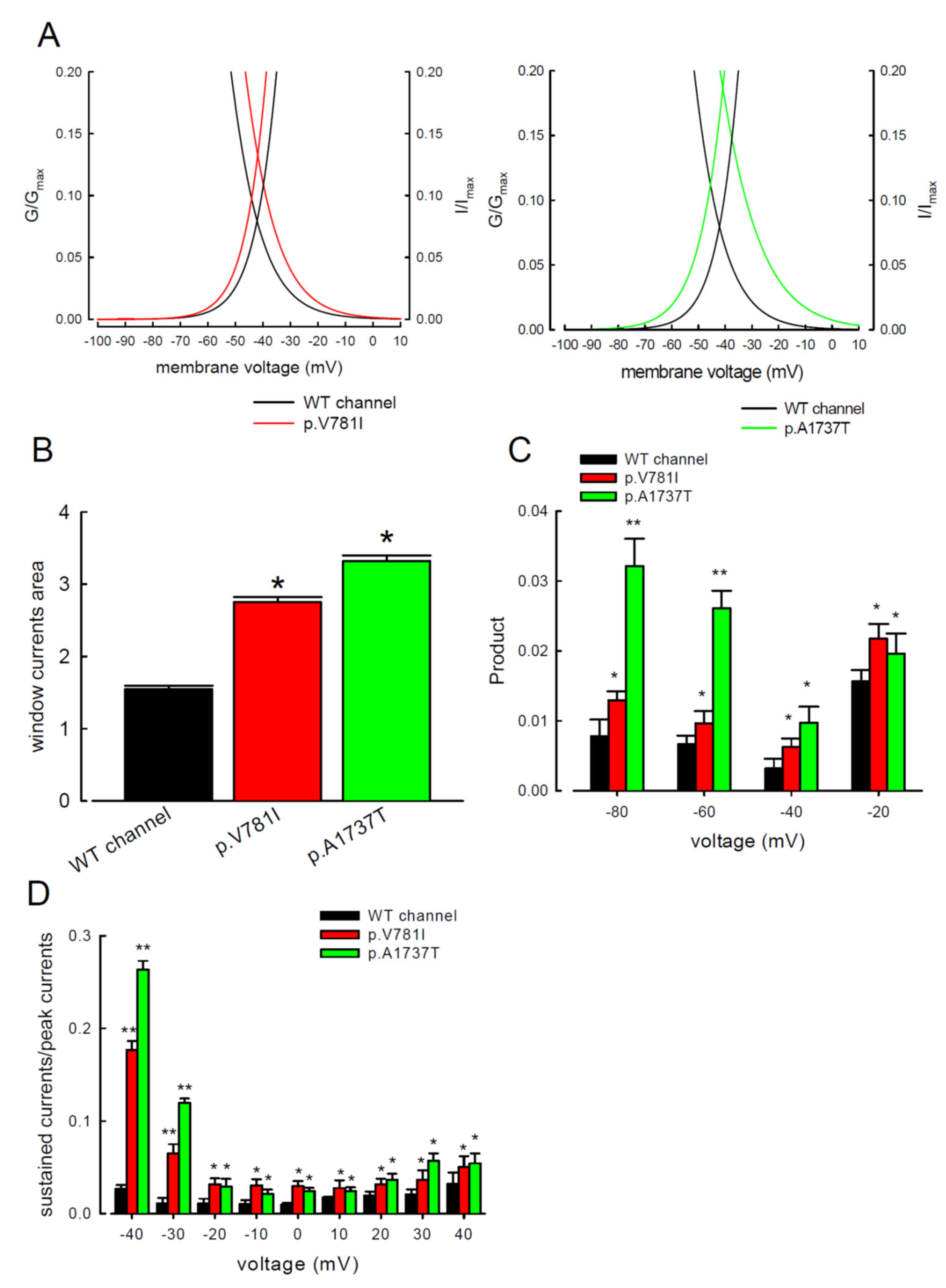
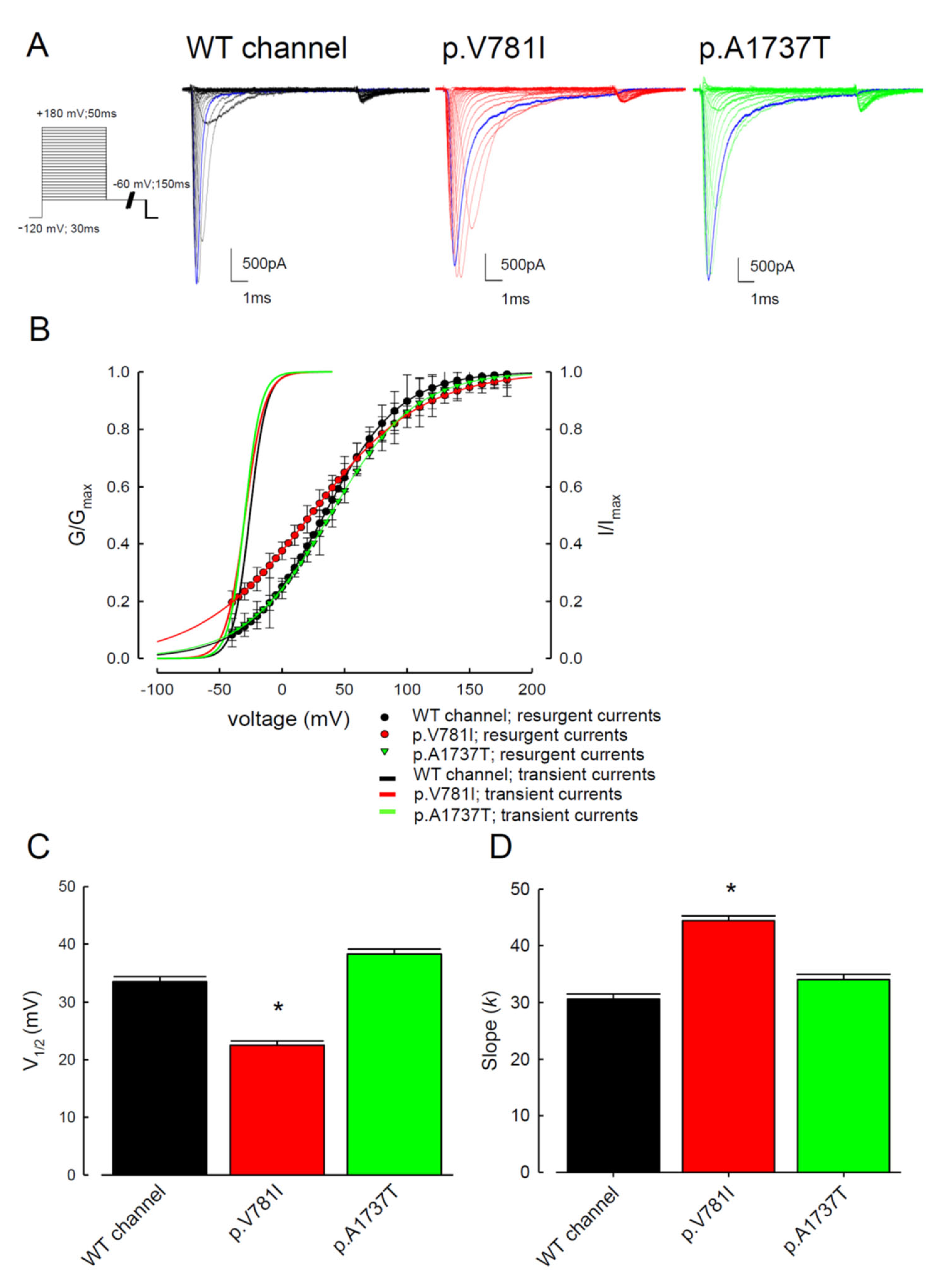
3.1. The p.V781I and p.A1737T Mutant Channels Showed Larger Sustained Na+ Currents Than the WT Nav1.4 Channel
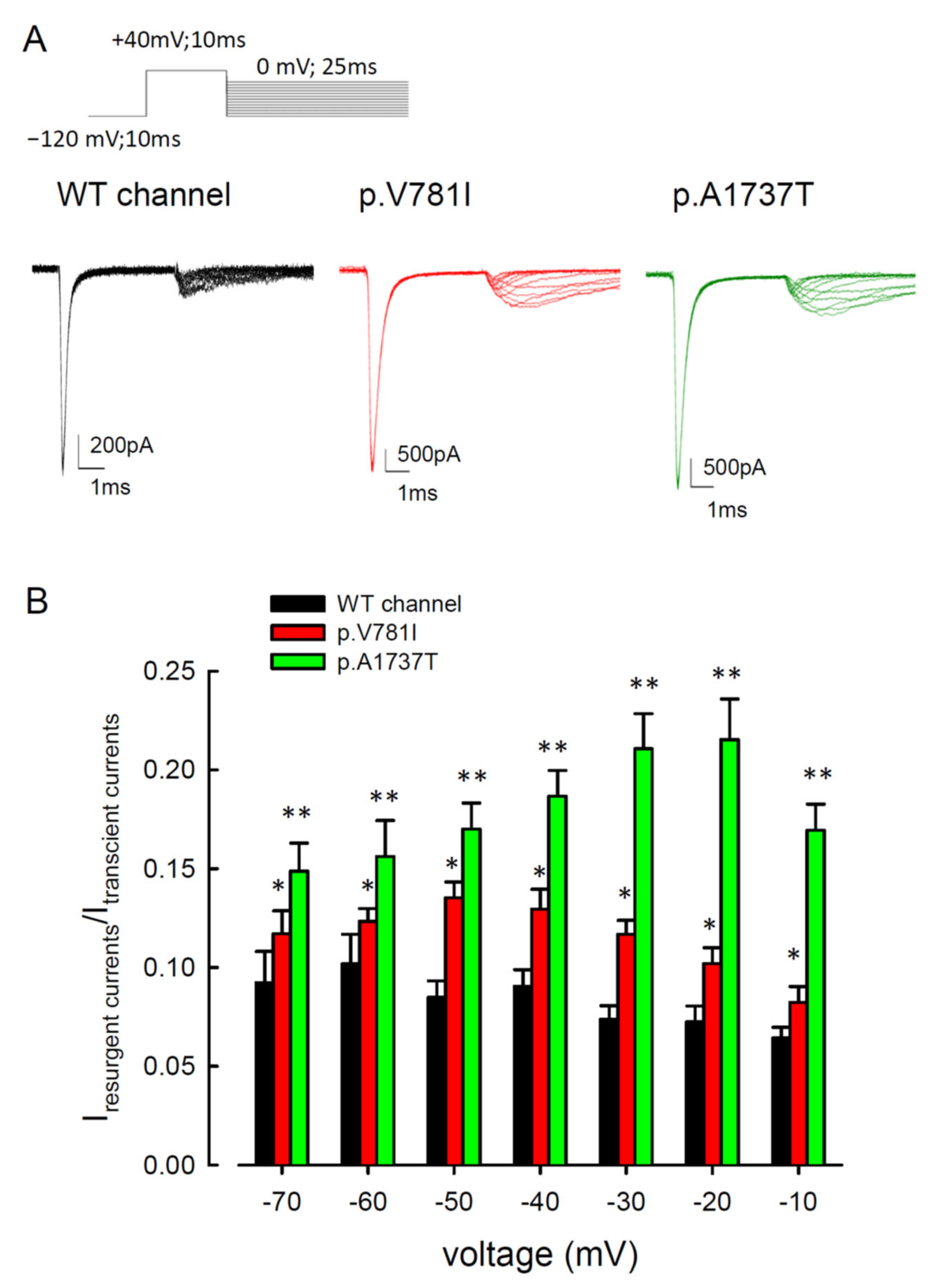
3.2. The Mutant Nav1.4 Channels Increase the Resurgent Na+ Currents
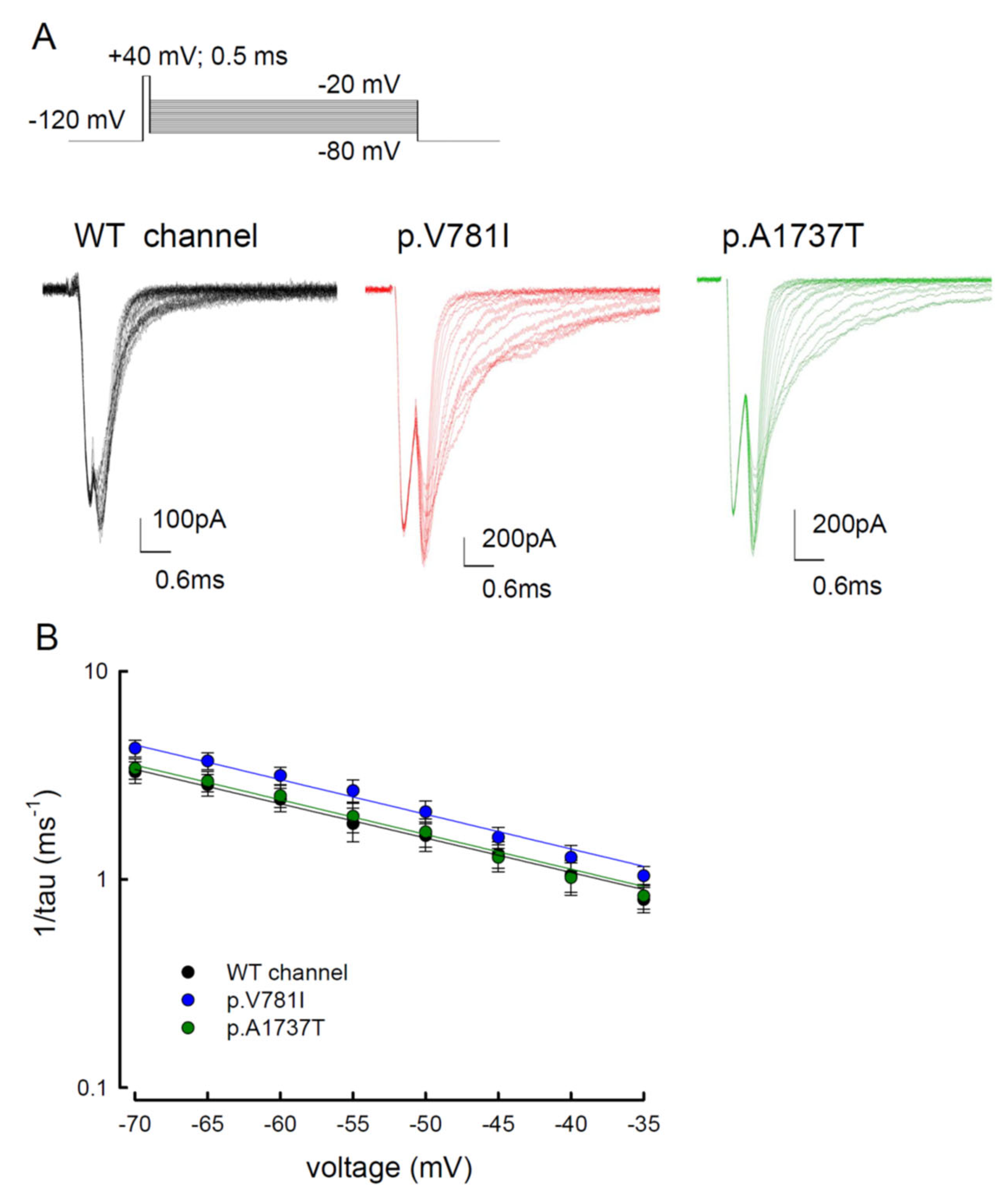
3.3. There Are Probably Two Individual Open States for Transient and Resurgent Na+ Currents in the Nav1.4 Channel
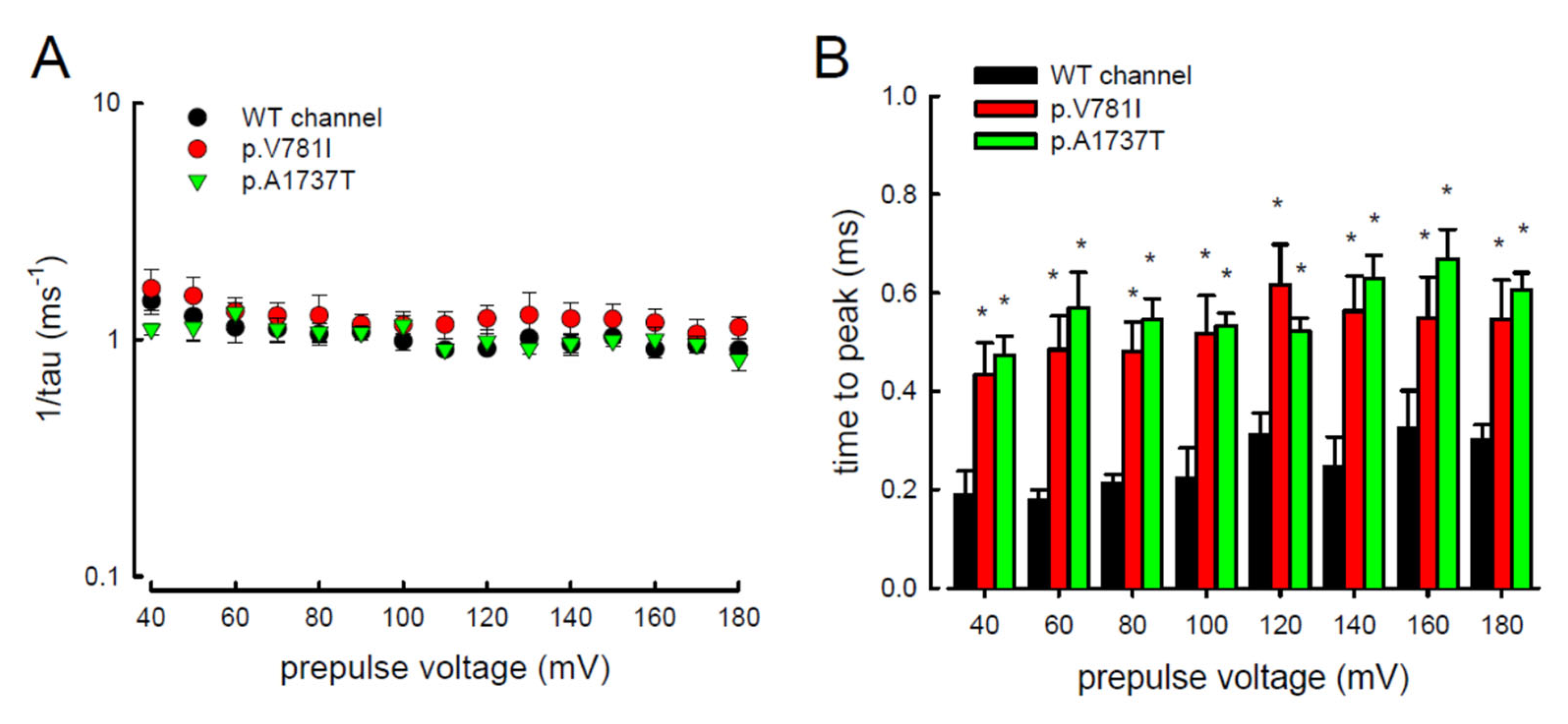
3.4. Resurgent Na+ Current Decay Rates and Time to Peak in the Mutant Channels
3.5. The Tail Currents in the WT and the p.V781I and p.A1737T Mutant Channels Were Unchanged
4. Discussion
4.1. Biophysical Changes Resulting from Two Mutant Nav1.4 Channel Proteins in Paramyotonia Congenita
4.2. Proposal of a New Open State Responsible for Resurgent Na+ Current
4.3. Sustained and Resurgent Na+ Currents Are Increased in the Mutant p.V781I and p.A1737T Channels
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mazon, M.J.; Barros, F.; De la Pena, P.; Quesada, J.F.; Escudero, A.; Cobo, A.M.; Pascual-Pascual, S.I.; Gutierrez-Rivas, E.; Guillen, E.; Arpa, J.; et al. Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul. Disord. 2012, 22, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Bonanno, S.; Altamura, C.; Desaphy, J.F. Ion Channel Gene Mutations Causing Skeletal Muscle Disorders: Pathomechanisms and Opportunities for Therapy. Cells 2021, 10, 1521. [Google Scholar] [CrossRef] [PubMed]
