
Inflammation and Oxidative Stress as Common Mechanisms of Pulmonary, Autonomic and Musculoskeletal Dysfunction after Spinal Cord Injury




{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
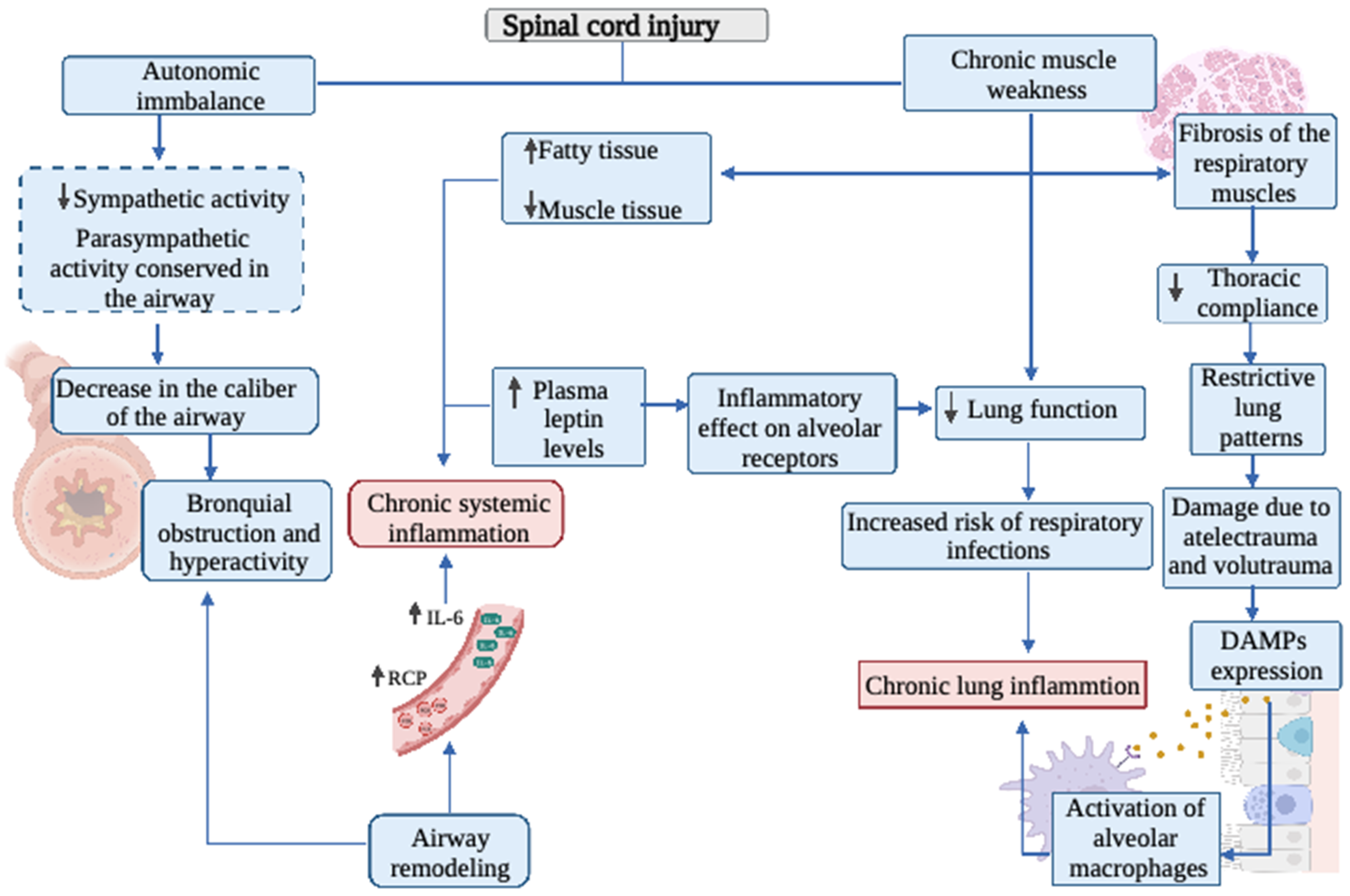
2. Respiratory System
3. Autonomic Nervous System
4. Muscular System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Spinal Cord Injury. 2013. Available online: https://www.who.int/news-room/fact-sheets/detail/spinal-cord-injury (accessed on 1 February 2020).