- Stunnenberg, B.C.; LoRusso, S.; Arnold, W.D.; Barohn, R.J.; Cannon, S.C.; Fontaine, B.; Griggs, R.C.; Hanna, M.G.; Matthews, E.; Meola, G.; et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve 2020, 62, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.F.; Altamura, C.; Vicart, S.; Fontaine, B. Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine. J. Neuromuscul. Dis. 2021, 8, 357–381. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, W.; Chang, X.; Guo, J. Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review. Channels 2019, 13, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.C.; Huang, Y.C.; Wang, C.W.; Hsueh, C.H.; Lai, L.P.; Yeh, J.H. Paralysis periodica paramyotonica caused by SCN4A Arg1448Cys mutation. J. Formos. Med. Assoc. 2006, 105, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Holzherr, B.; Lehmann-Horn, F.; Kuzmenkina, E.; Fan, C.; Jurkat-Rott, K. A gating model for wildtype and R1448H Nav1.4 channels in paramyotonia. Acta Myol. 2014, 33, 22–33. [Google Scholar]
- Kol, S.; Turrell, B.R.; de Keyzer, J.; van der Laan, M.; Nouwen, N.; Driessen, A.J. YidC-mediated membrane insertion of assembly mutants of subunit c of the F1F0 ATPase. J. Biol. Chem. 2006, 281, 29762–29768. [Google Scholar] [CrossRef] [Green Version]
- David, M.; Martinez-Marmol, R.; Gonzalez, T.; Felipe, A.; Valenzuela, C. Differential regulation of Na(v)beta subunits during myogenesis. Biochem. Biophys. Res. Commun. 2008, 368, 761–766. [Google Scholar] [CrossRef]