- Bastien, D.; Lacroix, S. Cytokine pathways regulating glial and leukocyte function after spinal cord and peripheral nerve injury. Exp. Neurol. 2014, 258, 62–77. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.W.; Sadowsky, C. Spinal-cord injury. Lancet 2002, 359, 417–425. [Google Scholar] [CrossRef]
- Krassioukov, A. Autonomic function following cervical spinal cord injury. Respir. Physiol. Neurobiol. 2009, 169, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Raguindin, P.F.; Bertolo, A.; Zeh, R.M.; Fränkl, G.; Itodo, O.A.; Capossela, S.; Bally, L.; Minder, B.; Brach, M.; Eriks-Hoogland, I.; et al. Body Composition According to Spinal Cord Injury Level: A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 3911. [Google Scholar] [CrossRef]
- Dinh, A.; Bouchand, F.; Davido, B.; Duran, C.; Denys, P.; Lortat-Jacob, A.; Rottman, M.; Salomon, J.; Bernard, L. Management of established pressure ulcer infections in spinal cord injury patients. Med. Mal. Infect. 2019, 49, 9–16. [Google Scholar] [CrossRef]
- Popovich, P.G.; Stokes, B.T.; Whitacre, C.C. Concept of autoimmunity following spinal cord injury: Possible roles for T lymphocytes in the traumatized central nervous system. J. Neurosci. Res. 1996, 45, 349–363. [Google Scholar] [CrossRef]
- Gorgey, A.S.; Dolbow, D.R.; Dolbow, J.D.; Khalil, R.K.; Castillo, C.; Gater, D.R. Effects of spinal cord injury on body composition and metabolic profile—Part I. J. Spinal. Cord. Med. 2014, 37, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Schilero, G.J.; Spungen, A.M.; Bauman, W.A.; Radulovic, M.; Lesser, M. Pulmonary function and spinal cord injury. Respir. Physiol. Neurobiol. 2009, 166, 129–141. [Google Scholar] [CrossRef]
- Schilero, G.J.; Grimm, D.R.; Bauman, W.A.; Lenner, R.; Lesser, M. Assessment of airway caliber and bronchodilator responsiveness in subjects with spinal cord injury. Chest 2005, 127, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, M.; Fedullo, A.L.; Di Giacinto, B.; Squeo, M.R.; Aiello, P.; Dante, D.; Romano, S.; Magaudda, L.; Peluso, I.; Palmery, M.; et al. Cardiovascular Risk Factors and Haematological Indexes of Inflammation in Paralympic Athletes with Different Motor Impairments. Oxid. Med. Cell Longev. 2019, 2019, 6798140. [Google Scholar] [CrossRef] [Green Version]
- Schilero, G.J.; Bauman, W.A.; Radulovic, M. Traumatic Spinal Cord Injury: Pulmonary Physiologic Principles and Management. Clin. Chest. Med. 2018, 39, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Martínez Rodríguez, L.; Pérez, A.M.; del Mar López Rodríguez, M. Heart rate variability biofeedback. THERAPEÍA 2019, 11, 95–119. [Google Scholar]
- Phillips, A.A.; Krassioukov, A.V. Contemporary Cardiovascular Concerns after Spinal Cord Injury: Mechanisms, Maladaptations, and Management. J. Neurotrauma. 2015, 32, 1927–1942. [Google Scholar] [CrossRef] [PubMed]
- Griggs, K.E.; Price, M.J.; Goosey-Tolfrey, V.L. Cooling athletes with a spinal cord injury. Sports Med. 2015, 45, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mironets, E.; Fischer, R.; Bracchi-Ricard, V.; Saltos, T.M.; Truglio, T.S.; O’Reilly, M.L.; Swanson, K.A.; Bethea, J.R.; Tom, V.J. Attenuating Neurogenic Sympathetic Hyperreflexia Robustly Improves Antibacterial Immunity After Chronic Spinal Cord Injury. J. Neurosci. 2020, 40, 478–492. [Google Scholar] [CrossRef]
- Biering-Sørensen, B.; Kristensen, I.B.; Kjaer, M.; Biering-Sørensen, F. Muscle after spinal cord injury. Muscle Nerve. 2009, 40, 499–519. [Google Scholar] [CrossRef]
- Yarar-Fisher, C.; Bickel, C.S.; Kelly, N.A.; Stec, M.J.; Windham, S.T.; McLain, A.B.; Oster, R.A.; Bamman, M.M. Heightened TWEAK-NF-κB signaling and inflammation-associated fibrosis in paralyzed muscles of men with chronic spinal cord injury. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E754–E761. [Google Scholar] [CrossRef]
- Antonioni, A.; Fantini, C.; Dimauro, I.; Caporossi, D. Redox homeostasis in sport: Do athletes really need antioxidant support? Res. Sports Med. 2019, 27, 147–165. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox. Signal. 2011, 15, 2519–2528. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.; Di Marco, A.F.; Hoit, J.D.; Garshick, E. Respiratory dysfunction and management in spinal cord injury. Respir. Care 2006, 51, 853–868. [Google Scholar]
- Berlowitz, D.J.; Wadsworth, B.; Ross, J. Respiratory problems and management in people with spinal cord injury. Breathe 2016, 12, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateus, S.R.M.; Beraldo, P.S.S.; Horan, T.A. Maximal static mouth respiratory pressure in spinal cord injured patients: Correlation with motor level. Spinal. Cord. 2007, 45, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.W.; Shin, J.C.; Park, C.I.; Moon, J.H.; Rha, D.W.; Cho, D.H. Relationship between inspiratory muscle strength and cough capacity in cervical spinal cord injured patients. Spinal. Cord. 2006, 44, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malas, F.Ü.; Köseoğlu, F.; Kara, M.; Ece, H.; Aytekin, M.; Öztürk, G.T.; Özçakar, L.; Ulaşlı, A.M. Diaphragm ultrasonography and pulmonary function tests in patients with spinal cord injury. Spinal. Cord. 2019, 57, 679–683. [Google Scholar] [CrossRef]
- Kelley, A.; Garshick, E.; Gross, E.R.; Lieberman, S.L.; Tun, C.G.; Brown, R. Spirometry testing standards in spinal cord injury. Chest 2003, 123, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Forner, J.V. Lung volumes and mechanics of breathing in tetraplegics. Paraplegia 1980, 18, 258–266. [Google Scholar] [CrossRef]
- Anke, A.; Aksnes, A.K.; Stanghelle, J.K.; Hjeltnes, N. Lung volumes in tetraplegic patients according to cervical spinal cord injury level. Scand. J. Rehabil. Med. 1993, 25, 73–77. [Google Scholar]
- Baydur, A.; Adkins, R.H.; Milic-Emili, J. Lung mechanics in individuals with spinal cord injury: Effects of injury level and posture. J. Appl. Physiol. 2001, 90, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Terson de Paleville, D.G.; Sayenko, D.G.; Aslan, S.C.; Folz, R.J.; McKay, W.B.; Ovechkin, A.V. Respiratory motor function in seated and supine positions in individuals with chronic spinal cord injury. Respir. Physiol. Neurobiol. 2014, 203, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Stolzmann, K.L.; Gagnon, D.R.; Brown, R.; Tun, C.G.; Garshick, E. Longitudinal change in FEV1 and FVC in chronic spinal cord injury. Am. J. Respir. Crit. Care Med. 2008, 177, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Van Silfhout, L.; Peters, A.E.J.; Berlowitz, D.J.; Schembri, R.; Thijssen, D.; Graco, M. Long-term change in respiratory function following spinal cord injury. Spinal. Cord. 2016, 54, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, M.H.; Alavinia, S.M.; Craven, B.C. Management of obesity after spinal cord injury: A systematic review. J. Spinal. Cord. Med. 2017, 40, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Farkas, G.J.; Gater, D.R. Neurogenic obesity and systemic inflammation following spinal cord injury: A review. J. Spinal. Cord. Med. 2018, 41, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Garshick, E.; Walia, P.; Goldstein, R.L.; Teylan, M.; Lazzari, A.A.; Tun, C.G.; Hart, J.E. Plasma Leptin and Reduced FEV1 and FVC in Chronic Spinal Cord Injury. PM&R 2018, 10, 276–285. [Google Scholar]