- Bouhours, M.; Sternberg, D.; Davoine, C.S.; Ferrer, X.; Willer, J.C.; Fontaine, B.; Tabti, N. Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J. Physiol. 2004, 554, 635–647. [Google Scholar] [CrossRef]
- Lossin, C.; Nam, T.S.; Shahangian, S.; Rogawski, M.A.; Choi, S.Y.; Kim, M.K.; Sunwoo, I.N. Altered fast and slow inactivation of the N440K Nav1.4 mutant in a periodic paralysis syndrome. Neurology 2012, 79, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kinoshita, M.; Sasaki, R.; Aoike, F.; Takahashi, M.P.; Sakoda, S.; Hirose, K. New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve 2009, 39, 666–673. [Google Scholar] [CrossRef] [PubMed]
- El-Bizri, N.; Kahlig, K.M.; Shyrock, J.C.; George, A.L., Jr.; Belardinelli, L.; Rajamani, S. Ranolazine block of human Na v 1.4 sodium channels and paramyotonia congenita mutants. Channels 2011, 5, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carle, T.; Fournier, E.; Sternberg, D.; Fontaine, B.; Tabti, N. Cold-induced disruption of Na+ channel slow inactivation underlies paralysis in highly thermosensitive paramyotonia. J. Physiol. 2009, 587, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Ye, J.; Tang, S.; Wang, J.; Luo, B.; Ji, F.; Zhang, X.; Yu, Y.; Cheng, X.; Li, Y. N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita. J. Physiol. 