- Bruno, A.; Chanez, P.; Chiappara, G.; Siena, L.; Giammanco, S.; Gjomarkaj, M.; Bonsignore, G.; Bousquet, J.; Vignola, A.M. Does leptin play a cytokine-like role within the airways of COPD patients? Eur. Respir. J. 2005, 26, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Vernooy, J.H.; Ubags, N.D.; Brusselle, G.G.; Tavernier, J.; Suratt, B.T.; Joos, G.F.; Wouters, E.F.; Bracke, K.R. Leptin as regulator of pulmonary immune responses: Involvement in respiratory diseases. Pulm. Pharmacol. Ther. 2013, 26, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Garshick, E.; Stolzmann, K.L.; Gagnon, D.R.; Morse, L.R.; Brown, R. Systemic inflammation and reduced pulmonary function in chronic spinal cord injury. PM&R 2011, 3, 433–439. [Google Scholar]
- West, C.R.; Campbell, I.G.; Shave, R.E.; Romer, L.M. Resting cardiopulmonary function in Paralympic athletes with cervical spinal cord injury. Med. Sci. Sports Exerc. 2012, 44, 323–329. [Google Scholar] [CrossRef]
- Hickling, K.G.; Walsh, J.; Henderson, S.; Jackson, R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: A prospective study. Crit. Care Med. 1994, 22, 1568–1578. [Google Scholar] [CrossRef]
- Hickling, K.G.; Henderson, S.J.; Jackson, R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive. Care Med. 1990, 16, 372–377. [Google Scholar] [CrossRef]
- Carrasco Loza, R.; Villamizar Rodríguez, G.; Medel Fernández, N. Ventilator-Induced Lung Injury (VILI) in acute respiratory distress syndrome (ARDS): Volutrauma and Molecular Effects. Open. Respir. Med. J. 2015, 9, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, T.; Lili, Y.; Wen, Y.; Xinwei, W.; Xuhui, Z. Pulmonary edema and hemorrhage, possible causes of pulmonary infection and respiratory failure in the early stage of lower spinal cord injury. Med. Hypotheses. 2012, 79, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.P. Acute Respiratory Infections in Persons with Spinal Cord Injury. Phys. Med. Rehabil. Clin. N. Am. 2007, 18, 203–216. [Google Scholar] [CrossRef]
- Almenoff, P.L.; Alexander, L.R.; Spungen, A.M.; Lesser, M.D.; Bauman, W.A. Bronchodilatory effects of ipratropium bromide in patients with tetraplegia. Paraplegia 1995, 33, 274–277. [Google Scholar] [CrossRef]
- DeLuca, R.V.; Grimm, D.R.; Lesser, M.; Bauman, W.A.; Almenoff, P.L. Effects of a β2-agonist on airway hyperreactivity in subjects with cervical spinal cord injury. Chest 1999, 115, 1533–1538. [Google Scholar] [CrossRef]
- Schilero, G.J.; Hobson, J.C.; Singh, K.; Spungen, A.M.; Bauman, W.A.; Radulovic, M. Bronchodilator effects of ipratropium bromide and albuterol sulfate among subjects with tetraplegia. J. Spinal. Cord Med. 2018, 41, 42–47. [Google Scholar] [CrossRef]
- Cockcroft, D.W.; Davis, B.E. Mechanisms of airway hyperresponsiveness. J. Allergy. Clin. Immunol. 2006, 118, 551–559. [Google Scholar] [CrossRef]
- Araneda, O.F.; Carbonell, T.; Tuesta, M. Update on the Mechanisms of Pulmonary Inflammation and Oxidative Imbalance Induced by Exercise. Oxid. Med. Cell Longev. 2016, 2016, 4868536. [Google Scholar] [CrossRef] [Green Version]
- Dicpinigaitis, P.V.; Spungen, A.M.; Bauman, W.A.; Absgarten, A.; Almenoff, P.L. Bronchial hyperresponsiveness after cervical spinal cord injury. Chest 1994, 105, 1073–1076. [Google Scholar] [CrossRef]
- Singas, E.; Lesser, M.; Spungen, A.M.; Bauman, W.A.; Almenoff, P.L. Airway hyperresponsiveness to methacholine in subjects with spinal cord injury. Chest 1996, 110, 911–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, J.E.; Morse, L.; Tun, C.G.; Brown, R.; Garshick, E. Cross-sectional associations of pulmonary function with systemic inflammation and oxidative stress in individuals with chronic spinal cord injury. J. Spinal. Cord. Med. 2016, 39, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, W.; Korsten, M.; Radulovic, M.; Schilero, G.; Wech, J.; Spungen, A. 31st G. Heiner sell lectureship: Secondary medical consequences of spinal cord injury. Top. Spinal. Cord. Inj. Rehabil. 2012, 18, 354–378. [Google Scholar] [CrossRef] [Green Version]
- Radulovic, M.; Schilero, G.J.; Wecht, J.M.; La Fountaine, M.; Rosado-Rivera, D.; Bauman, W.A. Exhaled nitric oxide levels are elevated in persons with tetraplegia and comparable to that in mild asthmatics. Lung 2010, 188, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Teasell, R.W.; Arnold, J.M.; Krassioukov, A.; Delaney, G.A. Cardiovascular consequences of loss of supraspinal control of the sympathetic nervous system after spinal cord injury. Arch. Phys. Med. Rehabil. 2000, 81, 506–516. [Google Scholar] [CrossRef]
- Hou, S.; Rabchevsky, A.G. Autonomic consequences of spinal cord injury. Compr. Physiol. 2014, 4, 1419–1453. [Google Scholar]
- Zhang, Y.; Guan, Z.; Reader, B.; Shawler, T.; Mandrekar-Colucci, S.; Huang, K.; Weil, Z.; Bratasz, A.; Wells, J.; Powell, N.D.; et al. Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. J. Neurosci. 2013, 33, 12970–12981. [Google Scholar] [CrossRef]
- Noller, C.M.; Groah, S.L.; Nash, M.S. Inflammatory stress effects on health and function after spinal cord injury. Top. Spinal. Cord. Inj. Rehabil. 2017, 23, 207–217. [Google Scholar] [CrossRef]
- Cowan, R.E.; Nash, M.S. Cardiovascular disease, SCI and exercise: Unique risks and focused countermeasures. Disabil. Rehabil. 2010, 32, 2228–2236. [Google Scholar] [CrossRef]
- Madden, K.S.; Sanders, V.M.; Felten, D.L. Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 417–448. [Google Scholar] [CrossRef]
- Köseoǧlu, B.F.; Safer, V.B.; Öken Akselim, S. Cardiovascular disease risk in people with spinal cord injury: Is there a possible association between reduced lung function and increased risk of diabetes and hypertension. Spinal. Cord. 2017, 55, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, S.P.; Leicht, C.A.; Kamijo, Y.-I.; Kinoshita, T.; Stephenson, B.T.; Goosey-Tolfrey, V.L.; Bishop, N.C.; Tajima, F. The inflammatory response to a wheelchair half-marathon in people with a spinal cord injury—The role of autonomic function. J. Sports Sci. 2019, 37, 1717–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miteva, K.; Madonna, R.; De Caterina, R.; Van Linthout, S. Innate and adaptive immunity in atherosclerosis. Vascul. Pharmacol. 2018. Online ahead of print. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Chang, X.; Zhao, Z.; Zhang, W.; Liu, D.; Ma, C.; Zhang, T.; Meng, Q.; Yan, P.; Zou, L.; Zhang, M. Natural Antioxidants Improve the Vulnerability of Cardiomyocytes and Vascular Endothelial Cells under Stress Conditions: A Focus on Mitochondrial Quality Control. Oxid. Med. Cell. Longev. 2021, 2021, 6620677. [Google Scholar] [CrossRef]
- Chen, X.P.; Xun, K.L.; Wu, Q.; Zhang, T.T.; Shi, J.S.; Du, G.H. Oxidized low density lipoprotein receptor-1 mediates oxidized low density lipoprotein-induced apoptosis in human umbilical vein endothelial cells: Role of reactive oxygen species. Vascul. Pharmacol. 2007, 47, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Popa, C.; Popa, F.; Grigorean, V.T.; Onose, G.; Sandu, A.M.; Popescu, M.; Burnei, G.; Strambu, V.; Sinescu, C. Vascular dysfunctions following spinal cord injury. J. Med. Life. 2010, 3, 275–285. [Google Scholar] [PubMed]