2017, 595, 6837–6850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinato, A.; Altamura, C.; Imbrici, P.; Maggi, L.; Bernasconi, P.; Mantegazza, R.; Pasquali, L.; Siciliano, G.; Lo Monaco, M.; Vial, C.; et al. Pharmacogenetics of myotonic hNav1.4 sodium channel variants situated near the fast inactivation gate. Pharmacol. Res. 2019, 141, 224–235. [Google Scholar] [CrossRef]
- Ferriby, D.; Stojkovic, T.; Sternberg, D.; Hurtevent, J.F.; Hurtevent, J.P.; Vermersch, P. A new case of autosomal dominant myotonia associated with the V1589M missense mutation in the muscle sodium channel gene and its phenotypic classification. Neuromuscul. Disord. 2006, 16, 321–324. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, E.J.; Jung, D.S.; Park, K.H.; Kim, I.J.; Kwak, K.Y.; Kim, C.M.; Ko, H.Y. A Korean family with Arg1448Cys mutation of SCN4A channel causing paramyotonia congenita: Electrophysiologic, histopathologic, and molecular genetic studies. J. Korean Med. Sci. 2002, 17, 856–860. [Google Scholar] [CrossRef] [Green Version]
- Nurputra, D.K.; Nakagawa, T.; Takeshima, Y.; Harahap, I.S.; Morikawa, S.; Sakaeda, T.; Lai, P.S.; Matsuo, M.; Takaoka, Y.; Nishio, H. Paramyotonia congenita: From clinical diagnosis to in silico protein modeling analysis. Pediatr. Int. 2012, 54, 602–612. [Google Scholar] [CrossRef]