- Meisel, C.; Schwab, J.M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.J.; Ditor, D.S. Immune dysfunction and chronic inflammation following spinal cord injury. Spinal. Cord. 2015, 53, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.; Huonker, M.; Stahl, F.; Barturen, J.-M.; König, D.; Heim, M.; Lehmann, M.; Keul, J. Free plasma catecholamines in spinal cord injured persons with different injury levels at rest and during exercise. J. Auton. Nerv. Syst. 1998, 68, 96–100. [Google Scholar] [CrossRef]
- Schwab, J.M.; Zhang, Y.; Kopp, M.A.; Brommer, B.; Popovich, P.G. The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp. Neurol. 2014, 258, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Riegger, T.; Conrad, S.; Liu, K.; Schluesener, H.J.; Adibzahdeh, M.; Schwab, J.M. Spinal cord injury-induced immune de- pression syndrome (SCI-IDS). Eur. J. Neurosci. 2007, 25, 1743–1747. [Google Scholar] [CrossRef]
- Ankeny, D.P.; Lucin, K.M.; Sanders, V.M.; McGaughy, V.M.; Popovich, P.G. Spinal cord injury triggers systemic autoimmunity: Evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. J. Neurochem. 2006, 99, 1073–1087. [Google Scholar] [CrossRef]
- Hailer, N.P. Immunosuppression after traumatic or ischemic CNS damage: It is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 2008, 84, 211–233. [Google Scholar] [CrossRef]
- Hayes, K.C.; Hull, T.C.; Delaney, G.A.; Potter, P.J.; Sequeira, K.A.; Campbell, K.; Popovich, P.G. Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J. Neurotrauma. 2002, 19, 753–761. [Google Scholar] [CrossRef]
- Kil, K.; Zang, Y.C.; Yang, D.; Markowski, J.; Fuoco, G.S.; Vendetti, G.C.; Rivera, V.M.; Zhang, J.Z. T cell responses to myelin basic protein in patients with spinal cord injury and multiple sclerosis. J. Neuroimmunol. 1999, 9, 201–207. [Google Scholar] [CrossRef]
- Ankeney, D.P.; Guan, Z.; Popovich, P.G. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J. Clin. Invest. 2009, 119, 2990–2999. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, D.P.; Popovich, P.G. B cells and autoantibodies: Complex roles in CNS injury. Trends Immunol. 2010, 31, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltzman, J.W.; Battaglino, R.A.; Salles, L.; Jha, P.; Sudhakar, S.; Garshick, E.; Stott, H.L.; Zafonte, R.; Morse, L.R. B-cell maturation antigen, a proliferation-inducing ligand, and B-cell activating factor are candidate mediators of spinal cord injury-induced autoimmunity. J. Neurotrauma. 2013, 30, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, F.; Silveira, P.A.; Brink, R. B cells and the BAFF/APRIL axis: Fast-forward on autoimmunity and signaling. Curr. Opin. Immunol. 2007, 19, 327–336. [Google Scholar] [CrossRef]
- Rickert, R.C.; Jellusova, J.; Miletic, A.V. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol. Rev. 2011, 244, 115–133. [Google Scholar] [CrossRef]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef]
- Vadacca, M.; Margiotta, D.; Sambataro, D.; Buzzulini, F.; Lo Vullo, M.; Rigon, A.; Afeltra, A. Pathway in Sjo¨gren syndrome and systemic lupus erythematosus: Relationship with chronic inflammation and disease activity. Reumatismo 2010, 62, 259–265. [Google Scholar]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Dekaban, G.A.; Thawer, S. Pathogenic antibodies are ac- tive participants in spinal cord injury. J. Clin. Invest. 2009, 119, 2881–2884. [Google Scholar] [CrossRef] [Green Version]
- Herman, P.E.; Bloom, O. Altered leukocyte gene expression after traumatic spinal cord injury: Clinical implications. Neural. Regen. Res. 2018, 13, 1524–1529. [Google Scholar] [PubMed]