- Yoshinaga, H.; Sakoda, S.; Good, J.M.; Takahashi, M.P.; Kubota, T.; Arikawa-Hirasawa, E.; Nakata, T.; Ohno, K.; Kitamura, T.; Kobayashi, K.; et al. A novel mutation in SCN4A causes severe myotonia and school-age-onset paralytic episodes. J. Neurol. Sci. 2012, 315, 15–19. [Google Scholar] [CrossRef]
- Fan, Z.; George, A.L., Jr.; Kyle, J.W.; Makielski, J.C. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. 1996, 496 Pt 1, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, J.E.; Featherstone, D.E.; Ruben, P.C. Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis oocytes. J. Physiol. 1997, 499 Pt 3, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, B.; Mitrovic, N.; Lehmann-Horn, F.; Dengler, R.; Bufler, J. Mechanisms of cold sensitivity of paramyotonia congenita mutation R1448H and overlap syndrome mutation M1360V. J. Physiol. 2003, 547, 691–698. [Google Scholar] [CrossRef]
- Heine, R.; Pika, U.; Lehmann-Horn, F. A novel SCN4A mutation causing myotonia aggravated by cold and potassium. Hum. Mol. Genet. 1993, 2, 1349–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaliq, Z.M.; Gouwens, N.W.; Raman, I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: An experimental and modeling study. J. Neurosci. 2003, 23, 4899–4912. [Google Scholar] [CrossRef] [PubMed]
- Grieco, T.M.; Raman, I.M. Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J. Neurosci. 2004, 24, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.W.; Lin, P.C.; Chen, J.L.; Lee, M.J. Cannabidiol Selectively Binds to the Voltage-Gated Sodium Channel Nav1.4 in Its Slow-Inactivated State and Inhibits Sodium Current. Biomedicines 2021, 9, 1141. [Google Scholar] [CrossRef]