- Foster, S.L.; Medzhitov, R. Gene-specific control of the TLR-in- duced inflammatory response. Clin. Immunol. 2009, 130, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: A jour- ney from molecular to nano therapeutics. Front Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Findlay, D. Musculoskeletal Health in the Context of Spinal Cord Injury. Curr. Osteoporos. Rep. 2017, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Druschel, C.; Curt, A.; Fehlings, M.G. Traumatic spinal cord injury. Nat. Rev. Dis. Prim. 2017, 3, 17018. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Judge, A.R. Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Menéndez, H.; Ferrero, C.; Martín-Hernández, J.; Figueroa, A.; Marín, P.; Herrero, A. Chronic effects of simultaneous electromyostimulation and vibration on leg blood flow in spinal cord injury. Spinal. Cord. 2016, 54, 1169–1175. [Google Scholar] [CrossRef]
- West, C.R.; Alyahya, A.; Laher, I.; Krassioukov, A. Peripheral vascular function in spinal cord injury: A systematic review. Spinal. Cord. 2013, 51, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Gorgey, A.; Witt, O.; O’Brien, L.; Cardozo, C.; Chen, Q.; Lesnefsky, E.; Graham, Z. Mitochondrial health and muscle plasticity after spinal cord injury. Eur. J. Appl. Physiol. 2019, 119, 315–331. [Google Scholar] [CrossRef]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2019, 662, 49–60. [Google Scholar] [CrossRef]
- Invernizzi, M.; Carda, S.; Rizzi, M.; Grana, E.; Squarzanti, D.F.; Cisari, C.; Molinari, C.; Renò, F. Evaluation of serum myostatin and sclerostin levels in chronic spinal cord injured patients. Spinal. Cord. 2015, 53, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, M.; Stein, A.; Sison, C.; Glazer, A.; Jassal, N.; McCarthy, D.; Shatzer, M.; Hahn, B.; Chugh, R.; Davies, P.; et al. Elevated circulating levels of the pro-inflammatory cytokine macrophage migration inhibitory factor in individuals with acute spinal cord injury. Arch. Phys. Med. Rehabil. 2015, 96, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.; Fontes-Oliveira, C.; Borges, J.; Kashiabara, J.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. Springerplus 2016, 5, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yune, T.; Chang, M.; Kim, S.; Lee, Y.; Shin, S.; Rhim, H.; Kim, Y.; Shin, M.; Oh, Y.; Han, C.; et al. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J. Neurotrauma. 2003, 20, 207–219. [Google Scholar] [CrossRef]
- Phillips, T.; Leeuwenburgh, C. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005, 19, 668–670. [Google Scholar] [CrossRef]
- Min, K.; Smuder, A.J.; Kwon, O.S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Kavazis, A.N.; McClung, J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. 2007, 102, 2389–2397. [Google Scholar] [CrossRef]
- Barker, T.; Traber, M.G. From animals to humans: Evidence linking oxidative stress as a causative factor in muscle atrophy. J. Physiol. 2007, 583, 421–422. [Google Scholar] [CrossRef]
- Kadoguchi, T.; Takada, S.; Yokota, T.; Furihata, T.; Matsumoto, J.; Tsuda, M.; Mizushima, W.; Fukushima, A.; Okita, K.; Kinugawa, S. Deletion of NAD(P)H Oxidase 2 Prevents Angiotensin II-Induced Skeletal Muscle Atrophy. Biomed. Res. Int. 2018, 2018, 3194917. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Miura, M.; Itokawa, Y. Oxidative stress in skeletal muscle atrophied by immobilization. Acta. Physiol. Scand. 1991, 142, 527–528. [Google Scholar] [CrossRef]