- Huang, C.W.; Lai, H.J.; Lin, P.C.; Lee, M.J. Changes of Resurgent Na(+) Currents in the Nav1.4 Channel Resulting from an SCN4A Mutation Contributing to Sodium Channel Myotonia. Int. J. Mol. Sci. 2020, 21, 2593. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Hsieh, J.Y.; Kuo, C.C. The external pore loop interacts with S6 and S3-S4 linker in domain 4 to assume an essential role in gating control and anticonvulsant action in the Na(+) channel. J. Gen. Physiol. 2009, 134, 95–113. [Google Scholar] [CrossRef]
- Huang, C.W.; Lai, H.J.; Huang, P.Y.; Lee, M.J.; Kuo, C.C. The Biophysical Basis Underlying Gating Changes in the p.V1316A Mutant Nav1.7 Channel and the Molecular Pathogenesis of Inherited Erythromelalgia. PLoS Biol. 2016, 14, e1002561. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Lai, H.J.; Huang, P.Y.; Lee, M.J.; Kuo, C.C. Anomalous enhancement of resurgent Na(+) currents at high temperatures by SCN9A mutations underlies the episodic heat-enhanced pain in inherited erythromelalgia. Sci. Rep. 2019, 9, 12251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.W.; Lai, H.J.; Lin, P.C.; Lee, M.J. Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita. Biomedicines 2021, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.H.; Raman, I.M. Resurgent current of voltage-gated Na(+) channels. J. Physiol. 2014, 592, 4825–4838. [Google Scholar] [CrossRef]
- Raman, I.M.; Bean, B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 1997, 17, 4517–4526. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Sprunger, L.K.; Meisler, M.H.; Bean, B.P. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 1997, 19, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Bean, B.P. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: Evidence for two mechanisms. Biophys. J. 2001, 80, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Kuo, C.C. The position of the fourth segment of domain 4 determines status of the inactivation gate in Na+ channels. J. Neurosci. 2003, 23, 4922–4930. [Google Scholar] [CrossRef] [Green Version]
- Goldschen-Ohm, M.P.; Capes, D.L.; Oelstrom, K.M.; Chanda, B. Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat. Commun. 2013, 4, 1350. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Lin, S.; Chang, P.C.; Lin, H.C.; Kuo, C.C. Functional extension of amino acid triads from the fourth transmembrane segment (S4) into its external linker in Shaker K(+) channels. J. Biol. Chem. 2011, 286, 37503–37514. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.S.; Dib-Hajj, S.D.; Cox, J.J.; Tyrrell, L.; Elmslie, F.V.; Clarke, A.A.; Drenth, J.P.; Woods, C.G.; Waxman, S.G. A new Nav1.7 sodium channel mutation I234T in a child with severe pain. Eur. J. Pain 2010, 14, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Estacion, M.; Yang, Y.; Dib-Hajj, S.D.; Tyrrell, L.; Lin, Z.; Waxman, S.G. A new Nav1.7 mutation in an erythromelalgia patient. Biochem. Biophys. Res. Commun. 