- Gram, M.; Vigelsø, A.; Yokota, T.; Helge, J.W.; Dela, F.; Hey-Mogensen, M. Skeletal muscle mitochondrial H2O2 emission increases with immobilization and decreases after aerobic training in young and older men. J. Physiol. 2015, 593, 4011–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.H.; Harlow, L.; Graham, Z.A.; Bauman, W.A.; Cardozo, C. Spinal Cord Injury Leads to Hyperoxidation and Nitrosylation of Skeletal Muscle Ryanodine Receptor-1 Associated with Upregulation of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 4. J. Neurotrauma. 2017, 34, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Kadoguchi, T.; Kinugawa, S.; Takada, S.; Fukushima, A.; Furihata, T.; Homma, T.; Masaki, Y.; Mizushima, W.; Nishikawa, M.; Takahashi, M.; et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp. Physiol. 2015, 100, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef]
- Savikj, M.; Kostovski, E.; Lundell, L.S.; Iversen, P.O.; Massart, J.; Widegren, U. Altered oxidative stress and antioxidant defence in skeletal muscle during the first year following spinal cord injury. Physiol. Rep. 2019, 7, e14218. [Google Scholar] [CrossRef] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 2011, 110, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Whidden, M.A.; Smuder, A.J.; Wu, M.; Hudson, M.B.; Bradley Nelson, W.; Powers, S.K. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J. Appl. Physiol. 2010, 108, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Arc-Chagnaud, C.; Py, G.; Fovet, T.; Roumanille, R.; Demangel, R.; Pagano, A.F.; Delobel, P.; Blanc, S.; Jasmin, B.J.; Blottner, D.; et al. Evaluation of an Antioxidant and Anti-inflammatory Cocktail Against Human Hypoactivity-Induced Skeletal Muscle Deconditioning. Front Physiol. 2020, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Coyoy-Salgado, A.; Segura-Uribe, J.J.; Guerra-Araiza, C.; Orozco-Suárez, S.; Salgado-Ceballos, H.; Feria-Romero, I.A.; Gallardo, J.M.; Orozco-Barrios, C.E. The Importance of Natural Antioxidants in the Treatment of Spinal Cord Injury in Animal Models: An Overview. Oxid. Med. Cell Longev. 2019, 2019, 3642491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Hölscher, C.; Ma, X. Therapeutic potential of flavonoids in spinal cord injury. Rev. Neurosci. 2017, 28, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Pannu, R.; Barbosa, E.; Singh, A.K.; Singh, I. Attenuation of acute inflammatory response by atorvastatin after spinal cord injury in rats. J. Neurosci. Res. 2005, 79, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, J.; Koda, M.; Hashimoto, M.; Fujiyoshi, T.; Furuya, T.; Endo, T.; Okawa, A.; Yamazaki, M. Neuroprotective effects of granulocyte colony-stimulating factor and relationship to promotion of angiogenesis after spinal cord injury in rats: Laboratory investigation. J. Neurosurg. Spine 2011, 15, 414–421. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosales-Antequera, C.; Viscor, G.; Araneda, O.F. Inflammation and Oxidative Stress as Common Mechanisms of Pulmonary, Autonomic and Musculoskeletal Dysfunction after Spinal Cord Injury. Biology 2022, 11, 550. https://doi.org/10.3390/biology11040550
Rosales-Antequera C, Viscor G, Araneda OF. Inflammation and Oxidative Stress as Common Mechanisms of Pulmonary, Autonomic and Musculoskeletal Dysfunction after Spinal Cord Injury. Biology. 2022; 11(4):550. https://doi.org/10.3390/biology11040550
Chicago/Turabian StyleRosales-Antequera, Cristián, Ginés Viscor, and Oscar F. Araneda. 2022. "Inflammation and Oxidative Stress as Common Mechanisms of Pulmonary, Autonomic and Musculoskeletal Dysfunction after Spinal Cord Injury" Biology 11, no. 4: 550. https://doi.org/10.3390/biology11040550