2013, 432, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Capes, D.L.; Goldschen-Ohm, M.P.; Arcisio-Miranda, M.; Bezanilla, F.; Chanda, B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 2013, 142, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.C.; Liao, S.Y. Facilitation of recovery from inactivation by external Na+ and location of the activation gate in neuronal Na+ channels. J. Neurosci. 2000, 20, 5639–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Cannon, S.C. Sodium Channelopathies of Skeletal Muscle. Handb. Exp. Pharmacol. 2018, 246, 309–330. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-J.; Lin, P.-C.; Lin, M.-H.; Chiou, H.-Y.C.; Wang, K.; Huang, C.-W. Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita. Biology 2022, 11, 613. https://doi.org/10.3390/biology11040613
Lee M-J, Lin P-C, Lin M-H, Chiou H-YC, Wang K, Huang C-W. Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita. Biology. 2022; 11(4):613. https://doi.org/10.3390/biology11040613
Chicago/Turabian StyleLee, Ming-Jen, Pi-Chen Lin, Ming-Hong Lin, Hsin-Ying Clair Chiou, Kai Wang, and Chiung-Wei Huang. 2022. "Kinetic Alterations in Resurgent Sodium Currents of Mutant Nav1.4 Channel in Two Patients Affected by Paramyotonia Congenita" Biology 11, no. 4: 613. https://doi.org/10.3390/biology11040613